Photodynamic Therapy Activity of New Porphyrin-Xylan-Coated Silica Nanoparticles in Human Colorectal Cancer

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
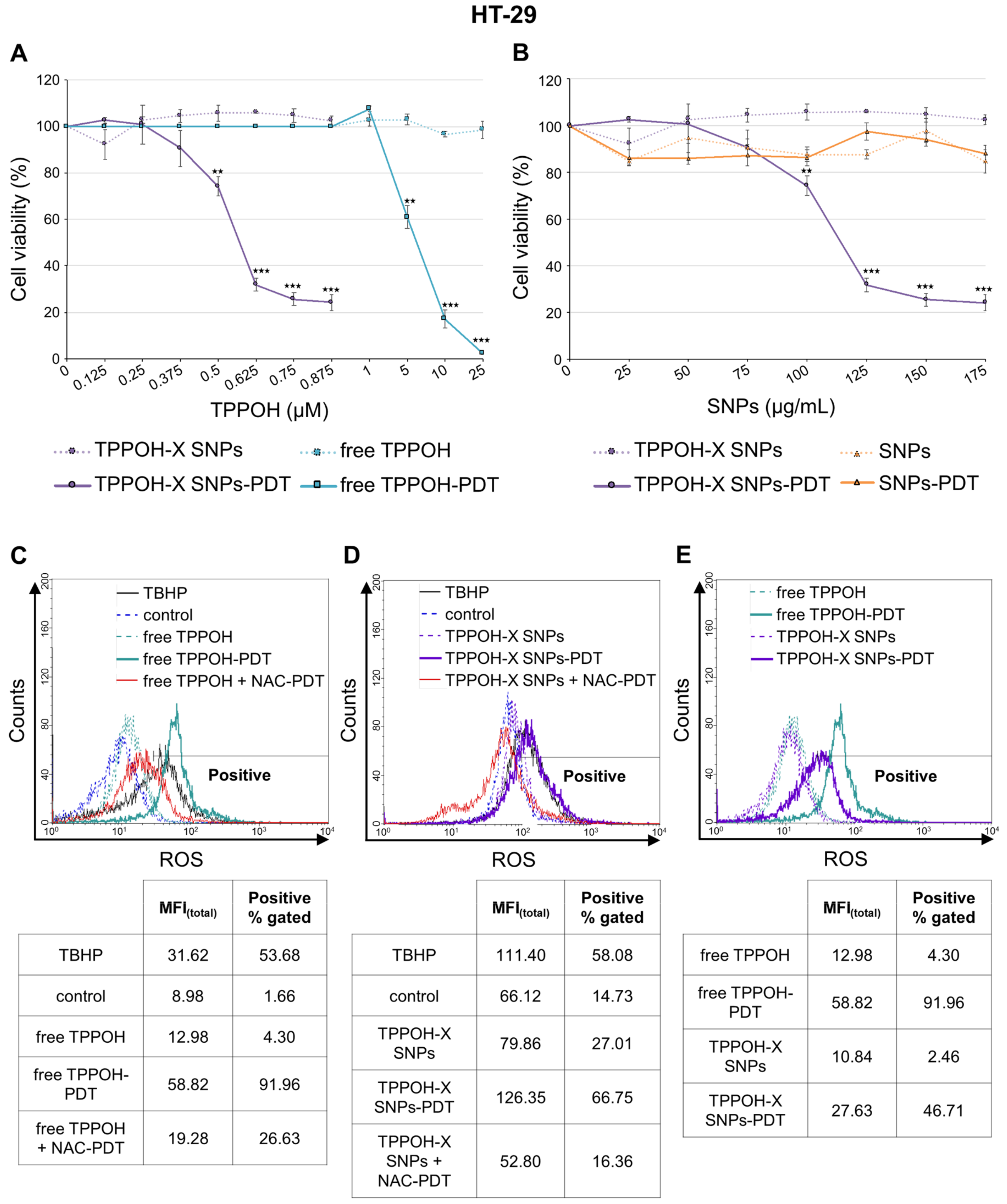
2.1. SNPs Vectorization Enhanced TPPOH-PDT Phototoxic Effects Mediated by ROS Production
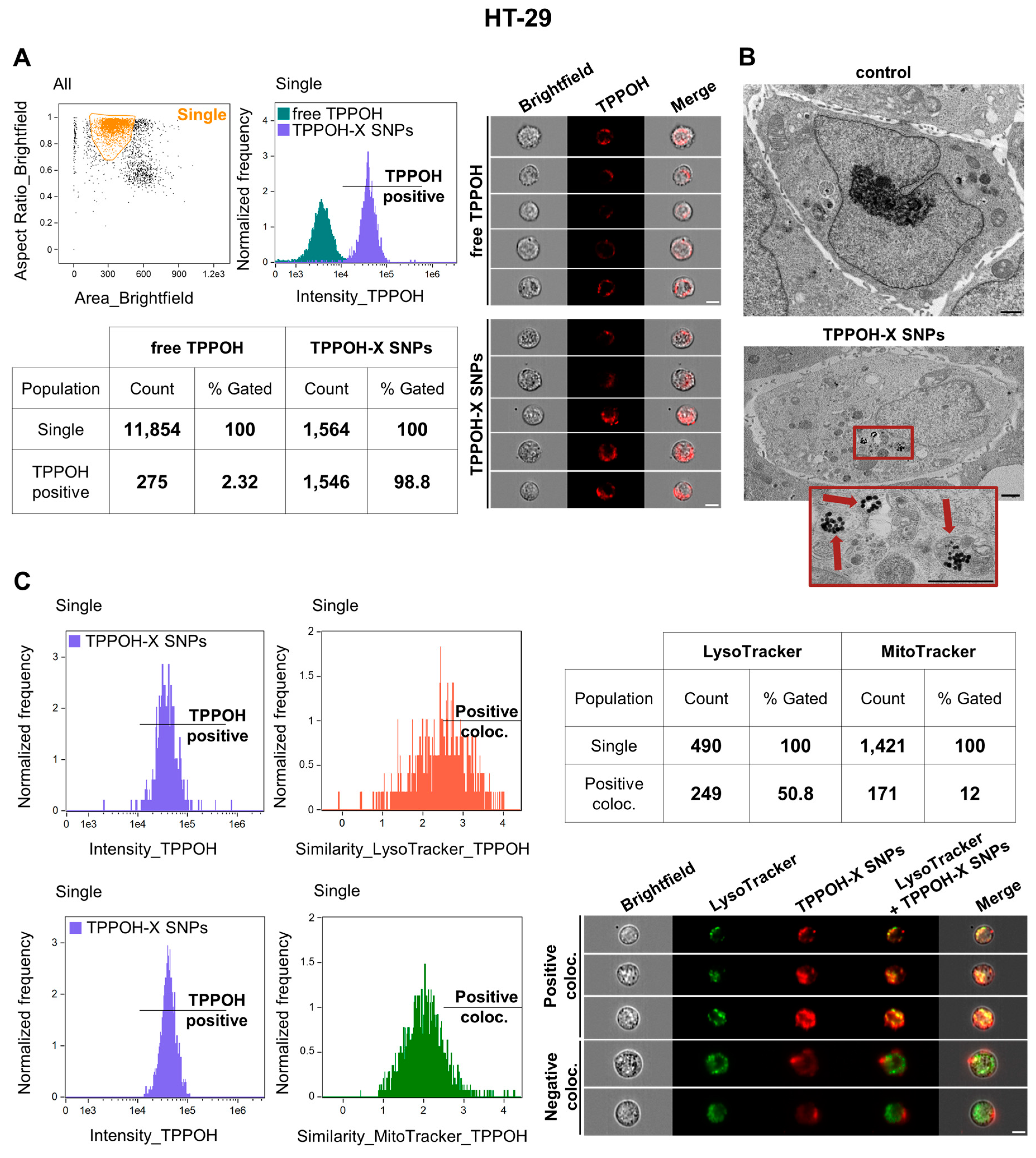
