Epigenetic Regulation of p21cip1/waf1 in Human Cancer

 , and
, and
Abstract
1. Introduction
2. Epigenetic Mechanisms
2.1. Epigenetic Writers
2.2. Epigenetic Erasers
2.3. Epigenetic Readers
3. Epigenetic Regulation of p21cip1/waf1
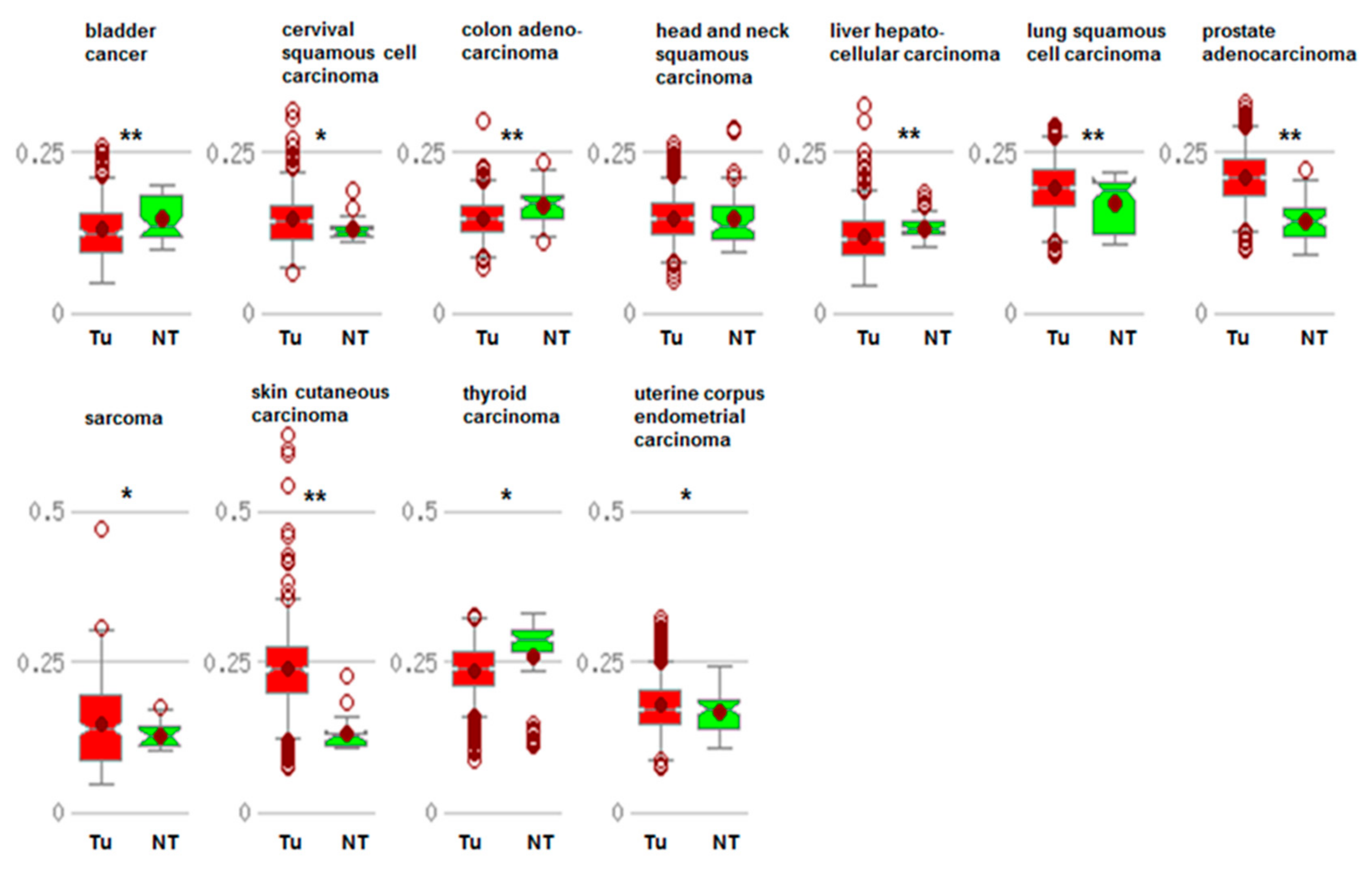
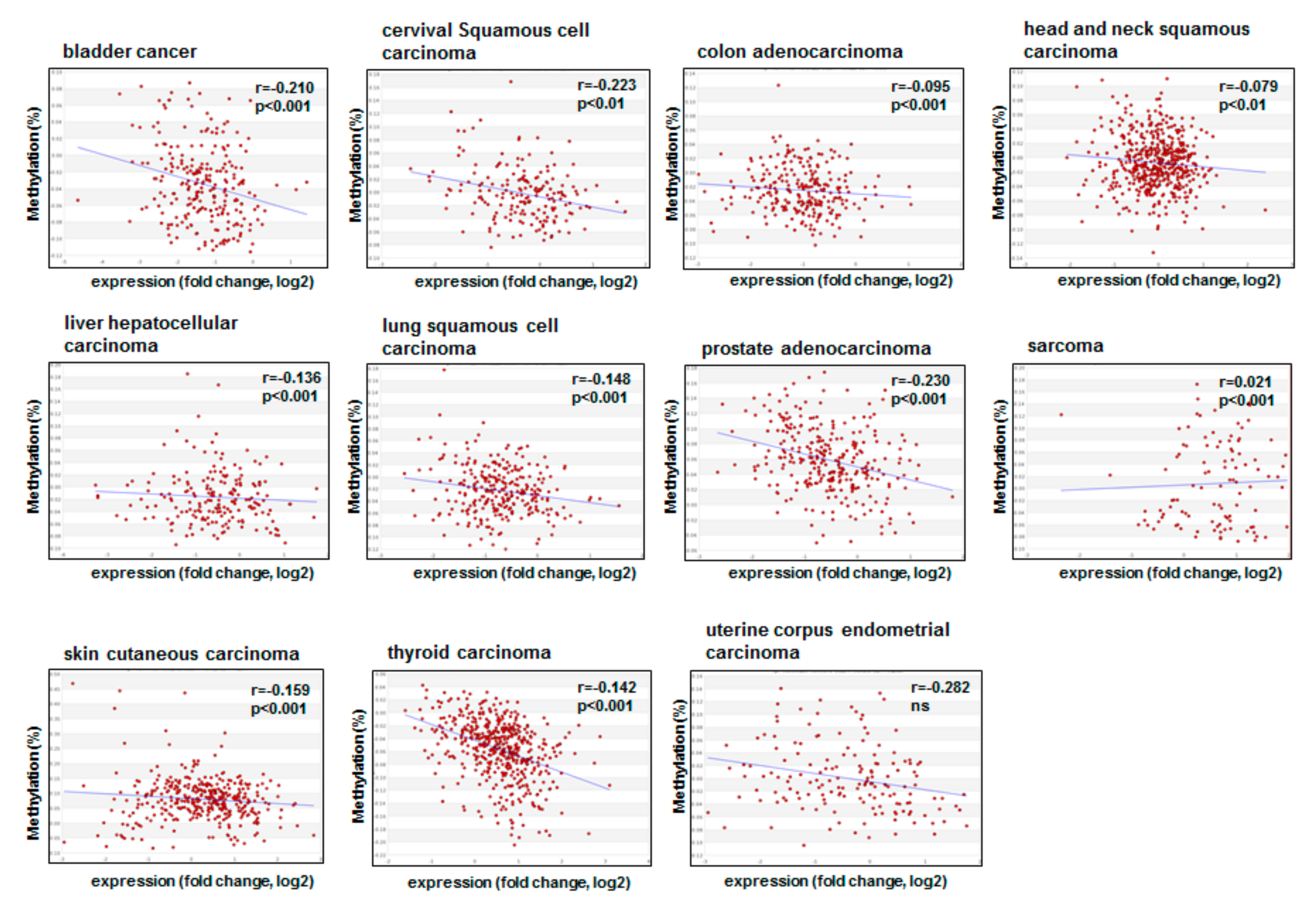
3.1. Promoter Methylation of CDKN1A
3.2. Writers and p21cip1/waf1
3.3. Erasers and p21cip1/waf1
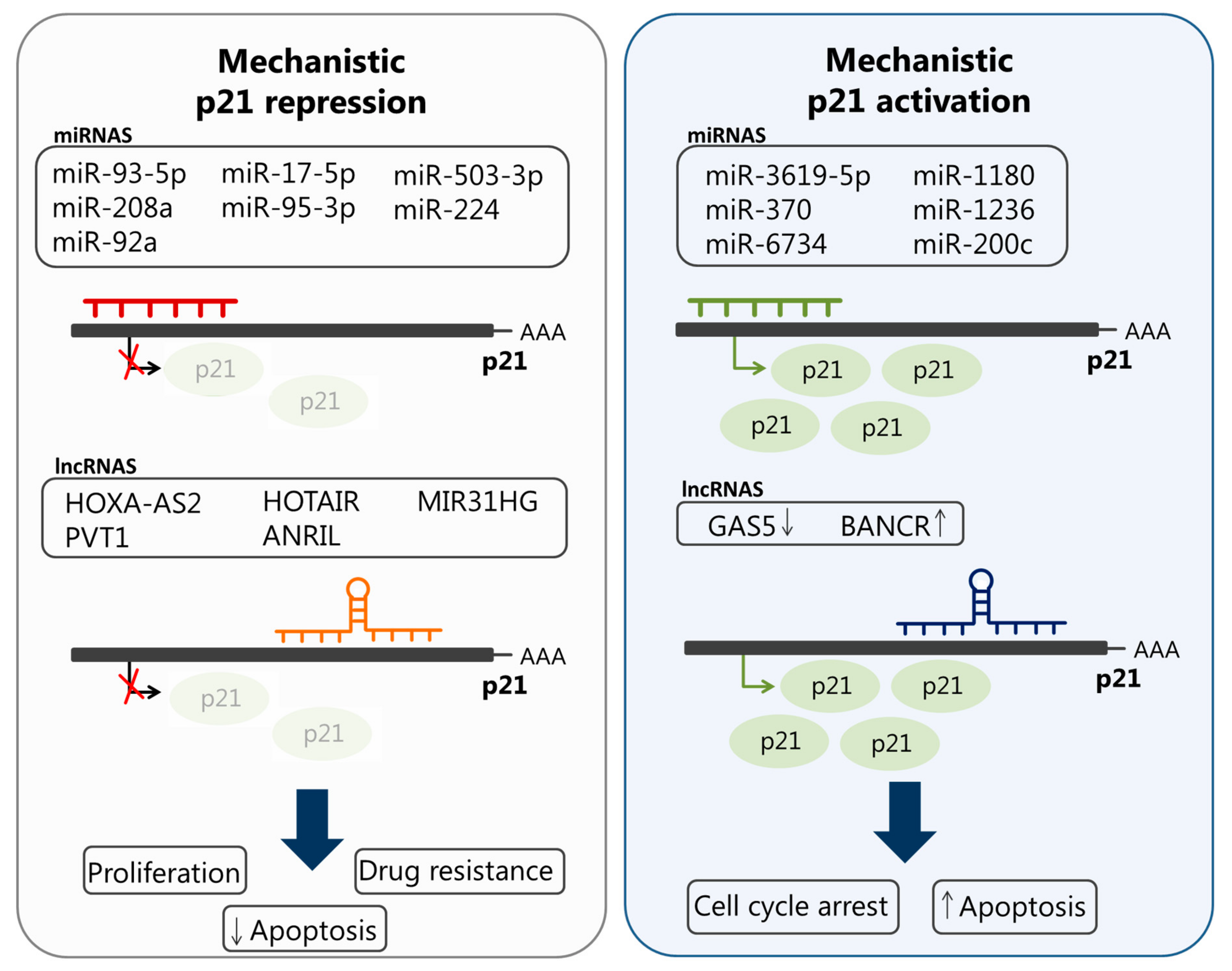
4. The Role of lncRNAs and miRNAs in the Deregulation of p21cip1/waf1 in Cancer

{kind=link}
{kind=link}
{kind=link}
| miRNA/LncRNA | Cancer Type | Expression Levels | Biological Functions | Ref. |
|---|---|---|---|---|
| HOXA cluster antisense RNA2 (HOXA-AS2) | GC | + | Epigenetically silences p21 transcription and promotes cancer cell growth | [102] |
| Plasmacytoma variant translocation 1 (PVT1) | PDAC | + | Promotes cancer cell proliferation and migration, and epithelial-to-mesenchymal transition (EMT), by downregulating p21 | [120] |
| HOX Antisense Intergenic RNA (HOTAIR) | Cervix | + | Promotes radio-resistance by silencing p21 expression | [121] |
| antisense noncoding RNA in the INK4 locus (ANRIL) | NSCLC | + | Promotes cancer cell proliferation and inhibits apoptosis by silencing p21 expression | [122] |
| LnCRNA MIR31HG | HNSCC | + | Promotes cell cycle progression and proliferation by targeting p21 | [123] |
| BRAF-activated noncoding RNA (BANCR) | CRC | + | Interacts with p21 and increases its expression to induce cell cycle arrest and apoptosis | [124] |
| miR-93-5p | Lung | + in plasma and tissue samples obtained from lung cancer patients | Suppression of senescence through reducing the expression of p21 | [130] |
| miR-208a | Lung | + in serum of lung cancer patients who were subjected to radiation therapy | Reduces the expression of p21 to promote cell proliferation and radio-resistance | [134] |
| miR-92a | Cervix | + in human cervical tissues and Hela cells | Promotes cell cycle progression and cell proliferation via inhibiting p21 expression | [135] |
| miR-17-5p | Multi-drug resistant human GC | + | Promotes drug resistance partially through downregulating p21 | [136] |
| miR-95-3p | OS, HCC | + in the serum of OS patients and in an HCC cell line | Promotes cell proliferation and growth by targeting p21 | [137,138] |
| miR-503-3p | Lung cancer | + | Induces apoptosis of lung cancer cells by downregulating p21 and CDK4 expression | [139] |
| miR-224 | Human lung adenocarcinoma | + in cisplatin (DDP; cis-diamminedichloroplatinum II)-resistant A549 cells | Promotes chemo-resistance through targeting p21 and modulating G1/S transition and apoptosis | [140] |
| miR-3619-5p | BC | − | Activates p21 expression by targeting its putative promotor. Reduced expression of this miRNA and p21 is associated with cancer progression and poor survival of patients with BC | [141] |
| miR-370, miR-1180, and miR-1236 | BC | – in BC tissues | Activates p21 and inhibits cancer | [142] |
| miR-6734 | CC | − | Modifies histones in the p21 promoter and induces cell cycle arrest in colon cancer cells | [143] |
| miR-200c | ESCC | − | Mediates sub-G1 and G2/M phase arrest and, consequently, enhances radio-resistance of esophageal squamous cancer cells through modulating the expression levels of several cell cycle regulators, including p21 | [144] |
