The Roles of Protein Tyrosine Phosphatases in Hepatocellular Carcinoma


_Kwok.png)
Abstract
:1. Introduction
2. PTPs Involved in HCC
2.1. The Role of Classical Receptor PTPs in Hepatocellular Carcinoma
2.2. The Role of Classical Non-Receptor PTPs in Hepatocellular Carcinoma
2.3. The Role of Dual Specificity Phosphatases in Hepatocellular Carcinoma
3. Targeting the PTPs for Therapy in Hepatocellular Carcinoma
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ursino, S.; Greco, C.; Cartei, F.; Colosimo, C.; Stefanelli, A.; Cacopardo, B.; Berretta, M.; Fiorica, F. Radiotherapy and hepatocellular carcinoma: Update and review of the literature. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1599–1604. [Google Scholar] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Kuban-Jankowska, A.; Gorska, M.; Knap, N.; Cappello, F.; Wozniak, M. Protein tyrosine phosphatases in pathological process. Front. Biosci. (Landmark Ed.) 2015, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Van Renne, N.; Roca Suarez, A.A.; Duong, F.H.; Gondeau, C.; Calabrese, D.; Fontaine, N.; Ababsa, A.; Bandiera, S.; Croonenborghs, T.; Pochet, N.; et al. miR-135a-5p-mediated downregulation of protein tyrosine phosphatase receptor delta is a candidate driver of HCV-associated hepatocarcinogenesis. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Acun, T.; Demir, K.; Oztas, E.; Arango, D.; Yakicier, M.C. PTPRD is homozygously deleted and epigenetically downregulated in human hepatocellular carcinomas. OMICS 2015, 19, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Bera, R.; Chiou, C.Y.; Yu, M.C.; Peng, J.M.; He, C.R.; Hsu, C.Y.; Huang, H.L.; Ho, U.Y.; Lin, S.M.; Lin, Y.J.; et al. Functional genomics identified a novel protein tyrosine phosphatase receptor type F-mediated growth inhibition in hepatocarcinogenesis. Hepatology 2014, 59, 2238–2250. [Google Scholar] [CrossRef] [PubMed]
- Nagano, H.; Noguchi, T.; Inagaki, K.; Yoon, S.; Matozaki, T.; Itoh, H.; Kasuga, M.; Hayashi, Y. Downregulation of stomach cancer-associated protein tyrosine phosphatase-1 (SAP-1) in advanced human hepatocellular carcinoma. Oncogene 2003, 22, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Xu, J.; Jiang, R.; Wang, Y.; Chen, C.; Deng, L.; Huang, X.; Wang, X.; Sun, B. Estrogen-sensitive PTPRO expression represses hepatocellular carcinoma progression by control of STAT3. Hepatology 2013, 57, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.C.; Gao, Q.; Shi, J.Y.; Guo, W.J.; Yang, L.X.; Liu, X.Y.; Liu, L.Z.; Ma, L.J.; Duan, M.; Zhao, Y.J.; et al. Protein tyrosine phosphatase receptor S acts as a metastatic suppressor in hepatocellular carcinoma by control of epithermal growth factor receptor-induced epithelial-mesenchymal transition. Hepatology 2015, 62, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; You, X.; Chi, X.; Wang, T.; Ye, L.; Niu, J.; Zhang, X. Hepatitis B virus X protein mutant HBxDelta127 promotes proliferation of hepatoma cells through up-regulating miR-215 targeting PTPRT. Biochem. Biophys. Res. Commun. 2014, 444, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Chen, Y.L.; Chu, P.Y.; Chen, L.J.; Hung, M.H.; Shiau, C.W.; Huang, J.W.; Tsai, M.H.; Chen, K.F. Protein tyrosine phosphatase 1B dephosphorylates PITX1 and regulates p120RasGAP in hepatocellular carcinoma. Hepatology 2016, 63, 1528–1543. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Yang, C.Y.; Hua, K.T.; Ho, M.C.; Johansson, G.; Jeng, Y.M.; Chen, C.N.; Chen, M.W.; Lee, W.J.; Su, J.L.; et al. Leukocyte cell-derived chemotaxin 2 antagonizes MET receptor activation to suppress hepatocellular carcinoma vascular invasion by protein tyrosine phosphatase 1B recruitment. Hepatology 2014, 59, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.Y.; Zhou, D.X.; Lu, J.; Zhang, W.J.; Zou, D.J. Down-regulated expression of the protein-tyrosine phosphatase 1B (PTP1B) is associated with aggressive clinicopathologic features and poor prognosis in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2012, 420, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Yan, X.; Liu, F.; Zhu, C.; Zhou, H.; Chen, Y.; Liu, J.; Gu, X.; Ni, R.; Zhang, T. Downregulated Expression of PTPN9 Contributes to Human Hepatocellular Carcinoma Growth and Progression. Pathol. Oncol. Res. 2016, 22, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.Z.; Cai, P.Q.; Li, M.; Fu, J.; Zhang, Z.Y.; Chen, J.W.; Cao, Y.; Yun, J.P.; Xie, D.; Cai, M.Y. Decreased expression of PTPN12 correlates with tumor recurrence and poor survival of patients with hepatocellular carcinoma. PLoS ONE 2014, 9, e85592. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Jiang, J.; Luo, C.; Sun, Q.; Ke, A.; Sun, C.; Hu, J.; Hu, Z.; Hu, B.; Zhu, K.; et al. Tumour-suppressive role of PTPN13 in hepatocellular carcinoma and its clinical significance. Tumour Biol. 2016, 37, 9691–9698. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.H.; Wu, D.C.; Tsai, C.Y.; Kuo, T.J.; Yu, W.C.; Chang, Y.S.; Chen, C.L.; Chang, C.F.; Chen, D.S.; Chen, P.J. Genetic characterization of fas-associated phosphatase-1 as a putative tumor suppressor gene on chromosome 4q21.3 in hepatocellular carcinoma. Clin. Cancer Res. 2006, 12, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.C.; Shiau, C.W.; Tai, W.T.; Hung, M.H.; Chu, P.Y.; Hsieh, F.S.; Lin, H.; Yu, H.C.; Chen, K.F. SHP-1 is a negative regulator of epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene 2015, 34, 5252–5263. [Google Scholar] [CrossRef] [PubMed]
