The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics


{kind=link}
{kind=link}
Abstract
:1. Introduction
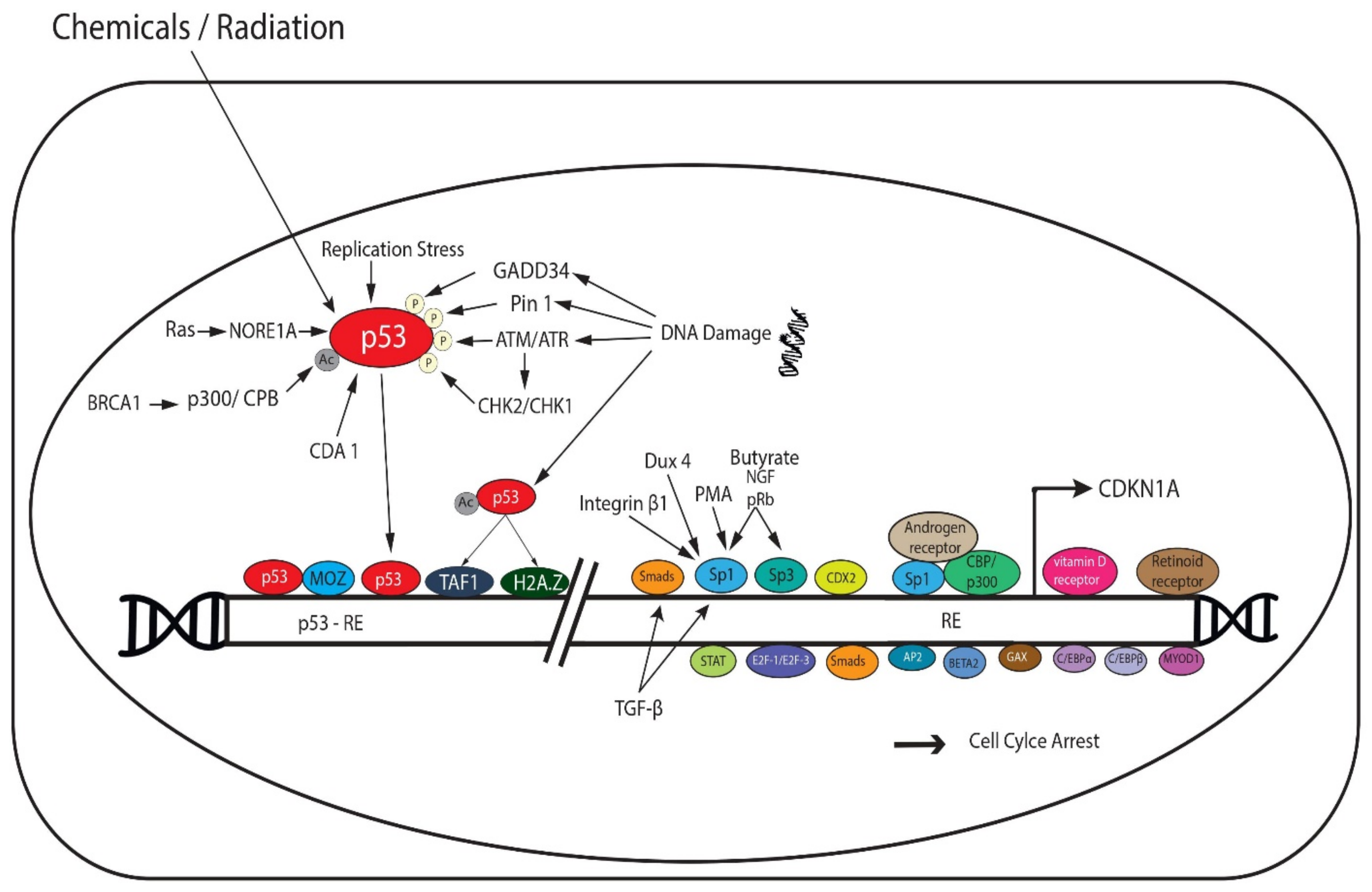
2. p53-Dependent and Independent Induction of p21
2.1. p53-Dependent Transcriptional Regulation of p21
2.2. p53-Independent Transcriptional Regulation of p21
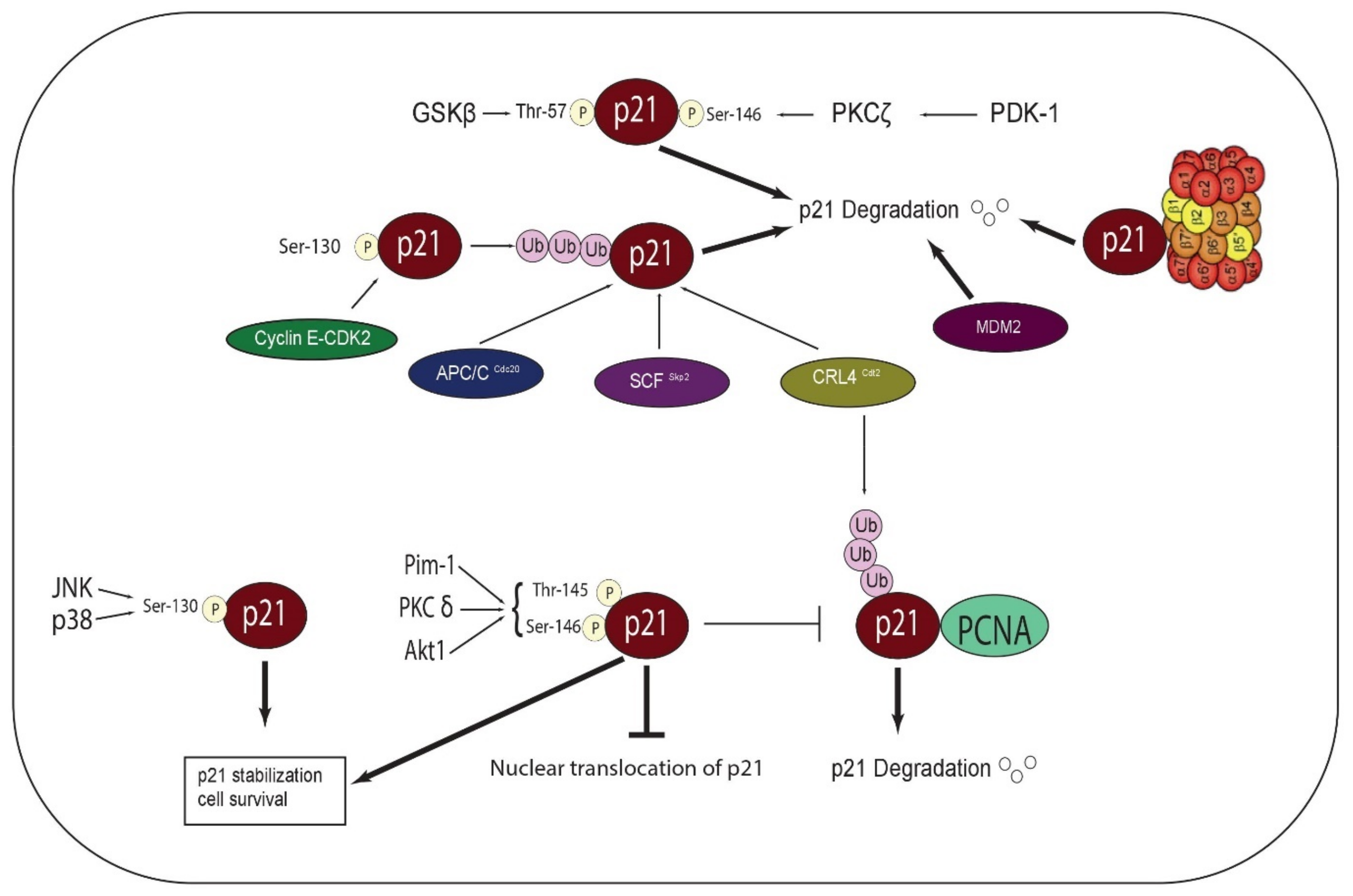
3. Post Transcriptional Regulation of p21
3.1. Phosphorylation, Stability, and Subcellular Localization of p21
3.2. Ubiquitin-Dependent Degradation of p21
3.3. Ubiquitin-Independent Degradation of p21
4. Deregulation of p21 in Cancer
5. Functions of p21
5.1. Role of p21 in Cell Cycle
5.2. Role of p21 in Apoptosis
5.3. Role of p21 in DNA Repair
5.4. Role of p21 in Transcriptional Regulation
6. Targeting p21 in Cancer Therapeutics
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Shimada, M.; Niida, H. Genetic instability in cancer cells by impaired cell cycle checkpoints. Cancer Sci. 2006, 97, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J. Cell Biochem. 2004, 91, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst.) 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Rollins, B.J. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L. Is p21 an oncogene? Mol. Cancer Ther. 2006, 5, 1385–1386. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.B., 3rd; Chen, X.; Smeets, M.; Hengst, L.; Prives, C.; Reed, S.I. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell Biol. 1998, 18, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Toufektchan, E.; Toledo, F. The Guardian of the Genome Revisited: p53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.D.; Watanabe, K.; Broude, E.V.; Fang, J.; Poole, J.C.; Kalinichenko, T.V.; Roninson, I.B. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: Implications for carcinogenesis, senescence, and age-related diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 4291–4296. [Google Scholar] [CrossRef]
- Parveen, A.; Akash, M.S.; Rehman, K.; Kyunn, W.W. Dual Role of p21 in the Progression of Cancer and Its Treatment. Crit. Rev. Eukaryot. Gene Expr. 2016, 26, 49–62. [Google Scholar] [CrossRef]
- Jung, Y.S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Harms, K.; Nozell, S.; Chen, X. The common and distinct target genes of the p53 family transcription factors. Cell Mol. Life Sci. 2004, 61, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Nozell, S.; Chen, X. p21B, a variant of p21(Waf1/Cip1), is induced by the p53 family. Oncogene 2002, 21, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Tyner, A.L. Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp. Cell Res. 1999, 246, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Li, A.G.; Piluso, L.G.; Cai, X.; Gadd, B.J.; Ladurner, A.G.; Liu, X. An acetylation switch in p53 mediates holo-TFIID recruitment. Mol. Cell 2007, 28, 408–421. [Google Scholar] [CrossRef]
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef]
- Gevry, N.; Chan, H.M.; Laflamme, L.; Livingston, D.M.; Gaudreau, L. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 2007, 21, 1869–1881. [Google Scholar] [CrossRef]
- Pauklin, S.; Kristjuhan, A.; Maimets, T.; Jaks, V. ARF and ATM/ATR cooperate in p53-mediated apoptosis upon oncogenic stress. Biochem. Biophys. Res. Commun. 2005, 334, 386–394. [Google Scholar] [CrossRef]
- Chai, Y.L.; Cui, J.; Shao, N.; Shyam, E.; Reddy, P.; Rao, V.N. The second BRCT domain of BRCA1 proteins interacts with p53 and stimulates transcription from the p21WAF1/CIP1 promoter. Oncogene 1999, 18, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Wulf, G.M.; Liou, Y.C.; Ryo, A.; Lee, S.W.; Lu, K.P. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J. Biol. Chem. 2002, 277, 47976–47979. [Google Scholar] [CrossRef] [PubMed]