2.2. SNPs Vectorization Increased TPPOH Accumulation in Lysosomes
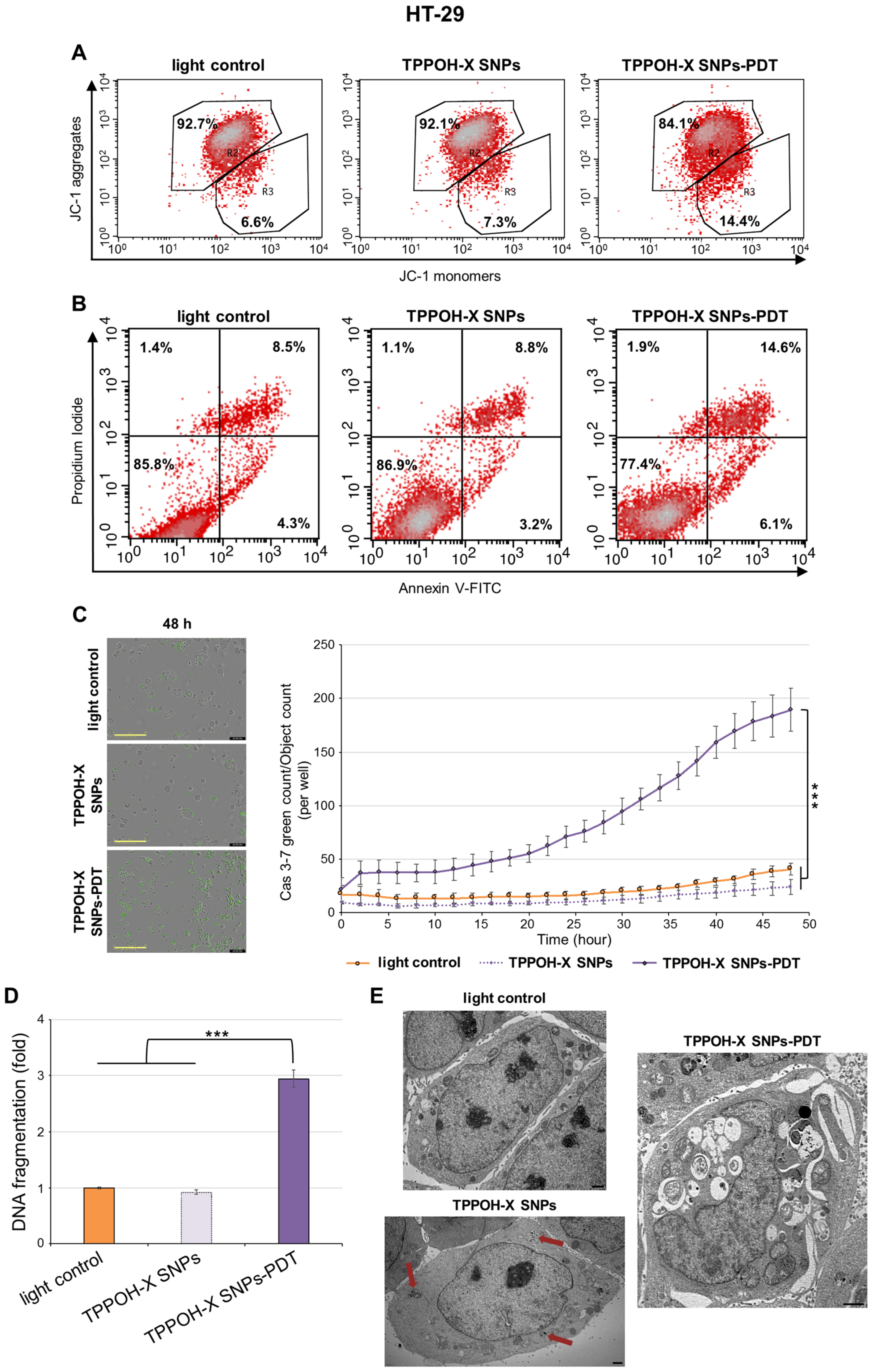
2.3. TPPOH-X SNPs-PDT Induced Apoptosis
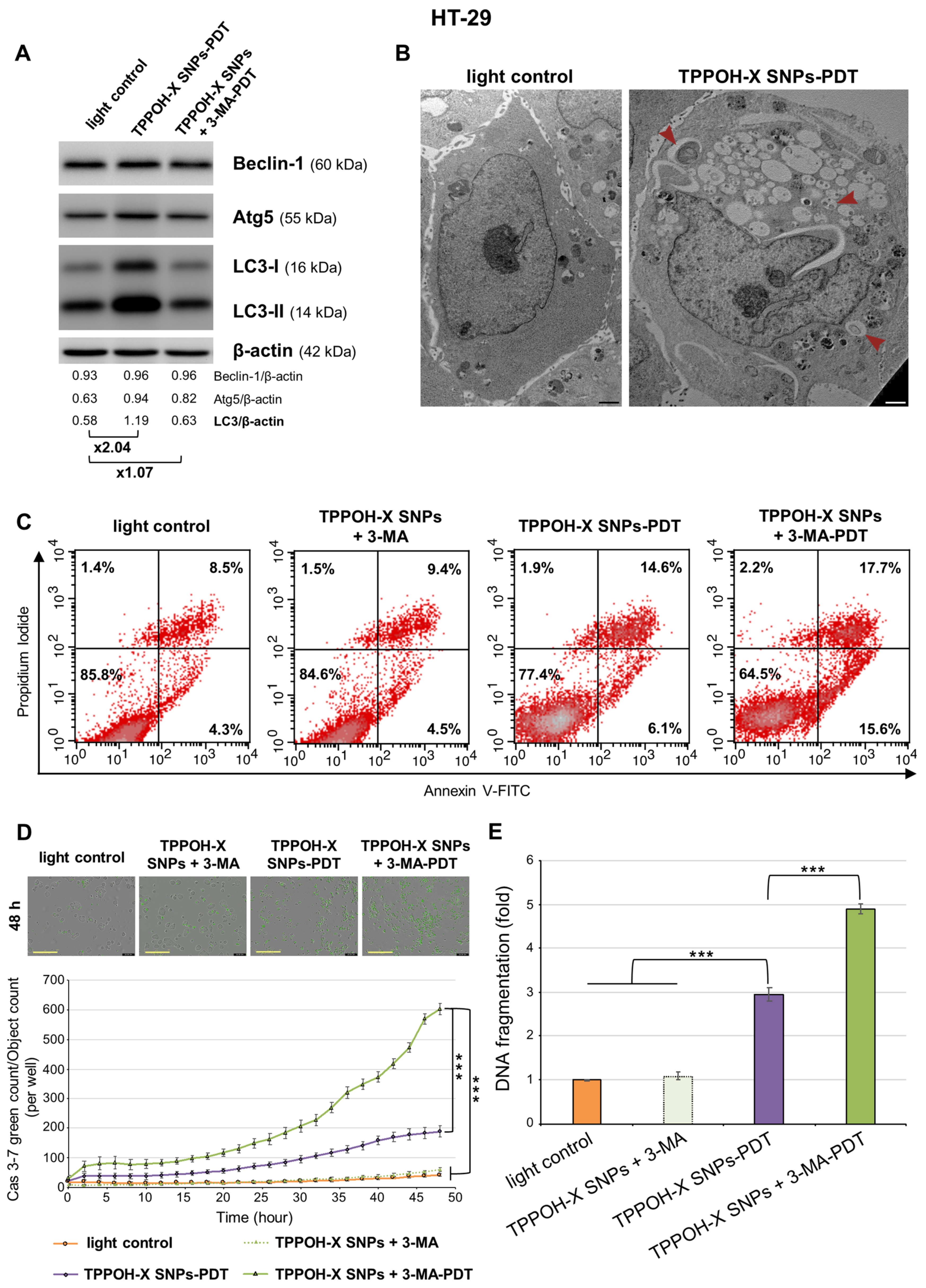
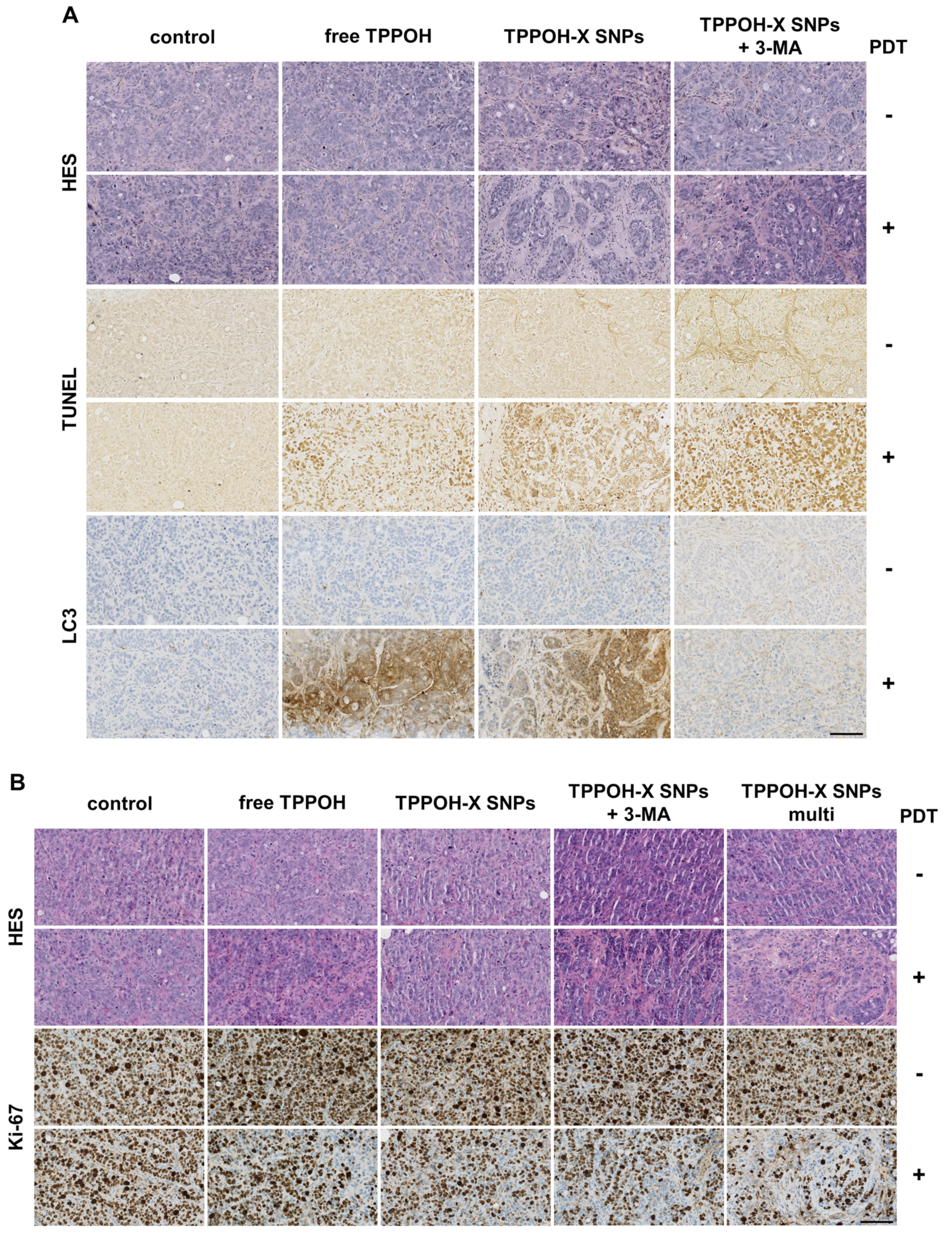
2.4. Autophagy Inhibition Enhanced TPPOH-X SNPs-PDT-Induced Apoptosis
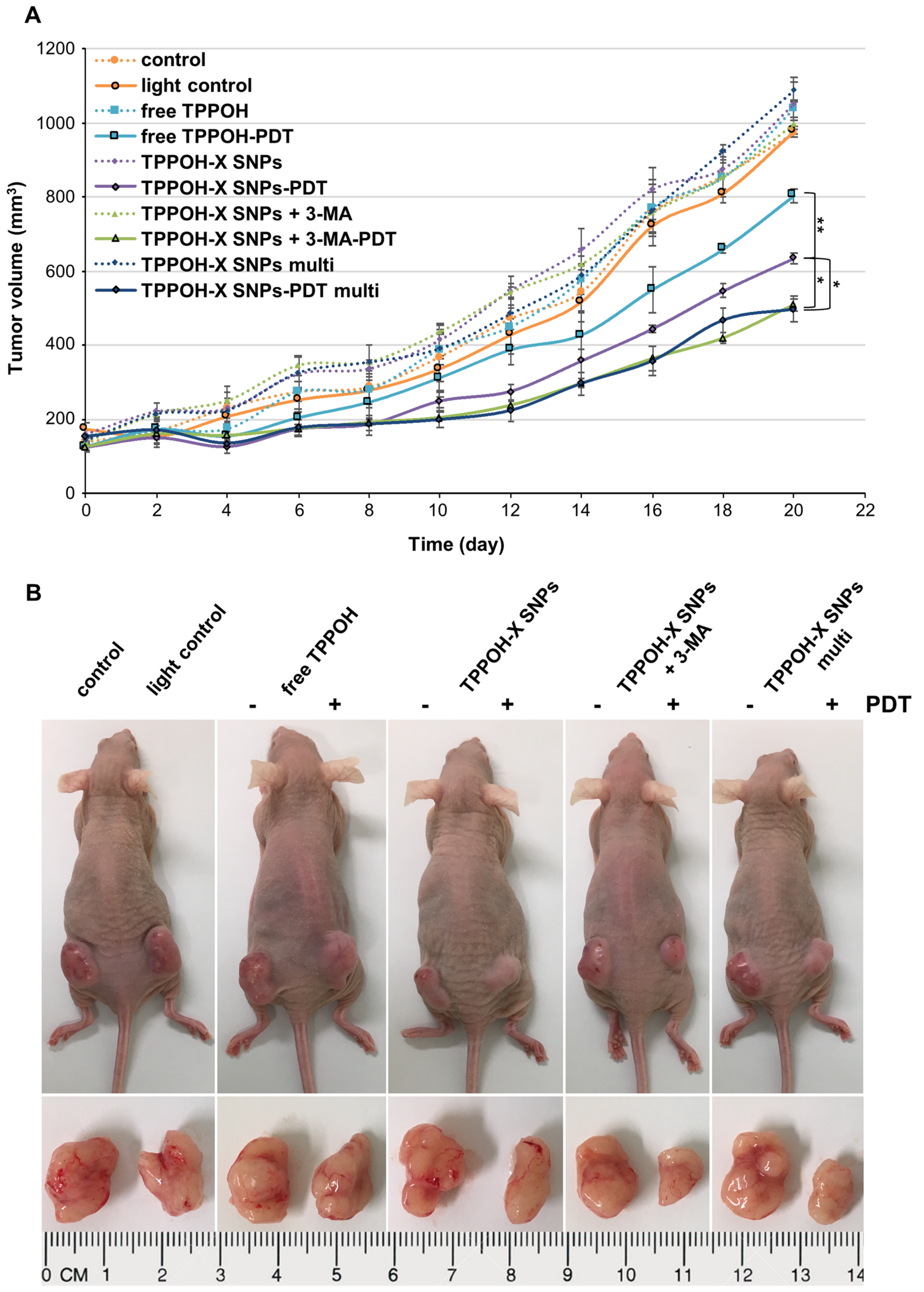