- Bard-Chapeau, E.A.; Li, S.W.; Ding, J.; Zhang, S.S.; Zhu, H.H.; Princen, F.; Fang, D.D.; Han, T.; Bailly-Maitre, B.; Poli, V.; et al. Ptpn11/Shp2 Acts as a Tumor Suppressor in Hepatocellular Carcinogenesis. Cancer Cell 2011, 19, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Hu, F.; Tai, Y.; Du, J.; Mao, B.; Yuan, Z.; Wang, Y.; Wei, L. The tumor suppressor role of Src homology phosphotyrosine phosphatase 2 in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2012, 138, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.W.; Chang, J.G.; Yeh, K.T.; Chen, R.M.; Tsai, J.J.; Su, W.W.; Hu, R.M. Increased expression of PRL-1 protein correlates with shortened patient survival in human hepatocellular carcinoma. Clin. Transl. Oncol. 2012, 14, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Wang, K.; Xu, K.; Xu, J.; Sun, J.; Chu, Z.; Lin, D.; Koeffler, P.H.; Wang, J.; Yin, D. Oncogenic function and prognostic significance of protein tyrosine phosphatase PRL-1 in hepatocellular carcinoma. Oncotarget 2014, 5, 3685–3696. [Google Scholar] [CrossRef] [PubMed]
- Mayinuer, A.; Yasen, M.; Mogushi, K.; Obulhasim, G.; Xieraili, M.; Aihara, A.; Tanaka, S.; Mizushima, H.; Tanaka, H.; Arii, S. Upregulation of protein tyrosine phosphatase type IVA member 3 (PTP4A3/PRL-3) is associated with tumor differentiation and a poor prognosis in human hepatocellular carcinoma. Ann. Surg. Oncol. 2013, 20, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Pinna, F.; Meloni, F.; Ladu, S.; Pellegrino, R.; Sini, M.; Daino, L.; Simile, M.M.; De Miglio, M.R.; Virdis, P.; et al. Dual-specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res. 2008, 68, 4192–4200. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Pinna, F.; Pellegrino, R.; Sanna, V.; Sini, M.; Daino, L.; Simile, M.M.; De Miglio, M.R.; Frau, M.; Tomasi, M.L.; et al. Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesis is under genetic control. Int. J. Cancer 2008, 123, 2057–2064. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Kim, J.S.; Cronin, J.C.; Sibenaller, Z.; Ryken, T.; Rosenberg, S.A.; Ressom, H.; Jean, W.; Bigner, D.; Yan, H.; et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008, 68, 10300–10306. [Google Scholar] [CrossRef] [PubMed]
- Veeriah, S.; Brennan, C.; Meng, S.; Singh, B.; Fagin, J.A.; Solit, D.B.; Paty, P.B.; Rohle, D.; Vivanco, I.; Chmielecki, J.; et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. USA 2009, 106, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, L.; Zhou, J.; Pan, J.; Qian, W.; Fu, J.; Zhang, G.; Zhu, Y.; Liu, C.; Wang, C.; et al. Reduced expression of PTPRD correlates with poor prognosis in gastric adenocarcinoma. PLoS ONE 2014, 9, e113754. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Otsuka, A.; Girard, L.; Sato, M.; Iwakawa, R.; Ogiwara, H.; Sanchez-Cespedes, M.; Minna, J.D.; Yokota, J. A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosomes Cancer 2010, 49, 342–352. [Google Scholar] [PubMed]
- Stallings, R.L.; Nair, P.; Maris, J.M.; Catchpoole, D.; McDermott, M.; O’Meara, A.; Breatnach, F. High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer Res. 2006, 66, 3673–3680. [Google Scholar] [CrossRef] [PubMed]
- Giefing, M.; Zemke, N.; Brauze, D.; Kostrzewska-Poczekaj, M.; Luczak, M.; Szaumkessel, M.; Pelinska, K.; Kiwerska, K.; Tonnies, H.; Grenman, R.; et al. High resolution ArrayCGH and expression profiling identifies PTPRD and PCDH17/PCH68 as tumor suppressor gene candidates in laryngeal squamous cell carcinoma. Genes Chromosomes Cancer 2011, 50, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Urushibara, N.; Karasaki, H.; Nakamura, K.; Mizuno, Y.; Ogawa, K.; Kikuchi, K. The selective reduction in PTP delta expression in hepatomas. Int. J. Oncol. 1998, 12, 603–607. [Google Scholar] [PubMed]
- Walia, V.; Prickett, T.D.; Kim, J.S.; Gartner, J.J.; Lin, J.C.; Zhou, M.; Rosenberg, S.A.; Elble, R.C.; Solomon, D.A.; Waldman, T.; et al. Mutational and functional analysis of the tumor-suppressor PTPRD in human melanoma. Hum. Mutat. 2014, 35, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Matozaki, T.; Suzuki, T.; Uchida, T.; Inazawa, J.; Ariyama, T.; Matsuda, K.; Horita, K.; Noguchi, H.; Mizuno, H.; Sakamoto, C.; et al. Molecular cloning of a human transmembrane-type protein tyrosine phosphatase and its expression in gastrointestinal cancers. J. Biol. Chem. 1994, 269, 2075–2081. [Google Scholar] [PubMed]