- Zacchi, P.; Gostissa, M.; Uchida, T.; Salvagno, C.; Avolio, F.; Volinia, S.; Ronai, Z.; Blandino, G.; Schneider, C.; Del Sal, G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 2002, 419, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Wu, W.; Wu, T.; Cao, Z.; Wilkins, R.; Toh, B.H.; Cooper, M.E.; Chai, Z. Antiproliferative autoantigen CDA1 transcriptionally up-regulates p21(Waf1/Cip1) by activating p53 and MEK/ERK1/2 MAPK pathways. J. Biol. Chem. 2007, 282, 11722–11731. [Google Scholar] [CrossRef] [PubMed]
- Rokudai, S.; Aikawa, Y.; Tagata, Y.; Tsuchida, N.; Taya, Y.; Kitabayashi, I. Monocytic leukemia zinc finger (MOZ) interacts with p53 to induce p21 expression and cell-cycle arrest. J. Biol. Chem. 2009, 284, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Donninger, H.; Vos, M.D.; Birrer, M.J.; Gordon, L.; Leaner, V.; Clark, G.J. NORE1A tumor suppressor candidate modulates p21CIP1 via p53. Cancer Res. 2009, 69, 4629–4637. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Johns, D.C.; Geiman, D.E.; Marban, E.; Dang, D.T.; Hamlin, G.; Sun, R.; Yang, V.W. Kruppel-like factor 4 (gut-enriched Kruppel-like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J. Biol. Chem. 2001, 276, 30423–30428. [Google Scholar] [CrossRef] [PubMed]
- Decesse, J.T.; Medjkane, S.; Datto, M.B.; Cremisi, C.E. RB regulates transcription of the p21/WAF1/CIP1 gene. Oncogene 2001, 20, 962–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Jin, S.; Hao, H.; Zheng, L.; Zhou, B.; Zhang, W.; Lv, H.; Yuan, Y. Dux4 induces cell cycle arrest at G1 phase through upregulation of p21 expression. Biochem. Biophys. Res. Commun. 2014, 446, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.Q.; Miyake, S.; Iwai, T.; Yuasa, Y. CDX2, a homeobox transcription factor, upregulates transcription of the p21/WAF1/CIP1 gene. Oncogene 2003, 22, 7942–7949. [Google Scholar] [CrossRef]
- Fang, Z.; Fu, Y.; Liang, Y.; Li, Z.; Zhang, W.; Jin, J.; Yang, Y.; Zha, X. Increased expression of integrin beta1 subunit enhances p21WAF1/Cip1 transcription through the Sp1 sites and p300-mediated histone acetylation in human hepatocellular carcinoma cells. J. Cell Biochem. 2007, 101, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yea, S.; Dolios, G.; Martignetti, J.A.; Narla, G.; Wang, R.; Walsh, M.J.; Friedman, S.L. Regulation of Kruppel-like factor 6 tumor suppressor activity by acetylation. Cancer Res. 2005, 65, 9216–9225. [Google Scholar] [CrossRef] [PubMed]
- Elston, R.; Inman, G.J. Crosstalk between p53 and TGF-beta Signalling. J. Signal Transduct. 2012, 2012, 294097. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.T.; Chun, K.H.; Park, B.D.; Choi, J.S.; Lee, S.K. Regulation of cyclin-dependent kinase inhibitor p21WAF1/CIP1 by protein kinase Cdelta-mediated phosphorylation. Apoptosis 2007, 12, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.T.; Ingram, A.; Ball, K.L. PDK1-dependent activation of atypical PKC leads to degradation of the p21 tumour modifier protein. EMBO J. 2002, 21, 6771–6780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornstein, G.; Bloom, J.; Sitry-Shevah, D.; Nakayama, K.; Pagano, M.; Hershko, A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J. Biol. Chem. 2003, 278, 25752–25757. [Google Scholar] [CrossRef] [PubMed]
- Amador, V.; Ge, S.; Santamaria, P.G.; Guardavaccaro, D.; Pagano, M. APC/C(Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Mol. Cell 2007, 27, 462–473. [Google Scholar] [CrossRef]
- Kim, Y.; Starostina, N.G.; Kipreos, E.T. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008, 22, 2507–2519. [Google Scholar] [CrossRef]
- Chen, X.; Chi, Y.; Bloecher, A.; Aebersold, R.; Clurman, B.E.; Roberts, J.M. N-acetylation and ubiquitin-independent proteasomal degradation of p21(Cip1). Mol. Cell 2004, 16, 839–847. [Google Scholar] [CrossRef]