| lncRNA growth arrest specific transcript 5 (GAS5) | GC | − | Increases p21 expression and enhances cell cycle arrest at the G1 phase | [145] |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Ocker, M.; Schneider-Stock, R. Histone deacetylase inhibitors: Signalling towards p21cip1/waf1. Int. J. Biochem. Cell Biol. 2007, 39, 1367–1374. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The Roles of Human DNA Methyltransferases and Their Isoforms in Shaping the Epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.F.; Tai, K.Y.; Chen, W.S.; Cheng, L.C.; Ho, H.N.; Lin, S.P. Functions of DNA methyltransferase 3-like in germ cells and beyond. Biol. Cell 2012, 104, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, S.; Jurkowski, T.P.; Kellner, S.; Schneider, D.; Jeltsch, A.; Helm, M. The RNA methyltransferase Dnmt2 methylates DNA in the structural context of a tRNA. RNA Biol. 2017, 14, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Jiemjit, A.; Fandy, T.E.; Carraway, H.; Bailey, K.A.; Baylin, S.; Herman, J.G.; Gore, S.D. p21(WAF1/CIP1) induction by 5-azacytosine nucleosides requires DNA damage. Oncogene 2008, 27, 3615–3623. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Subbaiah, V.K.; Zhang, Y.; Rajagopalan, D.; Abdullah, L.N.; Yeo-Teh, N.S.; Tomaic, V.; Banks, L.; Myers, M.P.; Chow, E.K.; Jha, S. E3 ligase EDD1/UBR5 is utilized by the HPV E6 oncogene to destabilize tumor suppressor TIP60. Oncogene 2016, 35, 2062–2074. [Google Scholar] [CrossRef]
- Mattera, L.; Escaffit, F.; Pillaire, M.J.; Selves, J.; Tyteca, S.; Hoffmann, J.S.; Gourraud, P.A.; Chevillard-Briet, M.; Cazaux, C.; Trouche, D. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene 2009, 28, 1506–1517. [Google Scholar] [CrossRef]
- Lu, Q.; Kamps, M.P. Heterodimerization of Hox proteins with Pbx1 and oncoprotein E2a-Pbx1 generates unique DNA-binding specifities at nucleotides predicted to contact the N-terminal arm of the Hox homeodomain—Demonstration of Hox-dependent targeting of E2a-Pbx1 in vivo. Oncogene 1997, 14, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ernst, P. Distinct functions of histone H3, lysine 4 methyltransferases in normal and malignant hematopoiesis. Curr. Opin. Hematol. 2017, 24, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Blazer, L.; Eram, M.S.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; Hajian, T. Targeting human SET1/MLL family of proteins. Protein Sci. 2017, 26, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.; Robert, F.; Young, R.A.; Struhl, K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell 2003, 11, 709–719. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Guo, J.; Dai, X.; Laurent, B.; Zheng, N.; Gan, W.; Zhang, J.; Guo, A.; Yuan, M.; Liu, P.; Asara, J.M.; et al. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat. Cell Biol. 2019, 21, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef]
- Xu, B.; Konze, K.D.; Jin, J.; Wang, G.G. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 2015, 43, 698–712. [Google Scholar] [CrossRef]
- Emran, A.A.; Chatterjee, A.; Rodger, E.J.; Tiffen, J.C.; Gallagher, S.J.; Eccles, M.R.; Hersey, P. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol. 2019, 40, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003, 17, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, Y. 5-Hydroxymethylcytosine: Generation, fate, and genomic distribution. Curr. Opin. Cell Biol. 2013, 25, 289–296. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011, 25, 2436–2452. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef]
- Li, W.; Xu, L. Epigenetic Function of TET Family, 5-Methylcytosine, and 5-Hydroxymethylcytosine in Hematologic Malignancies. Oncol. Res. Treat. 2019, 42, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Misawa, K.; Imai, A.; Mochizuki, D.; Mima, M.; Endo, S.; Misawa, Y.; Kanazawa, T.; Mineta, H. Association of TET3 epigenetic inactivation with head and neck cancer. Oncotarget 2018, 9, 24480–24493. [Google Scholar] [CrossRef] [PubMed]