- Noguchi, T.; Tsuda, M.; Takeda, H.; Takada, T.; Inagaki, K.; Yamao, T.; Fukunaga, K.; Matozaki, T.; Kasuga, M. Inhibition of cell growth and spreading by stomach cancer-associated protein-tyrosine phosphatase-1 (SAP-1) through dephosphorylation of p130cas. J. Biol. Chem. 2001, 276, 15216–15224. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.E.; Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Holzman, L.B.; Wiggins, R.C. GLEPP1, a renal glomerular epithelial cell (podocyte) membrane protein-tyrosine phosphatase. Identification, molecular cloning, and characterization in rabbit. J. Biol. Chem. 1994, 269, 19953–19962. [Google Scholar] [PubMed]
- Motiwala, T.; Majumder, S.; Kutay, H.; Smith, D.S.; Neuberg, D.S.; Lucas, D.M.; Byrd, J.C.; Grever, M.; Jacob, S.T. Methylation and silencing of protein tyrosine phosphatase receptor type O in chronic lymphocytic leukemia. Clin. Cancer Res. 2007, 13, 3174–3181. [Google Scholar] [CrossRef] [PubMed]
- Motiwala, T.; Kutay, H.; Ghoshal, K.; Bai, S.; Seimiya, H.; Tsuruo, T.; Suster, S.; Morrison, C.; Jacob, S.T. Protein tyrosine phosphatase receptor-type O (PTPRO) exhibits characteristics of a candidate tumor suppressor in human lung cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 13844–13849. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Majumder, S.; Roy, S.; Ghoshal, K.; Kutay, H.; Datta, J.; Younes, M.; Shapiro, C.L.; Motiwala, T.; Jacob, S.T. Estrogen-mediated suppression of the gene encoding protein tyrosine phosphatase PTPRO in human breast cancer: Mechanism and role in tamoxifen sensitivity. Mol. Endocrinol. 2009, 23, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Motiwala, T.; Roy, S.; Claus, R.; Mustafa, M.; Plass, C.; Freitas, M.A.; Ghoshal, K.; Jacob, S.T. Methylation of the PTPRO gene in human hepatocellular carcinoma and identification of VCP as its substrate. J. Cell. Biochem. 2013, 114, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yan, J.; Lin, H.; Hua, F.; Wang, X.; Liu, H.; Lv, X.; Yu, J.; Mi, S.; Wang, J.; et al. Toll-like receptor 4 activity protects against hepatocellular tumorigenesis and progression by regulating expression of DNA repair protein Ku70 in mice. Hepatology 2013, 57, 1869–1881. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, X.; Yan, S.; Yin, Y.; Hou, J.; Wang, X.; Sun, B. Interaction of PTPRO and TLR4 signaling in hepatocellular carcinoma. Tumour Biol. 2014, 35, 10267–10273. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Deng, L.; Zhuo, H.; Lin, Z.; Chen, Y.; Jiang, R.; Chen, D.; Zhang, X.; Huang, X.; Sun, B. PTPROt maintains T cell immunity in the microenvironment of hepatocellular carcinoma. J. Mol. Cell Biol. 2015, 7, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Streuli, M.; Krueger, N.X.; Hall, L.R.; Schlossman, S.F.; Saito, H. A new member of the immunoglobulin superfamily that has a cytoplasmic region homologous to the leukocyte common antigen. J. Exp. Med. 1988, 168, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Tenney, A.P.; Busch, S.A.; Horn, K.P.; Cuascut, F.X.; Liu, K.; He, Z.; Silver, J.; Flanagan, J.G. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 2009, 326, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Meathrel, K.; Adamek, T.; Batt, J.; Rotin, D.; Doering, L.C. Protein tyrosine phosphatase sigma-deficient mice show aberrant cytoarchitecture and structural abnormalities in the central nervous system. J. Neurosci. Res. 2002, 70, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhao, Y.J.; Wang, X.Y.; Guo, W.J.; Gao, S.; Wei, L.; Shi, J.Y.; Shi, G.M.; Wang, Z.C.; Zhang, Y.N.; et al. Activating mutations in PTPN3 promote cholangiocarcinoma cell proliferation and migration and are associated with tumor recurrence in patients. Gastroenterology 2014, 146, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.; Taylor, B.S.; Bivona, T.G.; Gong, Y.; Eng, S.; Brennan, C.W.; Kaufman, A.; Kastenhuber, E.R.; Banuchi, V.E.; Singh, B.; et al. Genomic dissection of the epidermal growth factor receptor (EGFR)/PI3K pathway reveals frequent deletion of the EGFR phosphatase PTPRS in head and neck cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 19024–19029. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, D.; Parsons, D.W.; Bardelli, A.; Sager, J.; Szabo, S.; Ptak, J.; Silliman, N.; Peters, B.A.; van der Heijden, M.S.; et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004, 304, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.; Peyser, N.D.; Ng, P.K.; Hritz, J.; Zeng, Y.; Lu, Y.; Li, H.; Wang, L.; Gilbert, B.R.; General, I.J.; et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.F.; Lau, S.H.; Hu, L.; Xie, D.; Wu, J.; Yang, J.; Wang, Y.; Wu, M.C.; Fung, J.; Bai, X.; et al. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin. Cancer Res. 2008, 14, 5061–5068. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Jeong, E.G.; Lee, S.H.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of PTPRT phosphatase domains in common human cancers. APMIS 2007, 115, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Seely, B.L.; Staubs, P.A.; Reichart, D.R.; Berhanu, P.; Milarski, K.L.; Saltiel, A.R.; Kusari, J.; Olefsky, J.M. Protein tyrosine phosphatase 1B interacts with the activated insulin receptor. Diabetes 1996, 45, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Zabolotny, J.M.; Bence-Hanulec, K.K.; Stricker-Krongrad, A.; Haj, F.; Wang, Y.; Minokoshi, Y.; Kim, Y.B.; Elmquist, J.K.; Tartaglia, L.A.; Kahn, B.B.; et al. PTP1B regulates leptin signal transduction in vivo. Dev. Cell 2002, 2, 489–495. [Google Scholar] [CrossRef]