- Touitou, R.; Richardson, J.; Bose, S.; Nakanishi, M.; Rivett, J.; Allday, M.J. A degradation signal located in the C-terminus of p21WAF1/CIP1 is a binding site for the C8 alpha-subunit of the 20S proteasome. EMBO J. 2001, 20, 2367–2375. [Google Scholar] [CrossRef]
- Jin, Y.; Lee, H.; Zeng, S.X.; Dai, M.S.; Lu, H. MDM2 promotes p21waf1/cip1 proteasomal turnover independently of ubiquitylation. EMBO J. 2003, 22, 6365–6377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, C.; Zhang, P.; Harper, J.W.; Elledge, S.J.; Leder, P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995, 82, 675–684. [Google Scholar] [CrossRef]
- Bhatia, K.; Fan, S.; Spangler, G.; Weintraub, M.; O’Connor, P.M.; Judde, J.G.; Magrath, I. A mutant p21 cyclin-dependent kinase inhibitor isolated from a Burkitt’s lymphoma. Cancer Res. 1995, 55, 1431–1435. [Google Scholar] [PubMed]
- Vidal, M.J.; Loganzo, F., Jr.; de Oliveira, A.R.; Hayward, N.K.; Albino, A.P. Mutations and defective expression of the WAF1 p21 tumour-suppressor gene in malignant melanomas. Melanoma Res. 1995, 5, 243–250. [Google Scholar] [CrossRef]
- Weiss, R.H. p21Waf1/Cip1 as a therapeutic target in breast and other cancers. Cancer Cell 2003, 4, 425–429. [Google Scholar] [CrossRef] [Green Version]
- Akhter, N.; Akhtar, M.S.; Ahmad, M.M.; Haque, S.; Siddiqui, S.; Hasan, S.I.; Shukla, N.K.; Husain, S.A. Association of mutation and hypermethylation of p21 gene with susceptibility to breast cancer: A study from north India. Mol. Biol. Rep. 2014, 41, 2999–3007. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Shchors, K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp. Cell Res. 2003, 283, 17–21. [Google Scholar] [CrossRef]
- Mukherjee, S.; Conrad, S.E. c-Myc suppresses p21WAF1/CIP1 expression during estrogen signaling and antiestrogen resistance in human breast cancer cells. J. Biol. Chem. 2005, 280, 17617–17625. [Google Scholar] [CrossRef]
- Jung, P.; Hermeking, H. The c-MYC-AP4-p21 cascade. Cell Cycle 2009, 8, 982–989. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef]
- Jung, P.; Menssen, A.; Mayr, D.; Hermeking, H. AP4 encodes a c-MYC-inducible repressor of p21. Proc. Natl. Acad. Sci. USA 2008, 105, 15046–15051. [Google Scholar] [CrossRef] [PubMed]
- Prince, S.; Carreira, S.; Vance, K.W.; Abrahams, A.; Goding, C.R. Tbx2 directly represses the expression of the p21(WAF1) cyclin-dependent kinase inhibitor. Cancer Res. 2004, 64, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.H.; Wang, C.Y.; Zhang, W.L.; Zhang, J.T.; Yuan, C.H.; Zhao, P.W.; Lin, Y.Y.; Hong, S.; Li, C.Y.; Wang, L. Histone deacetylase HDAC4 promotes gastric cancer SGC-7901 cells progression via p21 repression. PLoS ONE 2014, 9, e98894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Song, Y.; Chen, W.; Wang, X.; Miao, Z.; Cao, L.; Li, F.; Wang, G. By recruiting HDAC1, MORC2 suppresses p21 Waf1/Cip1 in gastric cancer. Oncotarget 2015, 6, 16461–16470. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Wu, J.; Pan, C.; Tan, X.; Lin, J.; Liu, R.; Chen, S.; Geng, R.; Huang, W. Downregulation of CDC27 inhibits the proliferation of colorectal cancer cells via the accumulation of p21Cip1/Waf1. Cell Death Dis 2016, 7, e2074. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Sivaprasad, U.; Terai, K.; Amador, V.; Pagano, M.; Dutta, A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008, 22, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.A.; Wang, J.Y. Ionizing radiation induces ATM-independent degradation of p21Cip1 in transformed cells. J. Biol. Chem. 2009, 284, 15061–15070. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.S.; Ruhul Amin, A.R.; Paul, R.K.; Gupta, K.; Hastak, K.; Agarwal, M.K.; Jackson, M.W.; Wald, D.N.; Mukhtar, H.; Agarwal, M.L. p53-Dependent p21-mediated growth arrest pre-empts and protects HCT116 cells from PUMA-mediated apoptosis induced by EGCG. Cancer Lett. 2010, 296, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Larrieu, D.; Ythier, D.; Brambilla, C.; Pedeux, R. ING2 controls the G1 to S-phase transition by regulating p21 expression. Cell Cycle 2010, 9, 3984–3990. [Google Scholar] [CrossRef]