- Bronowicka-Klys, D.E.; Roszak, A.; Pawlik, P.; Sajdak, S.; Sowinska, A.; Jagodzinski, P.P. Transcript levels of ten-eleven translocation type 1-3 in cervical cancer and non-cancerous cervical tissues. Oncol. Lett. 2017, 13, 3921–3927. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, T.B.; Veland, N.; Chen, T. Writers, Readers, and Erasers of Epigenetic Marks. In Epigenetic Cancer Therapy; Gray, S.G., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 31–66. [Google Scholar]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.H.; Perlin, J.R.; Leonelli, L.; Sonbuchner, L.S.; McDonald, C.H.; Cook, R.G.; Dou, Y.; et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.; O’Shea, M.; Hume, D.A.; Lengeling, A. Jmjd6, a JmjC Dioxygenase with Many Interaction Partners and Pleiotropic Functions. Front. Genet. 2017, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Gregoire, S. Class II histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell Biol. 2005, 25, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M. Deacetylase inhibitors—Focus on non-histone targets and effects. World J. Biol. Chem. 2010, 1, 55–61. [Google Scholar] [CrossRef]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef]
- Hayakawa, T.; Nakayama, J. Physiological roles of class I HDAC complex and histone demethylase. J. Biomed. Biotechnol. 2011, 2011, 129383. [Google Scholar] [CrossRef] [PubMed]
- Nemati, M.; Ajami, N.; Estiar, M.A.; Rezapour, S.; Gavgani, R.R.; Hashemzadeh, S.; Kafil, H.S.; Sakhinia, E. Deregulated expression of HDAC3 in colorectal cancer and its clinical significance. Adv. Clin. Exp. Med. 2018, 27, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Yasuda, M.; Sakaki, M.; Nagata, K.; Fujino, T.; Arai, E.; Hasebe, T.; Miyazawa, M.; Ogane, N.; Hasegawa, K.; et al. Association of histone deacetylase expression with histology and prognosis of ovarian cancer. Oncol. Lett 2018, 15, 3524–3531. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Min, S.K.; Park, H.R.; Kim, D.H.; Kwon, M.J.; Kim, L.S.; Ju, Y.S. Expression of Histone Deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in Invasive Ductal Carcinomas of the Breast. J. Breast Cancer 2014, 17, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Quint, K.; Agaimy, A.; Di Fazio, P.; Montalbano, R.; Steindorf, C.; Jung, R.; Hellerbrand, C.; Hartmann, A.; Sitter, H.; Neureiter, D.; et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011, 459, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Tallant, C.; Valentini, E.; Fedorov, O.; Overvoorde, L.; Ferguson, F.M.; Filippakopoulos, P.; Svergun, D.I.; Knapp, S.; Ciulli, A. Molecular basis of histone tail recognition by human TIP5 PHD finger and bromodomain of the chromatin remodeling complex NoRC. Structure 2015, 23, 80–92. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, H.; Guo, X.; Rong, N.; Song, Y.; Xu, Y.; Lan, W.; Zhang, X.; Liu, M.; Cao, C. The PHD1 finger of KDM5B recognizes unmodified H3K4 during the demethylation of histone H3K4me2/3 by KDM5B. Protein Cell 2014, 5, 837–850. [Google Scholar] [CrossRef]
- Couture, J.F.; Collazo, E.; Trievel, R.C. Molecular recognition of histone H3 by the WD40 protein WDR5. Nat. Struct. Mol. Biol. 2006, 13, 698–703. [Google Scholar] [CrossRef]
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W.; et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Berkyurek, A.C.; Suetake, I.; Arita, K.; Takeshita, K.; Nakagawa, A.; Shirakawa, M.; Tajima, S. The DNA methyltransferase Dnmt1 directly interacts with the SET and RING finger-associated (SRA) domain of the multifunctional protein Uhrf1 to facilitate accession of the catalytic center to hemi-methylated DNA. J. Biol. Chem. 2014, 289, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, M.; Ficz, G.; Oxley, D.; Raiber, E.A.; Bachman, M.; Booth, M.J.; Andrews, S.; Balasubramanian, S.; Reik, W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013, 14, R119. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef]