2.5. SNPs Vectorization and Autophagy Inhibition Enhanced TPPOH-PDT Effects on Suppressing CRC Tumor Growth In Vivo
2.6. TPPOH-X SNPs-PDT Induced Apoptosis In Vivo
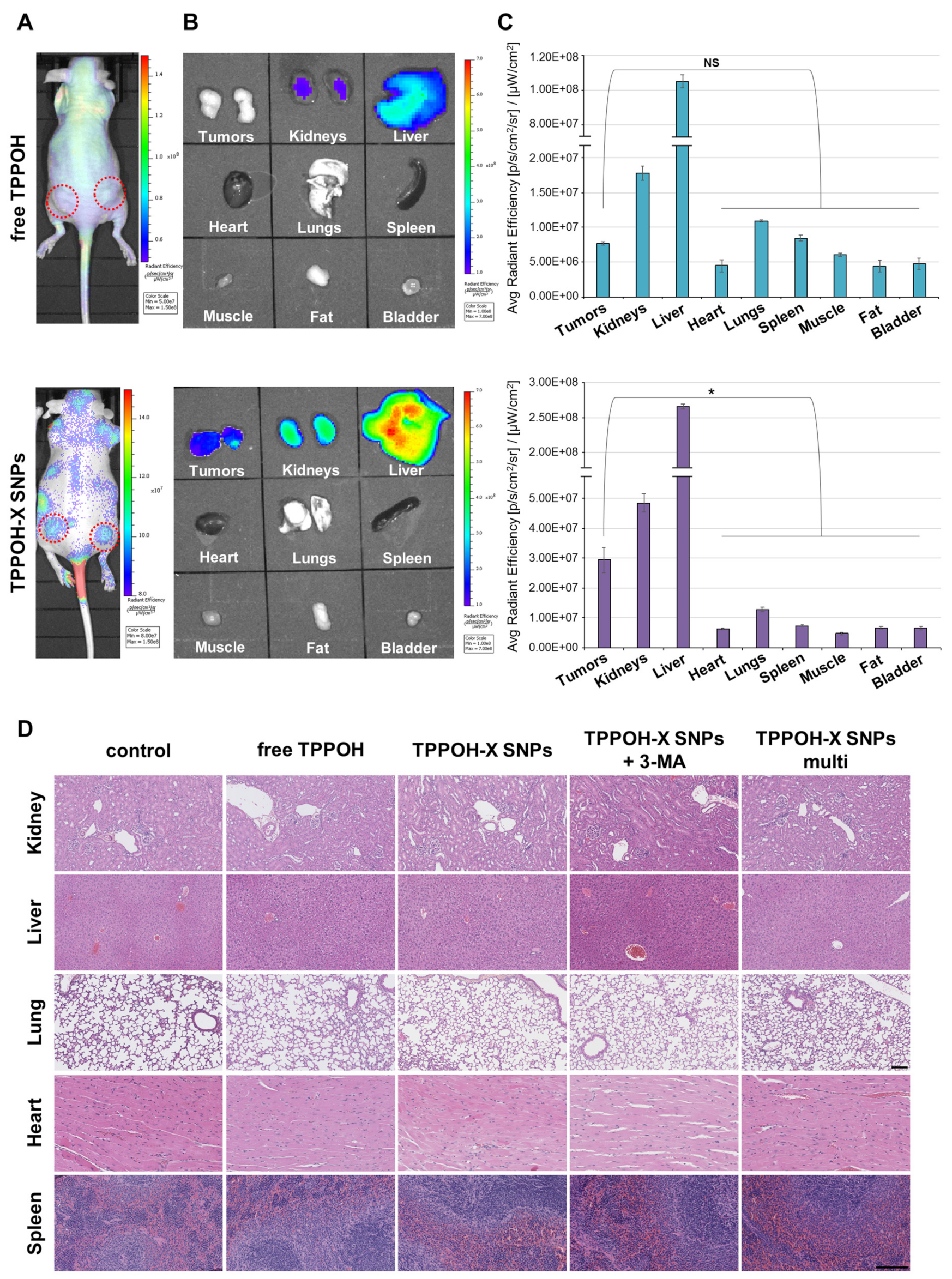
2.7. SNPs Vectorization Enhanced Tumor-Targeting without Systemic Toxicity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Free TPPOH and TPPOH-X SNPs
4.3. Cell Culture
4.4. In Vitro Phototoxicity of TPPOH-PDT
4.5. Intracellular ROS Generation by TPPOH-PDT
4.6. TPPOH Cellular Uptake and Localization
4.7. Transmission Electron Microscopy (TEM)
4.8. Autophagy Detection and Inhibition
4.9. In Vitro Apoptosis by TPPOH-X SNPs
4.10. Heterotopic CRC Model
4.11. In Vivo Antitumor Efficacy and Biosafety Evaluation of TPPOH-PDT
4.12. In Vivo Biodistribution of TPPOH-X SNPs
4.13. Ethical Statement
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Globocan. Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2018. Information and Online Prediction. WHO International Agency for Research of Cancer. 2018. Available online: http://gco.iarc.fr/today/home (accessed on 8 July 2019).