- Gartel, A.L.; Najmabadi, F.; Goufman, E.; Tyner, A.L. A role for E2F1 in Ras activation of p21(WAF1/CIP1) transcription. Oncogene 2000, 19, 961–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Wettersten, H.I.; Park, S.H.; Weiss, R.H. Small-molecule inhibitors of p21 as novel therapeutics for chemotherapy-resistant kidney cancer. Future Med. Chem. 2013, 5, 991–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanov, V.S.; Rudolph, K.L. p21 shapes cancer evolution. Nat. Cell Biol. 2016, 18, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Gawriluk, T.R.; Simkin, J.; Thompson, K.L.; Biswas, S.K.; Clare-Salzler, Z.; Kimani, J.M.; Kiama, S.G.; Smith, J.J.; Ezenwa, V.O.; Seifert, A.W. Comparative analysis of ear-hole closure identifies epimorphic regeneration as a discrete trait in mammals. Nat. Commun. 2016, 7, 11164. [Google Scholar] [CrossRef]
- Winters, Z.E.; Hunt, N.C.; Bradburn, M.J.; Royds, J.A.; Turley, H.; Harris, A.L.; Norbury, C.J. Subcellular localisation of cyclin B, Cdc2 and p21(WAF1/CIP1) in breast cancer. association with prognosis. Eur. J. Cancer 2001, 37, 2405–2412. [Google Scholar] [CrossRef]
- Ohata, M.; Nakamura, S.; Fujita, H.; Isemura, M. Prognostic implications of p21 (Waf1/Cip1) immunolocalization in multiple myeloma. Biomed. Res. 2005, 26, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Li, X.; Sellers, W.R.; Baker, K.B.; Leng, X.; Harper, J.W.; Taya, Y.; Kaelin, W.G., Jr. Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol. Cell Biol. 1999, 19, 1068–1080. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Hilton, M.B.; Kaldis, P. p21 Inhibits Cdk1 in the absence of Cdk2 to maintain the G1/S phase DNA damage checkpoint. Mol. Biol. Cell 2008, 19, 65–77. [Google Scholar] [CrossRef]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef]
- Gottifredi, V.; McKinney, K.; Poyurovsky, M.V.; Prives, C. Decreased p21 levels are required for efficient restart of DNA synthesis after S phase block. J. Biol. Chem. 2004, 279, 5802–5810. [Google Scholar] [CrossRef]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Smits, V.A.; Klompmaker, R.; Vallenius, T.; Rijksen, G.; Makela, T.P.; Medema, R.H. p21 inhibits Thr161 phosphorylation of Cdc2 to enforce the G2 DNA damage checkpoint. J. Biol. Chem. 2000, 275, 30638–30643. [Google Scholar] [CrossRef] [PubMed]
- Kreis, N.N.; Sanhaji, M.; Rieger, M.A.; Louwen, F.; Yuan, J. p21Waf1/Cip1 deficiency causes multiple mitotic defects in tumor cells. Oncogene 2014, 33, 5716–5728. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Lukas, J.; Strauss, M.; Bartek, J. p21WAF1/CIP1 mutants deficient in inhibiting cyclin-dependent kinases (CDKs) can promote assembly of active cyclin D/CDK4(6) complexes in human tumor cells. Cancer Res. 1998, 58, 5053–5056. [Google Scholar] [PubMed]
- Cheng, M.; Olivier, P.; Diehl, J.A.; Fero, M.; Roussel, M.F.; Roberts, J.M.; Sherr, C.J. The p21(Cip1) and p27(Kip1) CDK ’inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999, 18, 1571–1583. [Google Scholar] [CrossRef]