- Kolfschoten, I.G.; van Leeuwen, B.; Berns, K.; Mullenders, J.; Beijersbergen, R.L.; Bernards, R.; Voorhoeve, P.M.; Agami, R. A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell 2005, 121, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Conner, E.A.; Seo, D.; Hsieh, J.T.; Factor, V.M.; Thorgeirsson, S.S. Inactivation of Ras GTPase-activating proteins promotes unrestrained activity of wild-type Ras in human liver cancer. J. Hepatol. 2011, 54, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Lessard, L.; Stuible, M.; Tremblay, M.L. The two faces of PTP1B in cancer. Biochim. Biophys. Acta 2010, 1804, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Bjorge, J.D.; Fujita, D.J. PTP1B contributes to the oncogenic properties of colon cancer cells through src activation. Cancer Res. 2007, 67, 10129–10137. [Google Scholar] [CrossRef] [PubMed]
- Cortesio, C.L.; Chan, K.T.; Perrin, B.J.; Burton, N.O.; Zhang, S.; Zhang, Z.Y.; Huttenlocher, A. Calpain 2 and PTP1B function in a novel pathway with Src to regulate invadopodia dynamics and breast cancer cell invasion. J. Cell Biol. 2008, 180, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Dube, N.; Cheng, A.; Tremblay, M.L. The role of protein tyrosine phosphatase 1B in Ras signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Guan, Q.; Wang, Y.; Zhao, Z.J.; Zhou, G.W. SHP-1 suppresses cancer cell growth by promoting degradation of JAK kinases. J. Cell. Biochem. 2003, 90, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Eberl, H.C.; Renkawitz, J.; Olsen, J.V.; Tremblay, M.L.; Mann, M.; Ullrich, A.; Daub, H. Investigation of protein-tyrosine phosphatase 1B function by quantitative proteomics. Mol. Cell. Proteom. 2008, 7, 1763–1777. [Google Scholar] [CrossRef] [PubMed]
- Balavenkatraman, K.K.; Aceto, N.; Britschgi, A.; Mueller, U.; Bence, K.K.; Neel, B.G.; Bentires-Alj, M. Epithelial protein-tyrosine phosphatase 1B contributes to the induction of mammary tumors by HER2/Neu but is not essential for tumor maintenance. Mol. Cancer Res. 2011, 9, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Bentires-Alj, M.; Neel, B.G. Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 2007, 67, 2420–2424. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Hill, D.E.; Chernoff, J. Direct binding of the proline-rich region of protein tyrosine phosphatase 1B to the Src homology 3 domain of p130(Cas). J. Biol. Chem. 1996, 271, 31290–31295. [Google Scholar] [CrossRef] [PubMed]
- LaMontagne, K.R., Jr.; Hannon, G.; Tonks, N.K. Protein tyrosine phosphatase PTP1B suppresses p210 bcr-abl-induced transformation of rat-1 fibroblasts and promotes differentiation of K562 cells. Proc. Natl. Acad. Sci. USA 1998, 95, 14094–14099. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.P.; Andersen, J.N.; Cheng, A.; Tremblay, M.L.; Horvath, C.M.; Parisien, J.P.; Salmeen, A.; Barford, D.; Tonks, N.K. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J. Biol. Chem. 2001, 276, 47771–47774. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Matsuda, T. A cytosolic protein-tyrosine phosphatase PTP1B specifically dephosphorylates and deactivates prolactin-activated STAT5a and STAT5b. J. Biol. Chem. 2000, 275, 39718–39726. [Google Scholar] [CrossRef] [PubMed]
- Balsamo, J.; Leung, T.; Ernst, H.; Zanin, M.K.; Hoffman, S.; Lilien, J. Regulated binding of PTP1B-like phosphatase to N-cadherin: Control of cadherin-mediated adhesion by dephosphorylation of beta-catenin. J. Cell Biol. 1996, 134, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Tai, W.T.; Hsu, C.Y.; Huang, J.W.; Liu, C.Y.; Chen, P.J.; Kim, I.; Shiau, C.W. Blockade of STAT3 activation by sorafenib derivatives through enhancing SHP-1 phosphatase activity. Eur. J. Med. Chem. 2012, 55, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ruiz, P.; Rodriguez-Ubreva, J.; Cariaga, A.E.; Cortes, M.A.; Colas, B. SHP-1 in cell-cycle regulation. Anticancer Agents Med. Chem. 2011, 11, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.; Raghunath, P.; Wasik, A.; Junkins-Hopkins, J.M.; Jones, D.; Zhang, Q.; Odum, N.; Wasik, M.A. Loss of SHP-1 tyrosine phosphatase expression correlates with the advanced stages of cutaneous T-cell lymphoma. Hum. Pathol. 2007, 38, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Beldi-Ferchiou, A.; Skouri, N.; Ben Ali, C.; Safra, I.; Abdelkefi, A.; Ladeb, S.; Mrad, K.; Ben Othman, T.; Ben Ahmed, M. Abnormal repression of SHP-1, SHP-2 and SOCS-1 transcription sustains the activation of the JAK/STAT3 pathway and the progression of the disease in multiple myeloma. PLoS ONE 2017, 12, e0174835. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, H.; Yang, J.J.; Chen, J.; Jie, J.; Li, L.; Zhang, Y.; Liu, Z.R. Reciprocal regulation of protein kinase and pyruvate kinase activities of pyruvate kinase M2 by growth signals. J. Biol. Chem. 2013, 288, 15971–15979. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2009, 2, ra73. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Hung, M.H.; Chu, P.Y.; Chen, Y.L.; Chen, L.J.; Tsai, M.H.; Chen, M.H.; Shiau, C.W.; Boo, Y.P.; Chen, K.F. SH2 domain-containing phosphatase 1 regulates pyruvate kinase M2 in hepatocellular carcinoma. Oncotarget 2016, 7, 22193–22205. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Warshawsky, I.; Majerus, P.W. Cloning and expression of a cytosolic megakaryocyte protein-tyrosine-phosphatase with sequence homology to retinaldehyde-binding protein and yeast SEC14p. Proc. Natl. Acad. Sci. USA 1992, 89, 2980–2984. [Google Scholar] [CrossRef] [PubMed]
- Kruger, J.M.; Fukushima, T.; Cherepanov, V.; Borregaard, N.; Loeve, C.; Shek, C.; Sharma, K.; Tanswell, A.K.; Chow, C.W.; Downey, G.P. Protein-tyrosine phosphatase MEG2 is expressed by human neutrophils. Localization to the phagosome and activation by polyphosphoinositides. J. Biol. Chem. 2002, 277, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Vachon, E.; Zhang, J.; Cherepanov, V.; Kruger, J.; Li, J.; Saito, K.; Shannon, P.; Bottini, N.; Huynh, H.; et al. Tyrosine phosphatase MEG2 modulates murine development and platelet and lymphocyte activation through secretory vesicle function. J. Exp. Med. 2005, 202, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Sui, X.W.; Zhao, R.X.; Dai, C.H.; Krantz, S.B.; Zhao, Z.Z.J. PTP-MEG2 is activated in polycythemia vera erythroid progenitor cells and is required for growth and expansion of erythroid cells. Blood 2003, 102, 4354–4360. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Su, F.; Wang, X.; Gao, H.; Lei, L.; Chang, N.; Wu, Q.; Hu, K.; Zhu, X.; Chang, Z.; et al. Protein tyrosine phosphatase PTPN9 regulates erythroid cell development through STAT3 dephosphorylation in zebrafish. J. Cell Sci. 2014, 127, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Bottini, N.; Williams, S.; Cherepanov, V.; Musumeci, L.; Saito, K.; Bruckner, S.; Vachon, E.; Wang, X.; Kruger, J.; et al. Control of vesicle fusion by a tyrosine phosphatase. Nat. Cell Biol. 2004, 6, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Marlin, M.C.; Liang, Z.; Ahmad, M.; Ashpole, N.M.; Sonntag, W.E.; Zhao, Z.J.; Li, G. The Protein Tyrosine Phosphatase MEG2 Regulates the Transport and Signal Transduction of Tropomyosin Receptor Kinase A. J. Biol. Chem. 2016, 291, 23895–23905. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, H.; Ma, J.; Huang, H.; Qin, J.; Li, Y. PTPN9 promotes cell proliferation and invasion in Eca109 cells and is negatively regulated by microRNA-126. Oncol. Lett. 2017, 14, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Wang, Y.; Zhao, Z.J.; Gu, H. Protein-tyrosine phosphatase PTPN9 negatively regulates ErbB2 and epidermal growth factor receptor signaling in breast cancer cells. J. Biol. Chem. 2010, 285, 14861–14870. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Ren, F.; Rong, Y.; Wang, Y.; Geng, Y.; Wang, Y.; Feng, M.; Ju, Y.; Li, Y.; Zhao, Z.J.; et al. Protein tyrosine phosphatase Meg2 dephosphorylates signal transducer and activator of transcription 3 and suppresses tumor growth in breast cancer. Breast Cancer Res. 2012, 14, R38. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.M., Jr.; Plutzky, J.; Neel, B.G. Identification of a human src homology 2-containing protein-tyrosine-phosphatase: A putative homolog of Drosophila corkscrew. Proc. Natl. Acad. Sci. USA 1992, 89, 11239–11243. [Google Scholar] [CrossRef] [PubMed]
- Niihori, T.; Aoki, Y.; Ohashi, H.; Kurosawa, K.; Kondoh, T.; Ishikiriyama, S.; Kawame, H.; Kamasaki, H.; Yamanaka, T.; Takada, F.; et al. Functional analysis of PTPN11/SHP-2 mutants identified in Noonan syndrome and childhood leukemia. J. Hum. Genet. 2005, 50, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Counelis, G.J.; O’Rourke, D.M. The protein tyrosine phosphatase SHP-2 is required for EGFRvIII oncogenic transformation in human glioblastoma cells. Exp. Cell Res. 2009, 315, 2343–2357. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Shin, O.R.; Kim, H.K.; Cho, Y.S.; An, C.H.; Lim, K.W.; Kim, S.S. Overexpression of protein phosphatase non-receptor type 11 (PTPN11) in gastric carcinomas. Dig. Dis. Sci. 2010, 55, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Matozaki, T.; Murata, Y.; Saito, Y.; Okazawa, H.; Ohnishi, H. Protein tyrosine phosphatase SHP-2: A proto-oncogene product that promotes Ras activation. Cancer Sci. 2009, 100, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Grinnell, K.L.; Casserly, B.; Harrington, E.O. Role of protein tyrosine phosphatase SHP2 in barrier function of pulmonary endothelium. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L361–L370. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.D.; Agazie, Y.M. Inhibition of SHP2 leads to mesenchymal to epithelial transition in breast cancer cells. Cell Death Differ. 2008, 15, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Gelb, B.D. Germ-line and somatic PTPN11 mutations in human disease. Eur. J. Med. Genet. 2005, 48, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.J.; Feng, G.S. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 2007, 109, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.J.; Yu, J.K.; Ge, W.T.; Hu, H.G.; Yuan, Y.; Zheng, S. SPARCL1, Shp2, MSH2, E-cadherin, p53, ADCY-2 and MAPK are prognosis-related in colorectal cancer. World J. Gastroenterol. 2011, 17, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.S. Conflicting roles of molecules in hepatocarcinogenesis: Paradigm or paradox. Cancer Cell 2012, 21, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Anakk, S.; Bhosale, M.; Schmidt, V.A.; Johnson, R.L.; Finegold, M.J.; Moore, D.D. Bile acids activate YAP to promote liver carcinogenesis. Cell Rep. 2013, 5, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hsu, D.D.; Li, B.; Luo, X.; Alderson, N.; Qiao, L.; Ma, L.; Zhu, H.H.; He, Z.; Suino-Powell, K.; et al. Cytoplasmic tyrosine phosphatase Shp2 coordinates hepatic regulation of bile acid and FGF15/19 signaling to repress bile acid synthesis. Cell Metab. 2014, 20, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Xiang, D.M.; Sun, W.; Liu, N.; Sun, H.L.; Wen, W.; Shen, W.F.; Wang, R.Y.; Chen, C.; Wang, X.; et al. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J. Hepatol. 2015, 63, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Zhao, X.; Qu, Y.; Chen, C.; Zhu, C.; Zhang, H.; Yuan, H.; Jin, H.; Liu, X.; Wang, Y.; et al. Shp2 SUMOylation promotes ERK activation and hepatocellular carcinoma development. Oncotarget 2015, 6, 9355–9369. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Cheng, Z.; Liu, H.; Wang, X.; Han, T.; Sun, W.; Li, X.; Yang, W.; Chen, C.; Xia, M.; et al. Shp2 promotes liver cancer stem cell expansion by augmenting beta-catenin signaling and predicts chemotherapeutic response of patients. Hepatology 2017, 65, 1566–1580. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Co, D.; Sommercorn, J.; Tonks, N.K. Cloning and expression of PTP-PEST. A novel, human, nontransmembrane protein tyrosine phosphatase. J. Biol. Chem. 1993, 268, 17650. [Google Scholar] [PubMed]
- Sun, T.; Aceto, N.; Meerbrey, K.L.; Kessler, J.D.; Zhou, C.; Migliaccio, I.; Nguyen, D.X.; Pavlova, N.N.; Botero, M.; Huang, J.; et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 2011, 144, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.M.; Davidson, D.; Rhee, I.; Gratton, J.P.; Davis, E.C.; Veillette, A. The phosphatase PTP-PEST/PTPN12 regulates endothelial cell migration and adhesion, but not permeability, and controls vascular development and embryonic viability. J. Biol. Chem. 2012, 287, 43180–43190. [Google Scholar] [CrossRef] [PubMed]
- Angers-Loustau, A.; Cote, J.F.; Tremblay, M.L. Roles of protein tyrosine phosphatases in cell migration and adhesion. Biochem. Cell Biol. 1999, 77, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Espejo, R.; Rengifo-Cam, W.; Schaller, M.D.; Evers, B.M.; Sastry, S.K. PTP-PEST controls motility, adherens junction assembly, and Rho GTPase activity in colon cancer cells. Am. J. Physiol. Cell Physiol. 2010, 299, C454–C463. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Q.; Hu, P.; Gao, J.; Wei, W.D.; Xiao, X.S.; Tang, H.L.; Li, X.; Ge, Q.D.; Jia, W.H.; Liu, R.B.; et al. Low expression of tyrosine-protein phosphatase nonreceptor type 12 is associated with lymph node metastasis and poor prognosis in operable triple-negative breast cancer. Asian Pac. J. Cancer Prev. 2013, 14, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Villa-Moruzzi, E. Tyrosine phosphatases in the HER2-directed motility of ovarian cancer cells: Involvement of PTPN12, ERK5 and FAK. Anal. Cell. Pathol. 2011, 34, 101–112. [Google Scholar] [CrossRef]
- Zhang, X.K.; Xu, M.; Chen, J.W.; Zhou, F.; Ling, Y.H.; Zhu, C.M.; Yun, J.P.; Cai, M.Y.; Luo, R.Z. The prognostic significance of tyrosine-protein phosphatase nonreceptor type 12 expression in nasopharyngeal carcinoma. Tumour Biol. 2015, 36, 5201–5208. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, Y.; Luo, R.Z.; He, L.R.; Yang, J.; Zeng, M.S.; Wen, Z.S. Tyrosine-Protein Phosphatase Nonreceptor Type 12 Is a Novel Prognostic Biomarker for Esophageal Squamous Cell Carcinoma. Ann. Thorac. Surg. 2012, 93, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Newberg, J.Y.; Kodama, M.; Rangel, R.; Yoshihara, K.; Tien, J.C.; Parsons, P.H.; Wu, H.; Finegold, M.J.; Copeland, N.G.; et al. Transposon mutagenesis identifies genes and cellular processes driving epithelial-mesenchymal transition in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, E3384–E3393. [Google Scholar] [CrossRef] [PubMed]
