Neurokinin-1 Receptor Antagonists against Hepatoblastoma




Abstract
1. Introduction
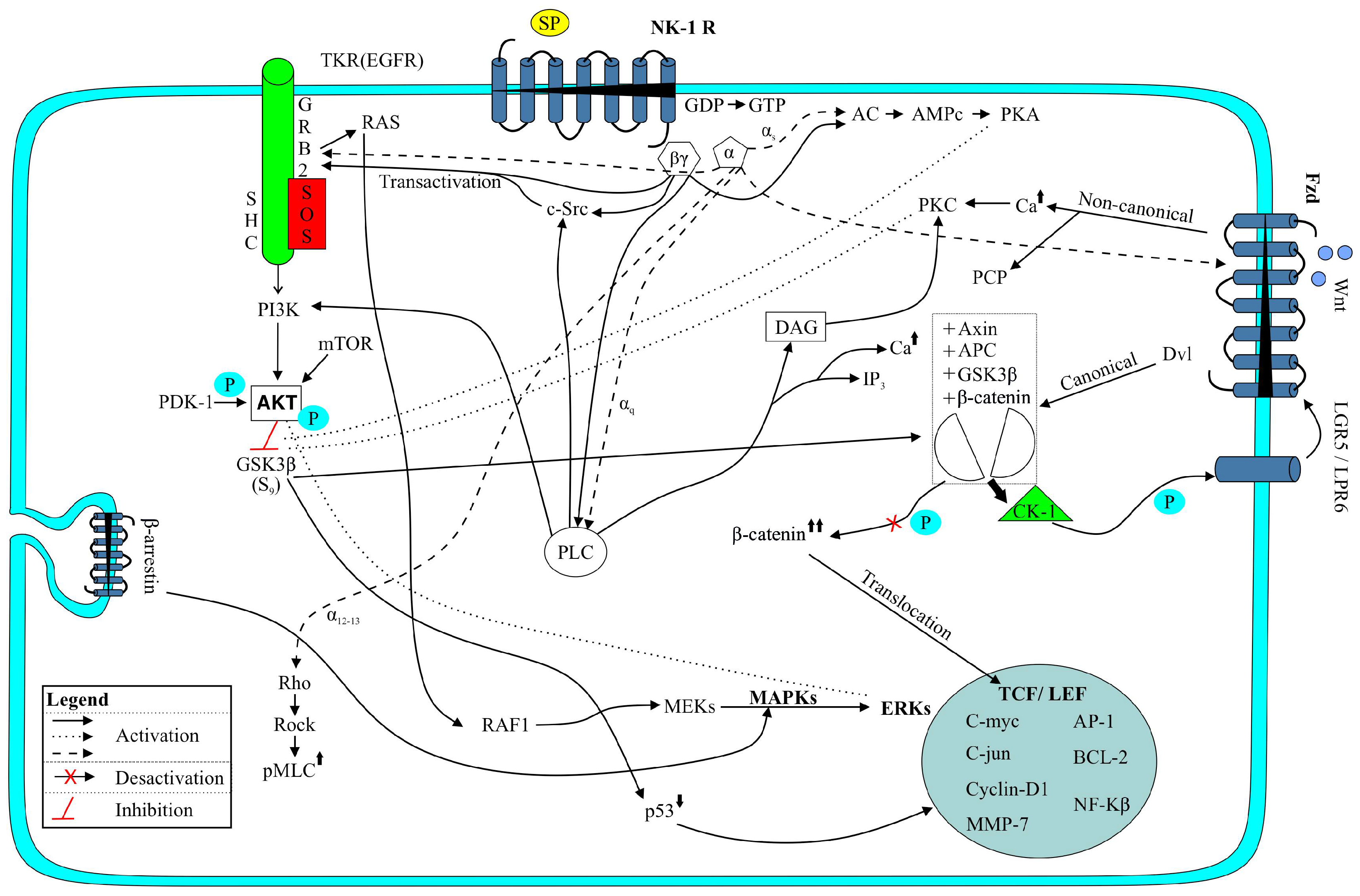
2. The SP/NK-1R System
3. Involvement of the SP/NK-1R System in HB Progression
3.1. PI3K/Akt/mTOR Signaling Pathway
3.2. Wnt Signaling Pathway
4. NK-1R Antagonists as Anti-HB Drugs: Mechanisms of Action and Therapeutic Properties
4.1. Antiproliferative Effect
4.2. Apoptotic Effect
4.3. Anti-Warburg Effect
4.4. Antiangiogenic Effect
4.5. Antimetastatic Effect
4.6. Combination Therapy Using NK-1R Antagonists
5. Safety Profile and Side-Effects of NK-1R Antagonists
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stiller, C.A.; Pritchard, J.; Steliarova-Foucher, E. Liver cancer in European children: Incidence and survival, 1978-1997. Report from the Automated Childhood Cancer Information System project. Eur. J. Cancer 2006, 42, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.L.; Maibach, R.; Hiyama, E.; Häberle, B.; Krailo, M.; Rangaswami, A.; Aronson, D.C.; Malogolowkin, M.H.; Perilongo, G.; von Schweinitz, D.; et al. Risk-stratified staging in paediatric hepatoblastoma: A unified analysis from the children’s hepatic tumors international collaboration. Lancet Oncol. 2017, 18, 122–131. [Google Scholar] [CrossRef]
- Bell, D.; Ranganathan, S.; Tao, J.; Monga, S.P. Novel advances in understanding of molecular pathogenesis of hepatoblastoma: A Wnt/β-catenin perspective. Gene Expr. 2017, 17, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides 2013, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Rosso, M.; Robles-Frías, M.J.; Salinas-Martín, M.V.; Coveñas, R. The NK-1 receptor is expressed in human melanoma and is involved in the antitumor action of the NK-1 receptor antagonist aprepitant on melanoma cell lines. Lab. Investig. 2010, 90, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Neth, O.; Ilmer, M.; Garnier, A.; Salinas-Martín, M.V.; de Agustin Asencio, J.C.; von Schweinitz, D.; Kappler, R.; Muñoz, M. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J. Hepatol. 2014, 60, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R.; Esteban, F.; Redondo, M. The substance P/NK-1 receptor system: NK-1 receptor antagonists as anti-cancer drugs. J. Biosci. 2015, 40, 441–463. [Google Scholar] [CrossRef]
- Roush, E.D.; Kwatra, M.M. Human substance P receptor expressed in Chinese hamster ovary cells directly activates g (alpha q/11), g (alpha s), g (alpha o). FEBS Lett. 1998, 428, 291–294. [Google Scholar] [CrossRef]
- Mitsuhashi, M.; Ohashi, Y.; Shichijo, S.; Christian, C.; Sudduth-Klinger, J.; Harrowe, G.; Payan, D.G. Multiple intracellular signaling pathways of the neuropeptide substance P receptor. J. Neurosci. Res. 1992, 32, 437–443. [Google Scholar] [CrossRef]
- Sagan, S.; Chassaing, G.; Pradier, L.; Lavielle, S. Tachykinin peptides affect differently the second messenger pathways after binding to CHO-expressed human NK-1 receptors. J. Pharmacol. Exp. Ther. 1996, 276, 1039–1048. [Google Scholar]