- Musselman, C.A.; Lalonde, M.E.; Cote, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Otani, J.; Nankumo, T.; Arita, K.; Inamoto, S.; Ariyoshi, M.; Shirakawa, M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009, 10, 1235–1241. [Google Scholar] [CrossRef]
- Rajakumara, E.; Wang, Z.; Ma, H.; Hu, L.; Chen, H.; Lin, Y.; Guo, R.; Wu, F.; Li, H.; Lan, F.; et al. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol. Cell 2011, 43, 275–284. [Google Scholar] [CrossRef]
- Dialynas, G.K.; Vitalini, M.W.; Wallrath, L.L. Linking Heterochromatin Protein 1 (HP1) to cancer progression. Mutat. Res. 2008, 647, 13–20. [Google Scholar] [CrossRef]
- Coles, A.H.; Jones, S.N. The ING gene family in the regulation of cell growth and tumorigenesis. J. Cell Physiol. 2009, 218, 45–57. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Barton, M.C. Bromodomain Histone Readers and Cancer. J. Mol. Biol. 2017, 429, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- French, C.A. NUT Carcinoma: Clinicopathologic features, pathogenesis, and treatment. Pathol. Int. 2018, 68, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Postel-Vinay, S.; Herbschleb, K.; Massard, C.; Woodcock, V.; Soria, J.C.; Walter, A.O.; Ewerton, F.; Poelman, M.; Benson, N.; Ocker, M.; et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: Emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur. J. Cancer 2019, 109, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Gelato, K.A.; Fernandez-Montalvan, A.; Siegel, S.; Haendler, B. Targeting BET bromodomains for cancer treatment. Epigenomics 2015, 7, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026534. [Google Scholar] [CrossRef]
- Fang, J.Y.; Lu, Y.Y. Effects of histone acetylation and DNA methylation on p21( WAF1) regulation. World J. Gastroenterol. 2002, 8, 400–405. [Google Scholar] [CrossRef]
- Chatterjee, B.; Ghosh, K.; Kanade, S.R. Curcumin-mediated demethylation of the proximal promoter CpG island enhances the KLF4 recruitment that leads to increased expression of p21Cip1 in vitro. J. Cell Biochem. 2019, 120, 809–820. [Google Scholar] [CrossRef]
- Zhu, W.G.; Srinivasan, K.; Dai, Z.; Duan, W.; Druhan, L.J.; Ding, H.; Yee, L.; Villalona-Calero, M.A.; Plass, C.; Otterson, G.A. Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter. Mol. Cell Biol. 2003, 23, 4056–4065. [Google Scholar] [CrossRef] [PubMed]
- Koutsodontis, G.; Moustakas, A.; Kardassis, D. The role of Sp1 family members, the proximal GC-rich motifs, and the upstream enhancer region in the regulation of the human cell cycle inhibitor p21WAF-1/Cip1 gene promoter. Biochemistry 2002, 41, 12771–12784. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Li, J.; Liu, Q.; Liu, C.; Li, C.; Song, G.; Zhu, H.; Gao, H.; Zhang, Y. P21(Waf1/Cip1) and p27(Kip1) are correlated with the development and invasion of prolactinoma. J. Neurooncol. 2018, 136, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Askari, M.; Sobti, R.C.; Nikbakht, M.; Sharma, S.C. Aberrant promoter hypermethylation of p21 (WAF1/CIP1) gene and its impact on expression and role of polymorphism in the risk of breast cancer. Mol. Cell Biochem. 2013, 382, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Castillejo, J.A.; Jimenez, A.; Gonzalez, M.G.; Moreno, F.; Rodriguez Mdel, C.; Barrios, M.; Maldonado, J.; Torres, A. 5′ CpG island hypermethylation is associated with transcriptional silencing of the p21(CIP1/WAF1/SDI1) gene and confers poor prognosis in acute lymphoblastic leukemia. Blood 2002, 99, 2291–2296. [Google Scholar] [CrossRef] [PubMed]