- Xue, L.; Williamson, A.; Gaines, S.; Andolfi, C.; Paul-Olson, T.; Neerukonda, A.; Steinhagen, E.; Smith, R.; Cannon, L.M.; Polite, B.; et al. An Update on Colorectal Cancer. Curr. Probl. Surg. 2018, 5555, 76–116. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Yamashita, K.; Hasegawa, H.; Oshikiri, T.; Hosono, M.; Higashino, N.; Yamamoto, M.; Matsuda, Y.; Kanaji, S.; Nakamura, T.; et al. Recent updates in the surgical treatment of colorectal cancer. Ann. Gastroenterol. Surg. 2018, 22, 129–136. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Mayer, R.J. Systemic Treatment of Colorectal Cancer. Gastroenterology 2008, 134134, 1296–1310. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeught, K.; Xu, H.-C.; Li, Y.-J.; Lu, X.-B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 2424, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Kawczyk-Krupka, A.; Bugaj, A.M.; Latos, W.; Zaremba, K.; Wawrzyniec, K.; Kucharzewski, M.; Sieroń, A. Photodynamic therapy in colorectal cancer treatment-The state of the art in preclinical research. Photodiagnosis Photodyn. Ther. 2016, 13, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy–mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–2107. [Google Scholar] [CrossRef]
- Hamblin, M.R.; Mroz, P. Advances in Photodynamic Therapy: Basic, Translational and Clinical, 1st ed.; Boston, Mass, Artech House Publishers: Norwood, MA, USA, 2008. [Google Scholar]
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 33, 380–387. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, M.C.; Crutchley, R.J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233, 351–371. [Google Scholar] [CrossRef]
- Mroz, P.; Yaroslavsky, A.; Kharkwal, G.B.; Hamblin, M.R. Cell Death Pathways in Photodynamic Therapy of Cancer. Cancers 2011, 33, 2516–2539. [Google Scholar] [CrossRef]
- Plaetzer, K.; Kiesslich, T.; Verwanger, T.; Krammer, B. The Modes of Cell Death Induced by PDT: An Overview. Med. Laser Appl. 2003, 1818, 7–19. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473473, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Chouhan, R.; Bagri, L.P.; Bajpai, A.K. Strategies of Targeting Tumors and Cancers. J. Cancer Res. Updates 2012, 1, 129–152. [Google Scholar]
- Huang, Y.Y.; Sharma, S.K.; Dai, T.; Chung, H.; Yaroslavsky, A.; Garcia-Diaz, M.; Chang, J.; Chiang, L.Y.; Hamblin, M.R. Can nanotechnology potentiate photodynamic therapy? Nanotechnol. Rev. 2012, 11, 111–146. [Google Scholar] [CrossRef]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug. Discov. Today 2006, 1111, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, X.; Dai, Z. Porphyrin-loaded nanoparticles for cancer theranostics. Nanoscale 2016, 8, 12394–12405. [Google Scholar] [CrossRef] [PubMed]
- Debele, T.A.; Peng, S.; Tsai, H.-C. Drug Carrier for Photodynamic Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 22094–22136. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wu, M.; Xiaojun Zhao, J. Recent Development of Silica Nanoparticles as Delivery Vectors for Cancer Imaging and Therapy. Nanomedicine 2014, 10, 297–312. [Google Scholar] [CrossRef]
- Couleaud, P.; Morosini, V.; Frochot, C.; Richeter, S.; Raehm, L.; Durand, J.-O. Silica-based nanoparticles for photodynamic therapy applications. Nanoscale 2010, 2, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, H.; Shi, J. In Vivo Bio-Safety Evaluations and Diagnostic/Therapeutic Applications of Chemically Designed Mesoporous Silica Nanoparticles. Adv. Mater. 2013, 25, 3144–3176. [Google Scholar] [CrossRef] [PubMed]
- Lemarchand, C.; Gref, R.; Couvreur, P. Polysaccharide-decorated nanoparticles. Eur. J. Pharm. Biopharm. 2004, 58, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Gref, R. Surface-engineered nanoparticles as drug carriers. In Synthesis, Functionalization and Surface Treatment of Nanoparticles, 1st ed.; Baraton, M.I., Ed.; American Scientific Publishers: California, CA, USA, 2002; pp. 233–256. [Google Scholar]
- Owens, D.E.; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Shrotri, A.; Kobayashi, H.; Fukuoka, A. Chapter Two-Catalytic Conversion of Structural Carbohydrates and Lignin to Chemicals. Adv. Catal. 2017, 60, 59–123. [Google Scholar]
- Ma, J.; Li, D.; Zhong, L.; Du, F.; Tan, J.; Yang, J.; Peng, X. Synthesis and characterization of biofunctional quaternized xylan-Fe2O3 core/shell nanocomposites and modification with polylysine and folic acid. Carbohydr. Polym. 2018, 199, 382–389. [Google Scholar] [CrossRef]
- Daus, S.; Heinze, T. Xylan-based nanoparticles: Prodrugs for ibuprofen release. Macromol. Biosci. 2010, 10, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Bouramtane, S.; Bretin, L.; Pinon, A.; Leger, D.; Liagre, B.; Richard, L.; Brégier, F.; Sol, V.; Chaleix, V. Porphyrin-xylan-coated silica nanoparticles for anticancer photodynamic therapy. Carbohydr. Polym. 2019, 213, 168–175. [Google Scholar] [CrossRef]
- Lai, K.; Killingsworth, M.C.; Lee, C.S. The significance of autophagy in colorectal cancer pathogenesis and implications for therapy. J. Clin. Pathol. 2014, 67, 854–858. [Google Scholar] [CrossRef]
- Duan, X.; Chen, B.; Cui, Y.; Zhou, L.; Wu, C.; Yang, Z.; Wen, Y.; Miao, X.; Li, Q.; Xiong, L.; et al. Ready player one? Autophagy shapes resistance to photodynamic therapy in cancers. Apoptosis 2018, 23, 587–606. [Google Scholar] [CrossRef]