- Takeda, Y.; Blount, P.; Sachais, B.S.; Hershey, A.D.; Raddatz, R.; Krause, J.E. Ligand binding kinetics of substance P and neurokinin A receptors stably expressed in Chinese hamster ovary cells and evidence for differential stimulation of inositol 1, 4, 5 trisphosphate and cyclic amp second messenger responses. J. Neurochem. 1992, 59, 740–745. [Google Scholar] [CrossRef]
- Garnier, A.; Ilmer, M.; Kappler, R.; Berger, M. Therapeutic innovations for targeting hepatoblastoma. Anticancer Res. 2016, 36, 5577–5592. [Google Scholar] [CrossRef]
- Ramkissoon, S.H.; Patel, H.J.; Taborga, M.; Rameshwar, P. G protein-coupled receptors in haematopoietic disruption. Expert Opin. Biol. Ther. 2006, 6, 109–120. [Google Scholar] [CrossRef]
- Nakajima, Y.; Tsuchida, K.; Negishi, M.; Ito, S.; Nakanishi, S. Direct linkage of three tachykinin receptors to stimulation of both phosphatidylinositol hydrolysis and cyclic AMP cascades in transfected Chinese hamster ovary cells. J. Biol. Chem. 1992, 267, 2437–2442. [Google Scholar]
- Holst, B.; Hastrup, H.; Raffetseder, U.; Martini, L.; Schwartz, T.W. Two active molecular phenotypes of the tachykinin NK1 receptor revealed by G-protein fusions and mutagenesis. J. Biol. Chem. 2001, 276, 19793–19799. [Google Scholar] [CrossRef]
- Khawaja, A.M.; Rogers, D.F. Tachykinins: Receptor to effector. Int. J. Biochem. Cell Biol. 1996, 28, 721–738. [Google Scholar] [CrossRef]
- Pennefather, J.N.; Lecci, A.; Candenas, M.L.; Patak, E.; Pinto, F.M.; Maggi, C.A. Tachykinins and tachykinin receptors: A growing family. Life Sci. 2004, 74, 1445–1463. [Google Scholar] [CrossRef]
- Kage, R.; Leeman, S.E.; Krause, J.E.; Costello, C.E.; Boyd, N.D. Identification of methionine as the site of covalent attachment of a p-benzoyl-phenylalanine-containing analogue of substance P on the substance P (NK-1) receptor. J. Biol. Chem. 1996, 271, 25797–25800. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Allen, C.J.; Ghilardi, J.R.; Rogers, S.D.; Mantyh, C.R.; Liu, H.; Basbaum, A.I.; Vigna, S.R.; Maggio, J.E. Rapid endocytosis of a G protein-coupled receptor: Substance P evoked internalization of its receptor in the rat striatum in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 2622–2626. [Google Scholar] [CrossRef]
- Roosterman, D.; Cottrell, G.S.; Schmidlin, F.; Steinhoff, M.; Bunnett, N.W. Recycling and resensitization of the neurokinin 1 receptor. Influence of agonist concentration and Rab GTPases. J. Biol. Chem. 2004, 279, 30670–30679. [Google Scholar] [CrossRef]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Déry, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta-arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef]
- Fong, T.M.; Anderson, S.A.; Yu, H.; Huang, R.R.; Strader, C.D. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol. Pharmacol. 1992, 41, 24–30. [Google Scholar]
- Page, N.M. New challenges in the study of the mammalian tachykinins. Peptides 2005, 26, 1356–1368. [Google Scholar] [CrossRef]