- Pinyol, M.; Hernandez, L.; Cazorla, M.; Balbin, M.; Jares, P.; Fernandez, P.L.; Montserrat, E.; Cardesa, A.; Lopez-Otin, C.; Campo, E. Deletions and loss of expression of p16INK4a and p21Waf1 genes are associated with aggressive variants of mantle cell lymphomas. Blood 1997, 89, 272–280. [Google Scholar] [PubMed]
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin. Exp. Med. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Hayashi, A.; Suenaga, N.; Shiomi, Y.; Nishitani, H. PCNA-dependent ubiquitination of Cdt1 and p21 in mammalian cells. Methods Mol. Biol. 2014, 1170, 367–382. [Google Scholar]
- Bloom, J.; Amador, V.; Bartolini, F.; DeMartino, G.; Pagano, M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell 2003, 115, 71–82. [Google Scholar] [CrossRef]
- Sesselmann, S.; Soder, S.; Voigt, R.; Haag, J.; Grogan, S.P.; Aigner, T. DNA methylation is not responsible for p21WAF1/CIP1 down-regulation in osteoarthritic chondrocytes. Osteoarthr. Cartil. 2009, 17, 507–512. [Google Scholar] [CrossRef]
- Love, I.M.; Sekaric, P.; Shi, D.; Grossman, S.R.; Androphy, E.J. The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle 2012, 11, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Hogarth, L.A.; Dietrich, P.A.; Bachmann, P.S.; Mackenzie, K.L.; Hall, A.G.; Lock, R.B. p53-independent epigenetic repression of the p21(WAF1) gene in T-cell acute lymphoblastic leukemia. J. Biol. Chem. 2011, 286, 37639–37650. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Seo, J.; Choi, D.Y.; Lee, E.W.; Ko, A.; Ha, N.C.; Yoon, J.B.; Lee, H.W.; Kim, K.P.; Song, J. Stabilization of p21 (Cip1/WAF1) following Tip60-dependent acetylation is required for p21-mediated DNA damage response. Cell Death Differ. 2013, 20, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, F.; Chang, C.K.; He, Q.; Wu, L.Y.; Zhang, Z.; Li, X. MYCN contributes to the malignant characteristics of erythroleukemia through EZH2-mediated epigenetic repression of p21. Cell Death Dis. 2017, 8, e3126. [Google Scholar] [CrossRef] [PubMed]
- Zang, C.; Nie, F.Q.; Wang, Q.; Sun, M.; Li, W.; He, J.; Zhang, M.; Lu, K.H. Long non-coding RNA LINC01133 represses KLF2, P21 and E-cadherin transcription through binding with EZH2, LSD1 in non small cell lung cancer. Oncotarget 2016, 7, 11696–11707. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, R.; Park, G.; Park, J.W.; Kim, J.E. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J. Biol. Chem. 2012, 287, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Rios, D.; Graca, I.; Vieira, F.Q.; Ramalho-Carvalho, J.; Pereira-Silva, E.; Martins, A.T.; Oliveira, J.; Goncalves, C.S.; Costa, B.M.; Henrique, R.; et al. Histone methyltransferase PRMT6 plays an oncogenic role of in prostate cancer. Oncotarget 2016, 7, 53018–53028. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Liu, M.; Nie, M.; Deng, Y.; Yao, B.; Gui, T.; Li, X.; Ma, L.; Guo, C.; et al. Suv4-20h1 promotes G1 to S phase transition by downregulating p21(WAF1/CIP1) expression in chronic myeloid leukemia K562 cells. Oncol. Lett. 2018, 15, 6123–6130. [Google Scholar]
- Bellucci, L.; Dalvai, M.; Kocanova, S.; Moutahir, F.; Bystricky, K. Activation of p21 by HDAC inhibitors requires acetylation of H2A.Z. PLoS ONE 2013, 8, e54102. [Google Scholar] [CrossRef]
- Gevry, N.; Chan, H.M.; Laflamme, L.; Livingston, D.M.; Gaudreau, L. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 2007, 21, 1869–1881. [Google Scholar] [CrossRef]