- Yue, F.; Cheng, Y.; Breschi, A.; Vierstra, J.; Wu, W.; Ryba, T.; Sandstrom, R.; Ma, Z.; Davis, C.; Pope, B.D.; et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014, 515, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, T.; Yan, G.; Song, X.; Xie, L.; Zhou, Y.; Li, J.; Hu, X.; Li, Z.; Hu, J.; Zhang, Y.; et al. Deubiquitylation and stabilization of p21 by USP11 is critical for cell-cycle progression and DNA damage responses. Proc. Natl. Acad. Sci. USA 2018, 115, 4678–4683. [Google Scholar] [CrossRef] [Green Version]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002, 1, 639–649. [Google Scholar]
- Suzuki, A.; Tsutomi, Y.; Miura, M.; Akahane, K. Caspase 3 inactivation to suppress Fas-mediated apoptosis: Identification of binding domain with p21 and ILP and inactivation machinery by p21. Oncogene 1999, 18, 1239–1244. [Google Scholar] [CrossRef]
- Gervais, J.L.; Seth, P.; Zhang, H. Cleavage of CDK inhibitor p21(Cip1/Waf1) by caspases is an early event during DNA damage-induced apoptosis. J. Biol. Chem. 1998, 273, 19207–19212. [Google Scholar] [CrossRef]
- Asada, M.; Yamada, T.; Ichijo, H.; Delia, D.; Miyazono, K.; Fukumuro, K.; Mizutani, S. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999, 18, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Baptiste-Okoh, N.; Barsotti, A.M.; Prives, C. Caspase 2 is both required for p53-mediated apoptosis and downregulated by p53 in a p21-dependent manner. Cell Cycle 2008, 7, 1133–1138. [Google Scholar] [CrossRef]
- Zhang, Y.; Fujita, N.; Tsuruo, T. Caspase-mediated cleavage of p21Waf1/Cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene 1999, 18, 1131–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneuchi, M.; Yamashita, T.; Shindoh, M.; Segawa, K.; Takahashi, S.; Furuta, I.; Fujimoto, S.; Fujinaga, K. Induction of apoptosis by the p53-273L (Arg --> Leu) mutant in HSC3 cells without transactivation of p21Waf1/Cip1/Sdi1 and bax. Mol. Carcinog. 1999, 26, 44–52. [Google Scholar] [CrossRef]
- Shaulian, E.; Schreiber, M.; Piu, F.; Beeche, M.; Wagner, E.F.; Karin, M. The mammalian UV response: C-Jun induction is required for exit from p53-imposed growth arrest. Cell 2000, 103, 897–907. [Google Scholar] [CrossRef]
- Canman, C.E.; Gilmer, T.M.; Coutts, S.B.; Kastan, M.B. Growth factor modulation of p53-mediated growth arrest versus apoptosis. Genes Dev. 1995, 9, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Helt, C.E.; Rancourt, R.C.; Staversky, R.J.; O’Reilly, M.A. p53-dependent induction of p21(Cip1/WAF1/Sdi1) protects against oxygen-induced toxicity. Toxicol. Sci. 2001, 63, 214–222. [Google Scholar] [CrossRef]
- Li, C.Y.; Suardet, L.; Little, J.B. Potential role of WAF1/Cip1/p21 as a mediator of TGF-beta cytoinhibitory effect. J. Biol. Chem. 1995, 270, 4971–4974. [Google Scholar] [CrossRef]
- Gartel, A.L. The conflicting roles of the cdk inhibitor p21(CIP1/WAF1) in apoptosis. Leuk. Res. 2005, 29, 1237–1238. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; McKinstry, R.; Gupta, S.; Gilfor, D.; Windle, J.J.; Hylemon, P.B.; Grant, S.; Fisher, P.B.; Dent, P. Cyclin kinase inhibitor p21 potentiates bile acid-induced apoptosis in hepatocytes that is dependent on p53. Hepatology 2002, 36, 39–48. [Google Scholar] [CrossRef]
- Kang, K.H.; Kim, W.H.; Choi, K.H. p21 promotes ceramide-induced apoptosis and antagonizes the antideath effect of Bcl-2 in human hepatocarcinoma cells. Exp. Cell Res. 1999, 253, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, R.; Bi, B.; Dao, T.; Bae, Y.; Matsuzawa, A.; Crispe, I.N. CD95/Fas signaling in T lymphocytes induces the cell cycle control protein p21cip-1/WAF-1, which promotes apoptosis. J. Immunol. 2000, 164, 4032–4036. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, L.; Steinman, R. A proapoptotic function of p21 in differentiating granulocytes. Leuk. Res. 2005, 29, 1315–1323. [Google Scholar] [CrossRef]