- Li, H.; Leeman, S.E.; Slack, B.E.; Hauser, G.; Saltsman, W.S.; Krause, J.E.; Blusztajn, J.K.; Boyd, N.D. A substance P (neurokinin-1) receptor mutant carboxyl-terminally truncated to resemble a naturally occurring receptor isoform displays enhanced responsiveness and resistance to desensitization. Proc. Natl. Acad. Sci. USA 1997, 94, 9475–9480. [Google Scholar] [CrossRef]
- Douglas, S.D.; Leeman, S.E. Neurokinin-1 receptor: Functional significance in the immune system in reference to selected infections and inflammation. Ann. N. Y. Acad. Sci. 2010, 1217, 83–95. [Google Scholar] [CrossRef]
- Muñoz, M.; González-Ortega, A.; Rosso, M.; Robles-Frías, M.J.; Carranza, A.; Salinas-Martín, M.V.; Coveñas, R. The substance P/neurokinin-1 receptor system in lung cancer: Focus on the antitumor action of neurokinin-1 receptor antagonists. Peptides 2012, 38, 318–325. [Google Scholar] [CrossRef]
- Friess, H.; Zhu, Z.; Liard, V.; Shi, X.; Shrikhande, S.V.; Wang, L.; Lieb, K.; Korc, M.; Palma, C.; Zimmermann, A.; et al. Neurokinin-1 receptor expression and its potential effects on tumor growth in human pancreatic cancer. Lab. Investig. 2003, 83, 731–742. [Google Scholar] [CrossRef]
- Hennig, I.M.; Laissue, J.A.; Horisberger, U.; Reubi, J.C. Substance-P receptors in human primary neoplasms: Tumoral and vascular localization. Int. J. Cancer 1995, 61, 786–792. [Google Scholar] [CrossRef]
- Singh, D.; Joshi, D.D.; Hameed, M.; Qian, J.; Gascón, P.; Maloof, P.B.; Mosenthal, A.; Rameshwar, P. Increased expression of preprotachykinin-I and neurokinin receptors in human breast cancer cells: Implications for bone marrow metastasis. Proc. Natl. Acad. Sci. USA 2000, 97, 1388–1393. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, L.; Xiong, T.; Chen, X.; Zhang, Y.; Yu, M.; Yang, J.; Yao, Z. Roles of full-length and truncated neurokinin-1 receptors on tumor progression and distant metastasis in human breast cancer. Breast Cancer Res. Treat. 2013, 140, 49–61. [Google Scholar] [CrossRef]
- Davoodian, M.; Boroumand, N.; Mehrabi Bahar, M.; Jafarian, A.H.; Asadi, M.; Hashemy, S.I. Evaluation of serum level of substance P and tissue distribution of NK-1 receptor in breast cancer. Mol. Biol. Rep. 2019, 46, 1285–1293. [Google Scholar] [CrossRef]
- Garnier, A.; Ilmer, M.; Becker, K.; Häberle, B.; von Schweinitz, D.; Kappler, R.; Berger, M. Truncated neurokinin-1 receptor is an ubiquitous antitumor target in hepatoblastoma, and its expression is independent of tumor biology and stage. Oncol. Lett. 2016, 11, 870–878. [Google Scholar] [CrossRef][Green Version]
- Muñoz, M.; Berger, M.; Rosso, M.; González-Ortega, A.; Carranza, A.; Coveñas, R. Antitumor activity of neurokinin-1 receptor antagonists in MG-63 human osteosarcoma xenografts. Int. J. Oncol. 2014, 44, 137–146. [Google Scholar] [CrossRef]