- Sirois, J.; Cote, J.F.; Charest, A.; Uetani, N.; Bourdeau, A.; Duncan, S.A.; Daniels, E.; Tremblay, M.L. Essential function of PTP-PEST during mouse embryonic vascularization, mesenchyme formation, neurogenesis and early liver development. Mech. Dev. 2006, 123, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Saras, J.; Claesson-Welsh, L.; Heldin, C.H.; Gonez, L.J. Cloning and characterization of PTPL1, a protein tyrosine phosphatase with similarities to cytoskeletal-associated proteins. J. Biol. Chem. 1994, 269, 24082–24089. [Google Scholar] [PubMed]
- Banville, D.; Ahmad, S.; Stocco, R.; Shen, S.H. A novel protein-tyrosine phosphatase with homology to both the cytoskeletal proteins of the band 4.1 family and junction-associated guanylate kinases. J. Biol. Chem. 1994, 269, 22320–22327. [Google Scholar] [PubMed]
- Maekawa, K.; Imagawa, N.; Nagamatsu, M.; Harada, S. Molecular cloning of a novel protein-tyrosine phosphatase containing a membrane-binding domain and GLGF repeats. FEBS Lett. 1994, 337, 200–206. [Google Scholar] [CrossRef]
- Revillion, F.; Puech, C.; Rabenoelina, F.; Chalbos, D.; Peyrat, J.P.; Freiss, G. Expression of the putative tumor suppressor gene PTPN13/PTPL1 is an independent prognostic marker for overall survival in breast cancer. Int. J. Cancer 2009, 124, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Mohn, K.L.; Laz, T.M.; Hsu, J.C.; Melby, A.E.; Bravo, R.; Taub, R. The immediate-early growth response in regenerating liver and insulin-stimulated H-35 cells: Comparison with serum-stimulated 3T3 cells and identification of 41 novel immediate-early genes. Mol. Cell. Biol. 1991, 11, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Diamond, R.H.; Cressman, D.E.; Laz, T.M.; Abrams, C.S.; Taub, R. PRL-1, a unique nuclear protein tyrosine phosphatase, affects cell growth. Mol. Cell. Biol. 1994, 14, 3752–3762. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.P.; Luo, Y.; Yu, X.; Wang, W.Q.; Zhou, B.; Liang, F.; Zhang, Z.Y. Phosphatase activity, trimerization, and the C-terminal polybasic region are all required for PRL1-mediated cell growth and migration. J. Biol. Chem. 2007, 282, 29043–29051. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Dong, J.M.; Guo, K.; Li, J.; Tan, H.X.; Koh, V.; Pallen, C.J.; Manser, E.; Hong, W. PRL-3 and PRL-1 promote cell migration, invasion, and metastasis. Cancer Res. 2003, 63, 2716–2722. [Google Scholar] [PubMed]
- Fiordalisi, J.J.; Keller, P.J.; Cox, A.D. PRL tyrosine phosphatases regulate rho family GTPases to promote invasion and motility. Cancer Res. 2006, 66, 3153–3161. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Bardelli, A.; Buckhaults, P.; Velculescu, V.E.; Rago, C.; St Croix, B.; Romans, K.E.; Choti, M.A.; Lengauer, C.; Kinzler, K.W.; et al. A phosphatase associated with metastasis of colorectal cancer. Science 2001, 294, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zeng, H.; Zhang, X.; Zhao, Y.; Sha, H.; Ge, X.; Zhang, M.; Gao, X.; Xu, Q. Phosphatase of regenerating liver-3 promotes motility and metastasis of mouse melanoma cells. Am. J. Pathol. 2004, 164, 2039–2054. [Google Scholar] [CrossRef]
- Radke, I.; Gotte, M.; Kersting, C.; Mattsson, B.; Kiesel, L.; Wulfing, P. Expression and prognostic impact of the protein tyrosine phosphatases PRL-1, PRL-2, and PRL-3 in breast cancer. Br. J. Cancer 2006, 95, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Miskad, U.A.; Semba, S.; Kato, H.; Yokozaki, H. Expression of PRL-3 phosphatase in human gastric carcinomas: Close correlation with invasion and metastasis. Pathobiology 2004, 71, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Ooki, A.; Yamashita, K.; Kikuchi, S.; Sakuramoto, S.; Katada, N.; Watanabe, M. Phosphatase of regenerating liver-3 as a convergent therapeutic target for lymph node metastasis in esophageal squamous cell carcinoma. Int. J. Cancer 2010, 127, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Liu, B.; Guo, X.; Shi, H.; Zhou, M.; Li, L.; Yang, S.; Tong, X.; Wang, H. Independent oncogenic and therapeutic significance of phosphatase PRL-3 in FLT3-ITD-negative acute myeloid leukemia. Cancer 2014, 120, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Polato, F.; Codegoni, A.; Fruscio, R.; Perego, P.; Mangioni, C.; Saha, S.; Bardelli, A.; Broggini, M. PRL-3 phosphatase is implicated in ovarian cancer growth. Clin. Cancer Res. 2005, 11, 6835–6839. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Rubio, T.; Kohn, M. Regulatory mechanisms of phosphatase of regenerating liver (PRL)-3. Biochem. Soc. Trans. 2016, 44, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.F.; Nathans, D. Identification of a set of genes expressed during the G0/G1 transition of cultured mouse cells. EMBO J. 1985, 4, 3145–3151. [Google Scholar] [PubMed]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Franklin, C.C.; Duke, R.C.; Kraft, R.S. Human DU145 prostate cancer cells overexpressing mitogen-activated protein kinase phosphatase-1 are resistant to Fas ligand-induced mitochondrial perturbations and cellular apoptosis. Mol. Cell. Biochem. 1999, 199, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Guo, J.; Kleeff, J.; Zimmermann, A.; Buchler, M.W.; Korc, M.; Friess, H. Down-regulation of the dual-specificity phosphatase MKP-1 suppresses tumorigenicity of pancreatic cancer cells. Gastroenterology 2003, 124, 1830–1845. [Google Scholar] [CrossRef]