- Tajima, K.; Matsuda, S.; Yae, T.; Drapkin, B.J.; Morris, R.; Boukhali, M.; Niederhoffer, K.; Comaills, V.; Dubash, T.; Nieman, L.; et al. SETD1A protects from senescence through regulation of the mitotic gene expression program. Nat. Commun. 2019, 10, 2854. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Tian, C.; An, L.; Nie, J.; Lu, K.; Xing, G.; Zhang, L.; He, F. Histone methyltransferase protein SETD2 interacts with p53 and selectively regulates its downstream genes. Cell Signal. 2008, 20, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Majello, B.; Gorini, F.; Sacca, C.D.; Amente, S. Expanding the Role of the Histone Lysine-Specific Demethylase LSD1 in Cancer. Cancers 2019, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.H.; Lim, S.; Schramm, A.; Friedrichs, N.; Koster, J.; Versteeg, R.; Ora, I.; Pajtler, K.; Klein-Hitpass, L.; Kuhfittig-Kulle, S.; et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: Implications for therapy. Cancer Res. 2009, 69, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, J.; Kong, W.; Huang, J.; Dong, B.; Huang, Y.; Xue, W.; Zhang, J. LSD1 inhibition suppresses the growth of clear cell renal cell carcinoma via upregulating P21 signaling. Acta Pharm. Sin. B 2019, 9, 324–334. [Google Scholar] [CrossRef]
- Nicholson, T.B.; Chen, T. LSD1 demethylates histone and non-histone proteins. Epigenetics 2009, 4, 129–132. [Google Scholar] [CrossRef]
- Xie, M.; Sun, M.; Zhu, Y.N.; Xia, R.; Liu, Y.W.; Ding, J.; Ma, H.W.; He, X.Z.; Zhang, Z.H.; Liu, Z.J.; et al. Long noncoding RNA HOXA-AS2 promotes gastric cancer proliferation by epigenetically silencing P21/PLK3/DDIT3 expression. Oncotarget 2015, 6, 33587–33601. [Google Scholar] [CrossRef]
- Battaglia, S.; Karasik, E.; Gillard, B.; Williams, J.; Winchester, T.; Moser, M.T.; Smiraglia, D.J.; Foster, B.A. LSD1 dual function in mediating epigenetic corruption of the vitamin D signaling in prostate cancer. Clin. Epigenet. 2017, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Urabe, M.; Gomi, A.; Nagai, M.; Yamaguchi, T.; Tsukahara, T.; Mizukami, H.; Kume, A.; Ozawa, K.; Watanabe, E. An R132H mutation in isocitrate dehydrogenase 1 enhances p21 expression and inhibits phosphorylation of retinoblastoma protein in glioma cells. Neurol. Med. Chir. 2013, 53, 645–654. [Google Scholar] [CrossRef][Green Version]
- Mateen, S.; Raina, K.; Jain, A.K.; Agarwal, C.; Chan, D.; Agarwal, R. Epigenetic modifications and p21-cyclin B1 nexus in anticancer effect of histone deacetylase inhibitors in combination with silibinin on non-small cell lung cancer cells. Epigenetics 2012, 7, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Han, D.; Sun, X.; Xie, Y.; Wu, Q.; Fu, C.; Yao, Y.; Li, H.; Li, Z.; Xu, K. Scriptaid inhibits cell survival, cell cycle, and promotes apoptosis in multiple myeloma via epigenetic regulation of p21. Exp. Hematol. 2018, 60, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Xiao, M.T.; Liu, L.X.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Goldman, S.L.; Hassan, C.; Khunte, M.; Soldatenko, A.; Jong, Y.; Afshinnekoo, E.; Mason, C.E. Epigenetic Modifications in Acute Myeloid Leukemia: Prognosis, Treatment, and Heterogeneity. Front. Genet. 2019, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Semin. Cancer Biol. 2018. [Google Scholar] [CrossRef]
- O’Rourke, C.J.; Munoz-Garrido, P.; Aguayo, E.L.; Andersen, J.B. Epigenome dysregulation in cholangiocarcinoma. Biochim. Biophys. Acta. Mol. Basis. Dis. 2018, 1864, 1423–1434. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martinez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Shenoy, N.; Bhagat, T.; Nieves, E.; Stenson, M.; Lawson, J.; Choudhary, G.S.; Habermann, T.; Nowakowski, G.; Singh, R.; Wu, X.; et al. Upregulation of TET activity with ascorbic acid induces epigenetic modulation of lymphoma cells. Blood Cancer J. 2017, 7, e587. [Google Scholar] [CrossRef]