- Luo, W.; Sharif, T.R.; Sharif, M. Substance P-induced mitogenesis in human astrocytoma cells correlates with activation of the mitogen-activated protein kinase signaling pathway. Cancer Res. 1995, 56, 4983–4991. [Google Scholar]
- Shibuya, H.; Yoneyama, M.; Ninomiya-Tsuji, J.; Matsumoto, K.; Taniguchi, T. IL-2 and EGF and EGF receptors stimulate the hematopoietic cell cycle via different signaling pathways: Demonstration of a novel role for c-myc. Cell 1992, 70, 57–67. [Google Scholar] [CrossRef]
- Wang, H.; Lu, J.; Edmunds, L.R.; Kulkarni, S.; Dolezal, J.; Tao, J.; Ranganathan, S.; Jackson, L.; Fromherz, M.; Beer-Stolz, D.; et al. Coordinated activities of multiple myc-dependent and myc-independent biosynthetic pathways in hepatoblastoma. J. Biol. Chem. 2016, 291, 26241–26251. [Google Scholar] [CrossRef]
- Castagliuolo, I.; Valenick, L.; Liu, J.; Pothoulakis, C. Epidermal growth factor receptor transactivation mediates substance P-induced mitogenic responses in U-373 MG cells. J. Biol. Chem. 2000, 275, 26545–26550. [Google Scholar] [CrossRef]
- Almendro, V.; García-Recio, S.; Gascón, P. Tyrosine kinase receptor transactivation associated to G protein-coupled receptors. Curr. Drug Targets 2010, 11, 1169–1180. [Google Scholar] [CrossRef]
- Molina, L.; Yang, H.; Adebayo Michael, A.O.; Oertel, M.; Bell, A.; Singh, S.; Chen, X.; Tao, J.; Monga, S.P.S. mTOR inhibition affects Yap1-β-catenin-induced hepatoblastoma growth and development. Oncotarget 2019, 10, 1475–1490. [Google Scholar] [CrossRef]
- Bockmann, S.; Seep, J.; Jonas, L. Delay of neutrophil apoptosis by the neuropeptide substance P: Involvement of caspase cascade. Peptides 2001, 22, 661–670. [Google Scholar] [CrossRef]
- Koon, H.W.; Zhao, D.; Zhan, Y.; Moyer, M.P.; Pothoulakis, C. Substance P mediates antiapoptotic responses in human colonocytes by Akt activation. Proc. Natl. Acad. Sci. USA 2007, 104, 2013–2018. [Google Scholar] [CrossRef]
- Lim, J.E.; Chung, E.; Son, Y. A neuropeptide, substance-P, directly induces tissue-repairing M2 like macrophages by activating the PI3K/Akt/mTOR pathway even in the presence of IFNγ. Sci. Rep. 2017, 7, 9417. [Google Scholar] [CrossRef]
- Hartmann, W.; Küchler, J.; Koch, A.; Friedrichs, N.; Waha, A.; Endl, E.; Czerwitzki, J.; Metzger, D.; Steiner, S.; Wurst, P.; et al. Activation of phosphatidylinositol-3′-kinase/AKT signaling is essential in hepatoblastoma survival. Clin. Cancer Res. 2009, 15, 4538–4545. [Google Scholar] [CrossRef]
- Ilmer, M.; Garnier, A.; Vykoukal, J.; Alt, E.; von Schweinitz, D.; Kappler, R.; Berger, M. Targeting the neurokinin-1 receptor compromises canonical Wnt signaling in hepatoblastoma. Mol. Cancer Ther. 2015, 14, 2712–2721. [Google Scholar] [CrossRef]
- Martelli, A.M.; Tabellini, G.; Bressanin, D.; Ognibene, A.; Goto, K.; Cocco, L.; Evangelisti, C. The emerging multiple roles of nuclear AKT. Biochim. Biophys. Acta 2012, 1823, 2168–2178. [Google Scholar] [CrossRef]
- Breuleux, M.; Klopfenstein, M.; Stephan, C.; Doughty, C.A.; Barys, L.; Maira, S.M.; Kwiatkowski, D.; Lane, H.A. Increased AKT s473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol. Cancer Ther. 2009, 8, 742–753. [Google Scholar] [CrossRef]