- Hackbarth, S.; Horneffer, V.; Wiehe, A.; Hillenkamp, F.; Röder, B. Photophysical properties of pheophorbide-a-substituted diaminobutane poly-propylene-imine dendrimer. Chem. Phys. 2001, 269, 339–346. [Google Scholar] [CrossRef]
- Li, Y.; Jang, W.D.; Nishiyama, N.; Kishimura, A.; Kawauchi, S.; Morimoto, Y.; Miake, S.; Yamashita, T.; Kikuchi, M.; Aida, T.; et al. Dendrimer Generation Effects on Photodynamic Efficacy of Dendrimer Porphyrins and Dendrimer-Loaded Supramolecular Nanocarriers. Chem. Mater. 2007, 19, 5557–5562. [Google Scholar] [CrossRef]
- Roy, I.; Ohulchanskyy, T.Y.; Pudavar, H.E.; Bergey, E.J.; Oseroff, A.R.; Morgan, J.; Dougherty, T.J.; Prasad, P.N. Ceramic-based nanoparticles entrapping water-insoluble photosensitizing anticancer drugs: A novel drug-carrier system for photodynamic therapy. J. Am. Chem. Soc. 2003, 125, 7860–7865. [Google Scholar] [CrossRef] [PubMed]
- Secret, E.; Maynadier, M.; Gallud, A.; Gary-Bobo, M.; Chaix, A.; Belamie, E.; Maillard, P.; Sailor, M.J.; Garcia, M.; Durand, J.O.; et al. Anionic porphyrin-grafted porous silicon nanoparticles for photodynamic therapy. Chem. Commun. (Camb.) 2013, 49, 4202–4204. [Google Scholar] [CrossRef] [PubMed]
- Youssef, Z.; Jouan-Hureaux, V.; Colombeau, L.; Arnoux, P.; Moussaron, A.; Baros, F.; Toufaily, J.; Hamieh, T.; Roques-Carmes, T.; Frochot, C. Titania and silica nanoparticles coupled to Chlorin e6 for anti-cancer photodynamic therapy. Photodiagnosis Photodyn. Ther. 2018, 22, 115–126. [Google Scholar] [CrossRef]
- Bharathiraja, S.; Moorthy, M.S.; Manivasagan, P.; Seo, H.; Lee, K.D.; Oh, J. Chlorin e6 conjugated silica nanoparticles for targeted and effective photodynamic therapy. Photodiagnosis Photodyn. Ther. 2017, 19, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Brezániová, I.; Záruba, K.; Králová, J.; Adámková, H.; Ulbrich, P.; Poučková, P.; Hrubý, M.; Štěpánek, P.; Král, V. Silica-based nanoparticles are efficient delivery systems for temoporfin. Photodiagnosis Photodyn. Ther. 2018, 21, 275–284. [Google Scholar]
- Simon, V.; Devaux, C.; Darmon, A.; Donnet, T.; Thiénot, E.; Germain, M.; Honnorat, J.; Duval, A.; Pottier, A.; Borghi, E.; et al. Pp IX silica nanoparticles demonstrate differential interactions with in vitro tumor cell lines and in vivo mouse models of human cancers. Photochem. Photobiol. 2010, 86, 213–222. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, B.; Deeba, F.; Bano, S.; Kulshreshtha, A.; Gopinath, P.; Negi, Y.S. Lipophilic 5-fluorouracil prodrug encapsulated xylan-stearic acid conjugates nanoparticles for colon cancer therapy. Int. J. Biol. Macromol. 2019, 128, 204–213. [Google Scholar]
- Sauraj Kumar, S.U.; Kumar, V.; Priyadarshi, R.; Gopinath, P.; Negi, Y.S. pH-responsive prodrug nanoparticles based on xylan-curcumin conjugate for the efficient delivery of curcumin in cancer therapy. Carbohydr. Polym. 2018, 188, 252–259. [Google Scholar] [CrossRef]
- Liu, X.; Sun, J. Endothelial cells dysfunction induced by silica nanoparticles through oxidative stress via JNK/P53 and NF-kB pathways. Biomaterials 2010, 31, 8198–8209. [Google Scholar] [CrossRef]
- Sergent, J.-A.; Paget, V.; Chevillard, S. Toxicity and genotoxicity of nano-SiO2 on human epithelial intestinal HT-29 cell line. Ann. Occup. Hyg. 2012, 56, 622–630. [Google Scholar]
- Cho, M.; Cho, W.S.; Choi, M.; Kim, S.J.; Han, B.S.; Kim, S.H.; Kim, H.O.; Sheen, Y.Y.; Jeong, J. The impact of size on tissue distribution and elimination by single intravenous injection of silica nanoparticles. Toxicol. Lett. 2009, 189, 177–183. [Google Scholar] [CrossRef]
- Kumar, R.; Roy, I.; Ohulchanskky, T.Y.; Vathy, L.A.; Bergey, E.J.; Sajjad, M.; Prasad, P.N. In vivo biodistribution and clearance studies using multimodal organically modified silica nanoparticles. ACS Nano 2010, 4, 699–708. [Google Scholar] [CrossRef]
- Chan, W.T.; Liu, C.C.; Chiau, J.S.C.; Tsai, S.T.; Liang, C.K.; Cheng, M.L.; Lee, H.C.; Yeung, C.Y.; Hou, S.Y. In vivo toxicologic study of larger silica nanoparticles in mice. Int. J. Nanomedicine 2017, 12, 3421–3432. [Google Scholar] [CrossRef]
- Liu, T.; Li, L.; Teng, X.; Huang, X.; Liu, H.; Chen, D.; Ren, J.; He, J.; Tang, F. Single and repeated dose toxicity of mesoporous hollow silica nanoparticles in intravenously exposed mice. Biomaterials 2011, 32, 1657–1668. [Google Scholar] [CrossRef]
- Zhou, C.N. Mechanisms of tumor necrosis induced by photodynamic therapy. J. Photochem. Photobiol. B 1989, 3, 299–318. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, F.; Jiang, J.; Sun, C.; Wang, X.; Shen, M.; Tian, R.; Shi, C.; Xu, M.; Peng, F.; et al. Synergistic antitumor activity of withaferin A combined with oxaliplatin triggers reactive oxygen species-mediated inactivation of the PI3K/AKT pathway in human pancreatic cancer cells. Cancer Lett. 2015, 357, 219–230. [Google Scholar] [CrossRef]
- Skulachev, V.P. Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells and organisms. Mol. Asp.S Med. 1999, 20, 139–184. [Google Scholar] [CrossRef]
- Costa, L.D.; de Silva, J.A.; Fonseca, S.M.; Arranja, C.T.; Urbano, A.M.; Sobral, A.J.F.N. Photophysical Characterization and in Vitro Phototoxicity Evaluation of 5,10,15,20-Tetra (quinolin-2-yl) porphyrin as a Potential Sensitizer for Photodynamic Therapy. Molecules 2016, 21, 439. [Google Scholar] [CrossRef]