- Lin, J.R.; Qin, H.H.; Wu, W.Y.; He, S.J.; Xu, J.H. Vitamin C protects against UV irradiation-induced apoptosis through reactivating silenced tumor suppressor genes p21 and p16 in a Tet-dependent DNA demethylation manner in human skin cancer cells. Cancer Biother. Radiopharm. 2014, 29, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Halaburkova, A.; Jendzelovsky, R.; Koval, J.; Herceg, Z.; Fedorocko, P.; Ghantous, A. Histone deacetylase inhibitors potentiate photodynamic therapy in colon cancer cells marked by chromatin-mediated epigenetic regulation of CDKN1A. Clin. Epigenet. 2017, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Wakimoto, N.; Yin, D.; Gery, S.; Kawamata, N.; Takai, N.; Komatsu, N.; Chumakov, A.; Imai, Y.; Koeffler, H.P. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int. J. Cancer 2007, 121, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, C.; Sui, X.; Cao, S.; Tang, F.; Sun, S.; Wang, S.; Chen, B. Histone deacetylase inhibitor chidamide induces growth inhibition and apoptosis in NK/T lymphoma cells through ATM-Chk2-p53-p21 signalling pathway. Invest. New Drugs 2018, 36, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kwa, F.A.A.; Cole-Sinclair, M.F.; Kapuscinski, M.K. Combination Treatment of p53-Null HL-60 cells with Histone Deacetylase Inhibitors and Chlorambucil Augments Apoptosis and Increases BCL6 and p21 Gene Expression. Curr. Mol. Pharmacol. 2019, 12, 72–81. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Dai, L.; Zhang, X.; Chen, D.; Wu, J.; Feng, X.; Zhang, Y.; Xie, H.; Zhou, L.; Zheng, S. The HDAC Inhibitor Quisinostat (JNJ-26481585) Supresses Hepatocellular Carcinoma alone and Synergistically in Combination with Sorafenib by G0/G1 phase arrest and Apoptosis induction. Int. J. Biol. Sci. 2018, 14, 1845–1858. [Google Scholar] [CrossRef]
- Natarajan, U.; Venkatesan, T.; Radhakrishnan, V.; Samuel, S.; Rathinavelu, A. Differential Mechanisms of Cell Death Induced by HDAC Inhibitor SAHA and MDM2 Inhibitor RG7388 in MCF-7 Cells. Cells 2018, 8, 8. [Google Scholar] [CrossRef]
- Wu, B.Q.; Jiang, Y.; Zhu, F.; Sun, D.L.; He, X.Z. Long Noncoding RNA PVT1 Promotes EMT and Cell Proliferation and Migration Through Downregulating p21 in Pancreatic Cancer Cells. Technol. Cancer Res. Treat. 2017, 1533034617700559. [Google Scholar] [CrossRef]
- Jing, L.; Yuan, W.; Ruofan, D.; Jinjin, Y.; Haifeng, Q. HOTAIR enhanced aggressive biological behaviors and induced radio-resistance via inhibiting p21 in cervical cancer. Tumour Biol. 2015, 36, 3611–3619. [Google Scholar] [CrossRef]
- Nie, F.Q.; Sun, M.; Yang, J.S.; Xie, M.; Xu, T.P.; Xia, R.; Liu, Y.W.; Liu, X.H.; Zhang, E.B.; Lu, K.H.; et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol. Cancer Ther. 2015, 14, 268–277. [Google Scholar] [CrossRef]
- Wang, R.; Ma, Z.; Feng, L.; Yang, Y.; Tan, C.; Shi, Q.; Lian, M.; He, S.; Ma, H.; Fang, J. LncRNA MIR31HG targets HIF1A and P21 to facilitate head and neck cancer cell proliferation and tumorigenesis by promoting cell-cycle progression. Mol. Cancer 2018, 17, 162. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, Y.; Wang, J.; Jie, D.; Yun, T.; Li, W.; Yan, L.; Wang, K.; Feng, J. Downregulated Long Noncoding RNA BANCR Promotes the Proliferation of Colorectal Cancer Cells via Downregualtion of p21 Expression. PLoS ONE 2015, 10, e0122679. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W. Gene regulation by microRNAs. Curr. Opin. Genet. Dev. 2006, 16, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Huang, V.; Place, R.F.; Portnoy, V.; Wang, J.; Qi, Z.; Jia, Z.; Yu, A.; Shuman, M.; Yu, J.; Li, L.C. Upregulation of Cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res. 2012, 40, 1695–1707. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, L.; Wang, Y.; Liu, N.; You, Y. Regulation of let-7 and its target oncogenes (Review). Oncol. Lett. 2012, 3, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Shin, H.J.; Bae, I.H. miR-93-5p suppresses cellular senescence by directly targeting Bcl-w and p21. Biochem. Biophys. Res. Commun. 2018, 505, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, J.S.; Tang, X.; Tucker, L.D.; Quesenberry, P.; Rigoutsos, I.; Ramratnam, B. MicroRNA 125a and its regulation of the p53 tumor suppressor gene. FEBS Lett. 2009, 583, 3725–3730. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef]