- Chen, K.F.; Chen, H.L.; Tai, W.T.; Feng, W.C.; Hsu, C.H.; Chen, P.J.; Cheng, A.L. Activation of phosphatidylinositol 3-kinase/AKT signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef]
- Yun, M.S.; Kim, S.E.; Jeon, S.H.; Lee, J.S.; Choi, K.Y. Both ERK and Wnt/beta-catenin pathways are involved in Wnt3a-induced proliferation. J. Cell Sci. 2005, 118, 313–322. [Google Scholar] [CrossRef]
- Mei, G.; Xia, L.; Zhou, J.; Zhang, Y.; Tuo, Y.; Fu, S.; Zou, Z.; Wang, Z.; Jin, D. Neuropeptide SP activates the Wnt signal transduction pathway and enhances the proliferation of bone marrow stromal stem cells. Cell Biol. Int. 2013, 37, 1225–1232. [Google Scholar] [CrossRef]
- Winn, R.A.; Bremnes, R.M.; Bemis, L.; Franklin, W.A.; Miller, Y.E.; Cool, C.; Heasley, L.E. Gamma-catenin expression is reduced or absent in a subset of human lung cancers and re-expression inhibits transformed cell growth. Oncogene 2002, 21, 7497–7506. [Google Scholar] [CrossRef]
- Fu, S.; Jin, D.; Liu, S.; Wang, L.; Wang, Z.; Mei, G.; Zou, Z.L.; Wu, J.Q.; Xu, Z.Y. Protective effect of neuropeptide substance P on bone marrow mesenchymal stem cells against apoptosis induced by serum deprivation. Stem Cells Int. 2015, 2015, 270328. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.T.; Lee, H.T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. Foxm1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef]
- Yu, Y.; Ramena, G.; Elble, R.C. The role of cancer stem cells in relapse of solid tumors. Front. Biosci. 2012, 4, 1528–1541. [Google Scholar] [CrossRef]
- Harrison, S.; Geppetti, P. Substance P. Int. J. Biochem. Cell Biol. 2001, 33, 555–576. [Google Scholar] [CrossRef]
- Hökfelt, T.; Pernow, B.; Wahren, J. Substance P: A pioneer amongst neuropeptides. J. Intern. Med. 2001, 249, 27–40. [Google Scholar] [CrossRef]
- Esteban, F.; Muñoz, M.; González-Moles, M.A.; Rosso, M. A role for substance P in cancer promotion and progression: A mechanism to counteract intracellular death signals following oncogene activation or DNA damage. Cancer Metastasis Rev. 2006, 25, 137–145. [Google Scholar] [CrossRef]
- Akazawa, T.; Kwatra, S.G.; Goldsmith, L.E.; Richardson, M.D.; Cox, E.A.; Sampson, J.H.; Kwatra, M.M. A constitutively active form of neurokinin 1 receptor and neurokinin 1 receptor-mediated apoptosis in glioblastomas. J. Neurochem. 2009, 109, 1079–1086. [Google Scholar] [CrossRef]
- Wang, J.G.; Yu, J.; Hu, J.L.; Yang, W.L.; Ren, H.; Ding, D.; Zhang, L.; Liu, X.P. Neurokinin-1activation affects EGFR related signal transduction in triple negative breast cancer. Cell Signal. 2015, 27, 1315–1324. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Medrano, S.; Gruenstein, E.; Dimlich, R.V. Substance P receptors on human astrocytoma cells are linked to glycogen breakdown. Neurosci. Lett. 1994, 167, 14–18. [Google Scholar] [CrossRef]