- Soria, G.; Speroni, J.; Podhajcer, O.L.; Prives, C.; Gottifredi, V. p21 differentially regulates DNA replication and DNA-repair-associated processes after UV irradiation. J. Cell Sci. 2008, 121, 3271–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, G.; Podhajcer, O.; Prives, C.; Gottifredi, V. P21Cip1/WAF1 downregulation is required for efficient PCNA ubiquitination after UV irradiation. Oncogene 2006, 25, 2829–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzalini, O.; Perucca, P.; Savio, M.; Necchi, D.; Bianchi, L.; Stivala, L.A.; Ducommun, B.; Scovassi, A.I.; Prosperi, E. Interaction of p21(CDKN1A) with PCNA regulates the histone acetyltransferase activity of p300 in nucleotide excision repair. Nucleic Acids Res. 2008, 36, 1713–1722. [Google Scholar] [CrossRef]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef]
- Fotedar, R.; Bendjennat, M.; Fotedar, A. Role of p21WAF1 in the cellular response to UV. Cell Cycle 2004, 3, 134–137. [Google Scholar] [CrossRef]
- Gratchev, A. The nucleotide excision repair of DNA in human cells and its association with xeroderma pigmentosum. Adv. Exp. Med. Biol. 2008, 637, 113–119. [Google Scholar]
- Stoyanova, T.; Yoon, T.; Kopanja, D.; Mokyr, M.B.; Raychaudhuri, P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol. Cell Biol. 2008, 28, 177–187. [Google Scholar] [CrossRef]
- Stivala, L.A.; Riva, F.; Cazzalini, O.; Savio, M.; Prosperi, E. p21(waf1/cip1)-null human fibroblasts are deficient in nucleotide excision repair downstream the recruitment of PCNA to DNA repair sites. Oncogene 2001, 20, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Tillhon, M.; Cazzalini, O.; Nardo, T.; Necchi, D.; Sommatis, S.; Stivala, L.A.; Scovassi, A.I.; Prosperi, E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair (Amst.) 2012, 11, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Scholz, M.; Taucher-Scholz, G. Characterization of CDKN1A (p21) binding to sites of heavy-ion-induced damage: Colocalization with proteins involved in DNA repair. Int. J. Radiat. Biol. 2002, 78, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Mauro, M.; Rego, M.A.; Boisvert, R.A.; Esashi, F.; Cavallo, F.; Jasin, M.; Howlett, N.G. p21 promotes error-free replication-coupled DNA double-strand break repair. Nucleic Acids Res. 2012, 40, 8348–8360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, M.; Yutoku, Y.; Koike, A. Accumulation of p21 proteins at DNA damage sites independent of p53 and core NHEJ factors following irradiation. Biochem. Biophys. Res. Commun. 2011, 412, 39–43. [Google Scholar] [CrossRef]
- Yaglom, J.A.; McFarland, C.; Mirny, L.; Sherman, M.Y. Oncogene-triggered suppression of DNA repair leads to DNA instability in cancer. Oncotarget 2014, 5, 8367–8378. [Google Scholar] [CrossRef] [Green Version]
- Mei, S.; Flemington, E.K.; Zhang, K. A computational framework for distinguishing direct versus indirect interactions in human functional protein-protein interaction networks. Integr. Biol. (Camb.) 2017, 9, 595–606. [Google Scholar] [CrossRef]
- Perkins, N.D. Not just a CDK inhibitor: Regulation of transcription by p21(WAF1/CIP1/SDI1). Cell Cycle 2002, 1, 39–41. [Google Scholar] [CrossRef]
- Perkins, N.D.; Felzien, L.K.; Betts, J.C.; Leung, K.; Beach, D.H.; Nabel, G.J. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science 1997, 275, 523–527. [Google Scholar] [CrossRef]
- Redeuilh, G.; Attia, A.; Mester, J.; Sabbah, M. Transcriptional activation by the oestrogen receptor alpha is modulated through inhibition of cyclin-dependent kinases. Oncogene 2002, 21, 5773–5782. [Google Scholar] [CrossRef]
- Coqueret, O. New roles for p21 and p27 cell-cycle inhibitors: A function for each cell compartment? Trends Cell Biol. 2003, 13, 65–70. [Google Scholar] [CrossRef]