- Jung, Y.S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Cui, Y.; Li, Z.; Jiao, Z.; Zhang, Y.; He, Y.; Chen, G.; Zhou, Q.; Wang, W.; Zhou, X.; et al. Radiation-induced miR-208a increases the proliferation and radioresistance by targeting p21 in human lung cancer cells. J. Exp. Clin. Cancer Res. 2016, 35, 7. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, H.; Zhao, M.; Wang, Y.; Deng, G.; Chen, R. MicroRNA-92a Promotes Cell Proliferation in Cervical Cancer via Inhibiting p21 Expression and Promoting Cell Cycle Progression. Oncol. Res. 2017, 25, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ji, F. Downregulation of microRNA-17-5p inhibits drug resistance of gastric cancer cells partially through targeting p21. Oncol. Lett. 2018, 15, 4585–4591. [Google Scholar] [CrossRef] [PubMed]



| Writers | Epigenetic Mark | Experimental Conditions | p21cip1/waf1 Regulation | Ref. |
|---|---|---|---|---|
| DNMTs | 5mC | Lung cancer cells | 5mC affects Sp1 and Sp3 binding activity | [82] |
| TIP60 | H2/H4ac | Decreased in colon and breast cancer | Acetylates the damage sensor ATM or the gate keeper p53 | [17,18] |
| PCAF | H3K9/H3K14 | Osteosarcoma and NSCLC cells | Expression ↓ | [83] |
| G9a-GLP | H3K9me | Cervical cancer and acute promyelocytic leukemia cells | Recruited by Gfi1, transcription ↓ | [84] |
| CTIP2 | H3K9me3 | Microglial cells | Induces transcriptional repression of p21 | [85] |
| EZH2 | H3K27me3 | Erythroleukemia and NSCLC cells | Induces transcriptional repression of p21 | [86,87] |
| DOT1L | H3K79 | NSCLC | DOT1L inhibition reduces H3K79me and increases p21 expression | [88] |
| PRMT6 | H3R2me2 | Prostate cancer | PRMT6 silencing leads to increased p21 expression | [89] |
| Suv4-20h1 | H4K20me | Myeloid leukemia cells | Induces transcriptional repression of p21 | [90] |
| H2A.Z | Osteosarcoma cells | Reduces binding of TIP60 to p21 promotor and activates p21 transcription | [91,92] |
| Erasers | Epigenetic Mark | Experimental Conditions | p21cip1/waf1 Regulation | Ref. |
|---|---|---|---|---|
| TET | 5hmC | Glioma cells | Expression ↑ | [104] |
| KDM1A/LSD1 | H3K4/H3K9me | Renal cells carcinoma and glioma | Regulates p21cip1/waf1 via demethylation of histone and non-histone targets | [100,101] |
| HDAC | Histone deacetylation | Multiple myeloma and NSCLC | Expression ↑ | [105,106] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ocker, M.; Al Bitar, S.; Monteiro, A.C.; Gali-Muhtasib, H.; Schneider-Stock, R. Epigenetic Regulation of p21cip1/waf1 in Human Cancer. Cancers 2019, 11, 1343. https://doi.org/10.3390/cancers11091343
Ocker M, Al Bitar S, Monteiro AC, Gali-Muhtasib H, Schneider-Stock R. Epigenetic Regulation of p21cip1/waf1 in Human Cancer. Cancers. 2019; 11(9):1343. https://doi.org/10.3390/cancers11091343
Chicago/Turabian StyleOcker, Matthias, Samar Al Bitar, Ana Carolina Monteiro, Hala Gali-Muhtasib, and Regine Schneider-Stock. 2019. "Epigenetic Regulation of p21cip1/waf1 in Human Cancer" Cancers 11, no. 9: 1343. https://doi.org/10.3390/cancers11091343
APA StyleOcker, M., Al Bitar, S., Monteiro, A. C., Gali-Muhtasib, H., & Schneider-Stock, R. (2019). Epigenetic Regulation of p21cip1/waf1 in Human Cancer. Cancers, 11(9), 1343. https://doi.org/10.3390/cancers11091343