- Ougolkov, A.V.; Fernández-Zapico, M.E.; Savoy, D.N.; Urrutia, R.A.; Billadeau, D.D. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005, 65, 2076–2081. [Google Scholar] [CrossRef]
- Zeng, J.; Liu, D.; Qiu, Z.; Huang, Y.; Chen, B.; Wang, L.; Xu, H.; Huang, N.; Liu, L.; Li, W. GSK3β overexpression indicates poor prognosis and its inhibition reduces cell proliferation and survival of non-small cell lung cancer cells. PLoS ONE 2014, 9, e91231. [Google Scholar] [CrossRef]
- Wiedermann, C.J.; Auer, B.; Sitte, B.; Reinisch, N.; Schratzberger, P.; Kähler, C.M. Induction of endothelial cell differentiation into capillary-like structures by substance P. Eur. J. Pharmacol. 1996, 298, 335–338. [Google Scholar] [CrossRef]
- Ho, W.Z.; Lai, J.P.; Zhu, X.H.; Uvaydova, M.; Douglas, S.D. Human monocytes and macrophages express substance P and neurokinin-1 receptor. J. Immunol. 1997, 159, 5654–5660. [Google Scholar]
- Walsh, D.A.; Hu, D.E.; Mapp, P.I.; Polak, J.M.; Blake, D.R.; Fan, T.P. Innervation and neurokinin receptors during angiogenesis in the rat sponge granuloma. Histochem. J. 1996, 28, 759–769. [Google Scholar] [CrossRef]
- Ziche, M.; Morbidelli, L.; Pacini, M.; Gepetti, P.; Alessandri, G.; Maggi, C.A. Substance P stimulates neovascularization in vivo and proliferation of cultured endothelial cells. Microvasc. Res. 1990, 40, 264–278. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Zhang, B.; Kempuraj, D.; Tagen, M.; Vasiadi, M.; Angelidou, A.; Alysandratos, K.D.; Kalogeromitros, D.; Asadi, S.; Stavrianeas, N.; et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. USA 2010, 107, 4448–4453. [Google Scholar] [CrossRef]
- Guha, S.; Eibl, G.; Kisfalvi, K.; Fan, R.S.; Burdick, M.; Reber, H.; Hines, O.J.; Strieter, R.; Rozengurt, E. Broad-spectrum G protein–coupled receptor antagonist, [D-Arg1, DTrp5,7,9, Leu11] SP: A dual inhibitor of growth and angiogenesis in pancreatic cancer. Cancer Res. 2005, 65, 2738–2745. [Google Scholar] [CrossRef]
- Fackler, O.T.; Grosse, R. Cell motility through plasma membrane blebbing. J. Cell Biol. 2008, 181, 879–884. [Google Scholar] [CrossRef]
- Lang, K.; Drell, T.L.; Lindecke, A.; Niggemann, B.; Kaltschmidt, C.; Zaenker, K.S.; Entschladen, F. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int. J. Cancer 2004, 112, 231–238. [Google Scholar] [CrossRef]
- Ma, J.; Yuan, S.; Cheng, J.; Kang, S.; Zhao, W.; Zhang, J. Substance P promotes the progression of endometrial adenocarcinoma. Int. J. Gynecol. Cancer 2016, 26, 845–850. [Google Scholar] [CrossRef]
- Meshki, J.; Douglas, S.D.; Lai, J.P.; Schwartz, L.; Kilpatrick, L.E.; Tuluc, F. Neurokinin 1 receptor mediates membrane blebbing in HEK293 cells through a Rho/Rho-associated coiled-coil kinase-dependent mechanism. J. Biol. Chem. 2009, 284, 9280–9289. [Google Scholar] [CrossRef]
- Meshki, J.; Douglas, S.D.; Hu, M.; Leeman, S.E.; Tuluc, F. Substance P induces rapid and transient membrane blebbing in U373MG cells in a p21-activated kinase-dependent manner. PLoS ONE 2011, 6, e25332. [Google Scholar] [CrossRef]