- Fritah, A.; Saucier, C.; Mester, J.; Redeuilh, G.; Sabbah, M. p21WAF1/CIP1 selectively controls the transcriptional activity of estrogen receptor alpha. Mol. Cell Biol. 2005, 25, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Broude, E.V.; Demidenko, Z.N.; Vivo, C.; Swift, M.E.; Davis, B.M.; Blagosklonny, M.V.; Roninson, I.B. p21 (CDKN1A) is a negative regulator of p53 stability. Cell Cycle 2007, 6, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- Coqueret, O.; Gascan, H. Functional interaction of STAT3 transcription factor with the cell cycle inhibitor p21WAF1/CIP1/SDI1. J. Biol. Chem. 2000, 275, 18794–18800. [Google Scholar] [CrossRef] [PubMed]
- Delavaine, L.; La Thangue, N.B. Control of E2F activity by p21Waf1/Cip1. Oncogene 1999, 18, 5381–5392. [Google Scholar] [CrossRef] [Green Version]
- Kitaura, H.; Shinshi, M.; Uchikoshi, Y.; Ono, T.; Iguchi-Ariga, S.M.; Ariga, H. Reciprocal regulation via protein-protein interaction between c-Myc and p21(cip1/waf1/sdi1) in DNA replication and transcription. J. Biol. Chem. 2000, 275, 10477–10483. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, A.; Cherier, J.; Barre, B.; Gamelin, E.; Coqueret, O. The cell cycle inhibitor p21waf1 binds to the myc and cdc25A promoters upon DNA damage and induces transcriptional repression. J. Biol. Chem. 2006, 281, 34742–34750. [Google Scholar] [CrossRef]
- Dai, M.; Al-Odaini, A.A.; Arakelian, A.; Rabbani, S.A.; Ali, S.; Lebrun, J.J. A novel function for p21Cip1 and acetyltransferase p/CAF as critical transcriptional regulators of TGFbeta-mediated breast cancer cell migration and invasion. Breast Cancer Res. 2012, 14, R127. [Google Scholar] [CrossRef]
- Zhu, H.; Chang, B.D.; Uchiumi, T.; Roninson, I.B. Identification of promoter elements responsible for transcriptional inhibition of polo-like kinase 1 and topoisomerase IIalpha genes by p21(WAF1/CIP1/SDI1). Cell Cycle 2002, 1, 59–66. [Google Scholar] [CrossRef]
- Ferrandiz, N.; Caraballo, J.M.; Garcia-Gutierrez, L.; Devgan, V.; Rodriguez-Paredes, M.; Lafita, M.C.; Bretones, G.; Quintanilla, A.; Munoz-Alonso, M.J.; Blanco, R.; et al. p21 as a transcriptional co-repressor of S-phase and mitotic control genes. PLoS ONE 2012, 7, e37759. [Google Scholar] [CrossRef]
- Wang, J.; Devgan, V.; Corrado, M.; Prabhu, N.S.; El-Deiry, W.S.; Riccardi, C.; Pandolfi, P.P.; Missero, C.; Dotto, G.P. Glucocorticoid-induced tumor necrosis factor receptor is a p21Cip1/WAF1 transcriptional target conferring resistance of keratinocytes to UV light-induced apoptosis. J. Biol. Chem. 2005, 280, 37725–37731. [Google Scholar] [CrossRef] [PubMed]
- Devgan, V.; Mammucari, C.; Millar, S.E.; Brisken, C.; Dotto, G.P. p21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev. 2005, 19, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Gregory, D.J.; Garcia-Wilson, E.; Poole, J.C.; Snowden, A.W.; Roninson, I.B.; Perkins, N.D. Induction of transcription through the p300 CRD1 motif by p21WAF1/CIP1 is core promoter specific and cyclin dependent kinase independent. Cell Cycle 2002, 1, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.H.; Porter, A.G. p21(WAF1) negatively regulates DNMT1 expression in mammalian cells. Biochem. Biophys. Res. Commun. 2009, 382, 171–176. [Google Scholar] [CrossRef]
- Kim, H.K.; Kang, M.A.; Kim, M.S.; Shin, Y.J.; Chi, S.G.; Jeong, J.H. Transcriptional Repression of High-Mobility Group Box 2 by p21 in Radiation-Induced Senescence. Mol. Cells 2018, 41, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Trakala, M.; Arias, C.F.; Garcia, M.I.; Moreno-Ortiz, M.C.; Tsilingiri, K.; Fernandez, P.J.; Mellado, M.; Diaz-Meco, M.T.; Moscat, J.; Serrano, M.; et al. Regulation of macrophage activation and septic shock susceptibility via p21(WAF1/CIP1). Eur. J. Immunol. 2009, 39, 810–819. [Google Scholar] [CrossRef]