- Baldea, I.; Olteanu, D.E.; Bolfa, P.; Ion, R.M.; Decea, N.; Cenariu, M.; Banciu, M.; Sesarman, A.V.; Filip, A.G. Efficiency of photodynamic therapy on WM35 melanoma with synthetic porphyrins: Role of chemical structure, intracellular targeting and antioxidant defense. J. Photochem. Photobiol. B 2015, 151, 142–152. [Google Scholar] [CrossRef]
- Liao, P.Y.; Gao, Y.H.; Wang, X.R.; Bao, L.L.; Bian, J.; Hu, T.S.; Zheng, M.Z.; Yan, Y.J.; Chen, Z.L. Tetraphenylporphyrin derivatives possessing piperidine group as potential agents for photodynamic therapy. J. Photochem. Photobiol. B 2016, 165, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Roby, A.; Erdogan, S.; Torchilin, V.P. Enhanced in vivo antitumor efficacy of poorly soluble PDT agent, meso-tetraphenylporphine, in PEG-PE-based tumor-targeted immunomicelles. Cancer Biol. Ther. 2007, 6, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wu, L.; Li, J.; Zhang, D.; Lan, S.; Zhang, X.; Lin, X.; Liu, G.; Liu, X.; Liu, J. Self-Luminescing Theranostic Nanoreactors with Intraparticle Relayed Energy Transfer for Tumor Microenvironment Activated Imaging and Photodynamic Therapy. Theranostics 2019, 9, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Wang, X.; Wang, P.; Zhang, K.; Liu, Q. Role of p38MAPK in apoptosis and autophagy responses to photodynamic therapy with Chlorin e6. Photodiagnosis Photodyn. Ther. 2015, 12, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Liu, Z.; Ouyang, G.; Lin, L.; Huang, H.; Kang, H.; Chen, W.; Miao, X.; Wen, Y. Autophagy inhibition enhances photocytotoxicity of Photosan-II in human colorectal cancer cells. Oncotarget 2017, 8, 6419–6432. [Google Scholar] [CrossRef]
- Wei, M.F.; Chen, M.W.; Chen, K.C.; Lou, P.J.; Lin, S.Y.F.; Hung, S.C.; Hsiao, M.; Yao, C.J.; Shieh, M.J. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy 2014, 10, 1179–1192. [Google Scholar] [CrossRef]
- Tu, P.; Huang, Q.; Ou, Y.; Du, X.; Li, K.; Tao, Y.; Yin, H. Aloe-emodin-mediated photodynamic therapy induces autophagy and apoptosis in human osteosarcoma cell line MG-63 through the ROS/JNK signaling pathway. Oncol. Rep. 2016, 35, 3209–3215. [Google Scholar] [CrossRef]
- Zhu, J.; Tian, S.; Li, K.T.; Chen, Q.; Jiang, Y.; Lin, H.D.; Yu, L.H.; Bai, D.Q. Inhibition of breast cancer cell growth by methyl pyropheophenylchlorin photodynamic therapy is mediated though endoplasmic reticulum stress-induced autophagy in vitro and vivo. Cancer Med. 2018, 7, 1908–1920. [Google Scholar] [CrossRef]
- Lepage, C.; Léger, D.Y.; Bertrand, J.; Martin, F.; Beneytout, J.L.; Liagre, B. Diosgenin induces death receptor-5 through activation of p38 pathway and promotes TRAIL-induced apoptosis in colon cancer cells. Cancer Lett. 2011, 301, 193–202. [Google Scholar] [CrossRef]
- Lu, J.; Liong, M.; Li, Z.; Zink, J.I.; Tamanoi, F. Biocompatibility, Biodistribution, and Drug-Delivery Efficiency of Mesoporous Silica Nanoparticles for Cancer Therapy in Animals. Small 2010, 6, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Guidance Document on Using In Vitro Data to Estimate In Vivo Starting Doses for Acute Toxicity. NIH Publication No. 01–4500. 2001. Available online: https://ntp.niehs.nih.gov/iccvam/docs/acutetox_docs/guidance0801/iv_guide.pdf (accessed on 3 April 2019).
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Kuwano, H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur. J. Cancer. 2010, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Fidanzi-Dugas, C.; Liagre, B.; Chemin, G.; Perraud, A.; Carrion, C.; Couquet, C.Y.; Granet, R.; Sol, V.; Léger, D.Y. Analysis of the in vitro and in vivo effects of photodynamic therapy on prostate cancer by using new photosensitizers, protoporphyrin IX-polyamine derivatives. Biochim. Biophys. Acta 2017, 1861, 1676–1690. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bretin, L.; Pinon, A.; Bouramtane, S.; Ouk, C.; Richard, L.; Perrin, M.-L.; Chaunavel, A.; Carrion, C.; Bregier, F.; Sol, V.; et al. Photodynamic Therapy Activity of New Porphyrin-Xylan-Coated Silica Nanoparticles in Human Colorectal Cancer. Cancers 2019, 11, 1474. https://doi.org/10.3390/cancers11101474
Bretin L, Pinon A, Bouramtane S, Ouk C, Richard L, Perrin M-L, Chaunavel A, Carrion C, Bregier F, Sol V, et al. Photodynamic Therapy Activity of New Porphyrin-Xylan-Coated Silica Nanoparticles in Human Colorectal Cancer. Cancers. 2019; 11(10):1474. https://doi.org/10.3390/cancers11101474
Chicago/Turabian StyleBretin, Ludovic, Aline Pinon, Soukaina Bouramtane, Catherine Ouk, Laurence Richard, Marie-Laure Perrin, Alain Chaunavel, Claire Carrion, Frédérique Bregier, Vincent Sol, and et al. 2019. "Photodynamic Therapy Activity of New Porphyrin-Xylan-Coated Silica Nanoparticles in Human Colorectal Cancer" Cancers 11, no. 10: 1474. https://doi.org/10.3390/cancers11101474
APA StyleBretin, L., Pinon, A., Bouramtane, S., Ouk, C., Richard, L., Perrin, M.-L., Chaunavel, A., Carrion, C., Bregier, F., Sol, V., Chaleix, V., Leger, D. Y., & Liagre, B. (2019). Photodynamic Therapy Activity of New Porphyrin-Xylan-Coated Silica Nanoparticles in Human Colorectal Cancer. Cancers, 11(10), 1474. https://doi.org/10.3390/cancers11101474