- Ranganathan, S.; Ningappa, M.; Ashokkumar, C.; Higgs, B.W.; Min, J.; Sun, Q.; Schmitt, L.; Subramaniam, S.; Hakonarson, H.; Sindhi, R. Loss of EGFR-ASAP1 signaling in metastatic and unresectable hepatoblastoma. Sci. Rep. 2016, 6, 38347. [Google Scholar] [CrossRef]
- Marín, J.J.G.; Cives-Losada, C.; Asensio, M.; Lozano, E.; Briz, O.; Macías, R.I.R. Mechanisms of anticancer drug resistance in hepatoblastoma. Cancers 2019, 11, 407. [Google Scholar] [CrossRef]
- Herzog, C.E.; Andrassy, R.J.; Eftekhari, F. Childhood cancers: Hepatoblastoma. Oncologist 2000, 5, 445–453. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef]
- Robinson, P.; Kasembeli, M.; Bharadwaj, U.; Engineer, N.; Eckols, K.T.; Tweardy, D.J. Substance P receptor signaling mediates doxorubicin-induced cardiomyocyte apoptosis and triple-negative breast cancer chemoresistance. BioMed Res. Int. 2016, 2016, 1959270. [Google Scholar] [CrossRef]
- Kitchens, C.A.; McDonald, P.R.; Pollack, I.F.; Wipf, P.; Lazo, J.S. Synergy between microtubule destabilizing agents and neurokinin 1 receptor antagonists identified by an siRNA synthetic lethal screen. FASEB J. 2009, 23, 756. [Google Scholar]
- Muñoz, M.; Crespo, J.C.; Crespo, J.P.; Coveñas, R. The neurokinin-1 receptor antagonist aprepitant and radiotherapy: A successful combination therapy in a patient with lung cancer. Mol. Clin. Oncol. 2019, 11, 50–54. [Google Scholar]
- Muñoz, M.; Parrilla, J.; Rosso, M.; Coveñas, R. Antipruritic vs. Antitumor action of aprepitant: A question of dose. Acta Derm. Venereol. 2019, 99, 620–621. [Google Scholar] [CrossRef]
- Paul, B.; Trovato, J.A.; Thompson, J.; Badros, A.Z.; Goloubeva, O. Efficacy of aprepitant in patients receiving high-dose chemotherapy with hematopoietic stem cell support. J. Oncol. Pharm. Pract. 2010, 16, 45–51. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. Safety of neurokinin-1 receptor antagonists. Expert Opin. Drug Saf. 2013, 12, 673–685. [Google Scholar] [CrossRef]
- Kramer, M.S.; Cutler, N.; Feighner, J.; Shrivastava, R.; Carman, J.; Sramek, J.J.; Reines, S.A.; Liu, G.; Snavely, D.; Wyatt-Knowles, E.; et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 1998, 281, 1640–1645. [Google Scholar] [CrossRef]
- Molinos-Quintana, A.; Trujillo-Hacha, P.; Piruat, J.I.; Bejarano-García, J.A.; García-Guerrero, E.; Pérez-Simón, J.A.; Muñoz, M. Human acute myeloid leukemia cells express neurokinin-1 receptor, which is involved in the antileukemic effect of neurokinin-1 receptor antagonists. Investig. New Drugs 2018, 37, 17–26. [Google Scholar] [CrossRef]


{kind=link}
| NK-1R | Express mRNA for the NK-1R |
| Expression of the TAC1R gene is increased | |
| Express full and truncated isoforms of the NK-1R. The HB cells express essentially the truncated form. Expression of the full form is higher in non-tumor cells | |
| SP | A universal mitogen (at nanomolar concentration) of tumor cells, including HB cells |