- Yao, H.; Yang, S.R.; Edirisinghe, I.; Rajendrasozhan, S.; Caito, S.; Adenuga, D.; O’Reilly, M.A.; Rahman, I. Disruption of p21 attenuates lung inflammation induced by cigarette smoke, LPS, and fMLP in mice. Am. J. Respir. Cell Mol. Biol. 2008, 39, 7–18. [Google Scholar] [CrossRef]
- Lapatas, V.; Stefanidakis, M.; Jimenez, R.C.; Via, A.; Schneider, M.V. Data integration in biological research: An overview. J. Biol. Res. (Thessalon.) 2015, 22, 9. [Google Scholar] [CrossRef]
- Xu, S.Q.; El-Deiry, W.S. p21(WAF1/CIP1) inhibits initiator caspase cleavage by TRAIL death receptor DR4. Biochem. Biophys. Res. Commun. 2000, 269, 179–190. [Google Scholar] [CrossRef]
- Gorospe, M.; Wang, X.; Guyton, K.Z.; Holbrook, N.J. Protective role of p21(Waf1/Cip1) against prostaglandin A2-mediated apoptosis of human colorectal carcinoma cells. Mol. Cell Biol. 1996, 16, 6654–6660. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Chang, J.K.; Smith, M.L.; Duba, D.; Fornace, A.J., Jr.; O’Connor, P.M. Cells lacking CIP1/WAF1 genes exhibit preferential sensitivity to cisplatin and nitrogen mustard. Oncogene 1997, 14, 2127–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, E.R., 3rd; Wu, G.S.; Waldman, T.; El-Deiry, W.S. Repair Defect in p21 WAF1/CIP1 -/- human cancer cells. Cancer Res. 1996, 56, 2250–2255. [Google Scholar] [PubMed]
- Wei, J.; Zhao, J.; Long, M.; Han, Y.; Wang, X.; Lin, F.; Ren, J.; He, T.; Zhang, H. p21WAF1/CIP1 gene transcriptional activation exerts cell growth inhibition and enhances chemosensitivity to cisplatin in lung carcinoma cell. BMC Cancer 2010, 10, 632. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Huang, H.; Chen, Y.N.; Deng, Y.T.; Zhang, B.; Xiong, X.D.; Yuan, Y.; Zhu, Y.; Huang, H.; Xie, L.; et al. DNA damage responsive miR-33b-3p promoted lung cancer cells survival and cisplatin resistance by targeting p21(WAF1/CIP1). Cell Cycle 2016, 15, 2920–2930. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, L.; Talmon, G.; Wang, J. MicroRNA-520g confers drug resistance by regulating p21 expression in colorectal cancer. J. Biol. Chem. 2015, 290, 6215–6225. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Geng, Y.; Liu, J.; Xie, Y.; Jiang, H.; Zuo, K.; Li, T.; Liu, Z. Trichostatin A promotes GLI1 degradation and P21 expression in multiple myeloma cells. Cancer Manag. Res. 2018, 10, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.K.; Liu, S.T.; Chang, C.C.; Huang, S.M. Regulatory mechanisms of fluvastatin and lovastatin for the p21 induction in human cervical cancer HeLa cells. PLoS ONE 2019, 14, e0214408. [Google Scholar] [CrossRef]
- Liu, J.; Shen, M.; Yue, Z.; Yang, Z.; Wang, M.; Li, C.; Xin, C.; Wang, Y.; Mei, Q.; Wang, Z. Triptolide inhibits colon-rectal cancer cells proliferation by induction of G1 phase arrest through upregulation of p21. Phytomedicine 2012, 19, 756–762. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Hoe, H.S.; Cho, H.J.; Park, K.K.; Kim, D.D.; Kim, C.H.; Magae, J.; Kang, D.W.; Lee, S.R.; Chang, Y.C. Suppression of c-Myc enhances p21(WAF1/CIP1) -mediated G1 cell cycle arrest through the modulation of ERK phosphorylation by ascochlorin. J. Cell Biochem. 2018, 119, 2036–2047. [Google Scholar] [CrossRef]
- Aasland, D.; Gotzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. https://doi.org/10.3390/cancers11101475
Al Bitar S, Gali-Muhtasib H. The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers. 2019; 11(10):1475. https://doi.org/10.3390/cancers11101475
Chicago/Turabian StyleAl Bitar, Samar, and Hala Gali-Muhtasib. 2019. "The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics" Cancers 11, no. 10: 1475. https://doi.org/10.3390/cancers11101475
APA StyleAl Bitar, S., & Gali-Muhtasib, H. (2019). The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers, 11(10), 1475. https://doi.org/10.3390/cancers11101475