| Non-peptide NK-1R antagonists | Antiproliferative action in a concentration-dependent manner: The higher the concentration, the greater the antitumor activity |
| Induce cell death by apoptosis, cleavage of caspase-3and proteolysis of poly (ADP-ribose) polymerase | |
| Aprepitant (IC50) for HepT1 (31.1 µM), HuH6 (33.18 µM), HepG2 (38.61 µM) | |
| L-732,138 (IC50) for HepT1 (42 µM), HuH6 (41 µM), HepG2 (110 µM) | |
| L-733,060 (IC50) for HepT1 (15 µM), HuH6 (14 µM), HepG2 (17 µM) | |
| Co-administration of aprepitant and cytostatics exerts a synergistic antitumor effect | |
| Pretreatment of non-tumor cells (human embryonic kidney (HEK)-293) with aprepitant, protected these cells from cytostatic toxicity |
| Items | HB Sample | HB Tumor |
|---|---|---|
| SP/NK-1R system | Expression of both SP and NK-1R | Truncated-TACR1 isoform is expressed ubiquitously among the different subsets of children with HB |
| Non-peptide NK-1R antagonists | In experimental animals: Dual effect of aprepitant (80 mg/kg/day for 24 days). The antagonist decreased the tumor volume (tumor cells die by apoptosis) and the angiogenic activity (decrease of the vascularized area, but not the microvascular density). Aprepitant lowered the tumor-specific alpha-fetoprotein serum level and decreased the number of Ki-67 positive cells |
| NK-1R | NK-1R: A new HB tumor marker |
| NK-1R: Therapeutic target in HB patients, independent of the clinical stage/tumor biology | |
| NK-1R: Involved in the viability of HB cells? | |
| The tr-NK-1R, but not the fl-NK-1R, is associated with cancer? | |
| The response of the NK-1R antagonists depends on the differential expression of the receptor (truncated/full)? | |
| tr-NK-1R: Responsible for a constitutive growth stimulus in the HB cells? | |
| Overexpression of the tr-NK-1R: Correlates with poorer prognosis and advance HB stages? | |
| tr-NK-1R: Not internalized in the HB cells? Involved in the malignant transformation of the HB cells? | |
| SP | Level of SP in the serum of HB patients |
| SP activates GSK-3β in the HB cells (Warburg effect)? | |
| SP is released from the HB cells to promote neovascularization? | |
| SP triggers the migration of HB cells? |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz, M.; Rosso, M.; Coveñas, R. Neurokinin-1 Receptor Antagonists against Hepatoblastoma. Cancers 2019, 11, 1258. https://doi.org/10.3390/cancers11091258
Muñoz M, Rosso M, Coveñas R. Neurokinin-1 Receptor Antagonists against Hepatoblastoma. Cancers. 2019; 11(9):1258. https://doi.org/10.3390/cancers11091258
Chicago/Turabian StyleMuñoz, Miguel, Marisa Rosso, and Rafael Coveñas. 2019. "Neurokinin-1 Receptor Antagonists against Hepatoblastoma" Cancers 11, no. 9: 1258. https://doi.org/10.3390/cancers11091258
APA StyleMuñoz, M., Rosso, M., & Coveñas, R. (2019). Neurokinin-1 Receptor Antagonists against Hepatoblastoma. Cancers, 11(9), 1258. https://doi.org/10.3390/cancers11091258