- Vicent, S.; Garayoa, M.; Lopez-Picazo, J.M.; Lozano, M.D.; Toledo, G.; Thunnissen, F.B.; Manzano, R.G.; Montuenga, L.M. Mitogen-activated protein kinase phosphatase-1 is overexpressed in non-small cell lung cancer and is an independent predictor of outcome in patients. Clin. Cancer Res. 2004, 10, 3639–3649. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Cheng, Z.; Malbon, C.C. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett. 2003, 191, 229–237. [Google Scholar] [CrossRef]
- Bang, Y.J.; Kwon, J.H.; Kang, S.H.; Kim, J.W.; Yang, Y.C. Increased MAPK activity and MKP-1 overexpression in human gastric adenocarcinoma. Biochem. Biophys. Res. Commun. 1998, 250, 43–47. [Google Scholar] [CrossRef] [PubMed]
- St John, M.A.; Dohadwala, M.; Luo, J.; Wang, G.; Lee, G.; Shih, H.; Heinrich, E.; Krysan, K.; Walser, T.; Hazra, S.; et al. Proinflammatory mediators upregulate snail in head and neck squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 6018–6027. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhou, S.; Shi, L.; Liu, X.; Lin, H.; Yu, H.; Tang, J.; Yu, T.; Cai, X. DUSP1 inhibits cell proliferation, metastasis and invasion and angiogenesis in gallbladder cancer. Oncotarget 2017, 8, 12133–12144. [Google Scholar] [PubMed]
- Tsujita, E.; Taketomi, A.; Gion, T.; Kuroda, Y.; Endo, K.; Watanabe, A.; Nakashima, H.; Aishima, S.; Kohnoe, S.; Maehara, Y. Suppressed MKP-1 is an independent predictor of outcome in patients with hepatocellular carcinoma. Oncology 2005, 69, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Muroni, M.R.; Calvisi, D.F.; Asara, G.; Casabona, D.; Frau, M.; Seddaiu, M.A.; Feo, F. Cell cycle deregulation in liver lesions of rats with and without genetic predisposition to hepatocarcinogenesis. Hepatology 2002, 35, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Slack, D.N.; Seternes, O.M.; Gabrielsen, M.; Keyse, S.M. Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J. Biol. Chem. 2001, 276, 16491–16500. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Yang, J.L. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J. Biol. Chem. 2006, 281, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Chuang, S.M.; Yang, J.L. ERK1/2 achieves sustained activation by stimulating MAPK phosphatase-1 degradation via the ubiquitin-proteasome pathway. J. Biol. Chem. 2003, 278, 21534–21541. [Google Scholar] [CrossRef] [PubMed]
- Galun, D.; Srdic-Rajic, T.; Bogdanovic, A.; Loncar, Z.; Zuvela, M. Targeted therapy and personalized medicine in hepatocellular carcinoma: Drug resistance, mechanisms, and treatment strategies. J. Hepatocell. Carcinoma 2017, 4, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.H.; Tai, W.T.; Shiau, C.W.; Chen, K.F. Downregulation of signal transducer and activator of transcription 3 by sorafenib: A novel mechanism for hepatocellular carcinoma therapy. World J. Gastroenterol. 2014, 20, 15269–15274. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Chien, D.S. BAY 43-9006: Preclinical data. Curr. Pharm. Des. 2002, 8, 2255–2257. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Yeh, S.H.; Shiau, C.W.; Chen, K.F.; Lin, W.H.; Tsai, T.F.; Teng, Y.C.; Chen, D.S.; Chen, P.J. Sorafenib Action in Hepatitis B Virus X-Activated Oncogenic Androgen Pathway in Liver through SHP-1. J. Natl. Cancer Inst. 2015, 107, 10. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Shiau, C.W.; Li, Y.S.; Chen, Y.L.; Chu, P.Y.; Huang, J.W.; Hsu, C.Y.; Hsu, Y.C.; Chen, P.J.; Chen, K.F. SC-60, a Dimer-Based Sorafenib Derivative, Shows a Better Anti-Hepatocellular Carcinoma Effect than Sorafenib in a Preclinical Hepatocellular Carcinoma Model. Mol. Cancer Ther. 2014, 13, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Shiau, C.W.; Chen, P.J.; Chu, P.Y.; Huang, H.P.; Liu, C.Y.; Huang, J.W.; Chen, K.F. Discovery of novel Src homology region 2 domain-containing phosphatase 1 agonists from sorafenib for the treatment of hepatocellular carcinoma. Hepatology 2014, 59, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Shiau, C.W.; Chen, H.L.; Liu, C.Y.; Lin, C.S.; Cheng, A.L.; Chen, P.J.; Chen, K.F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Cheng, A.L.; Shiau, C.W.; Huang, H.P.; Huang, J.W.; Chen, P.J.; Chen, K.F. Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma (vol 55, pg 1041, 2011). J. Hepatol. 2017, 66, 1334–1335. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Shiau, C.W.; Li, Y.S.; Chang, C.W.; Huang, J.W.; Hsueh, T.T.; Yu, H.C.; Chen, K.F. Nintedanib (BIBF-1120) inhibits hepatocellular carcinoma growth independent of angiokinase activity. J. Hepatol. 2014, 61, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Chu, P.Y.; Shiau, C.W.; Chen, Y.L.; Li, Y.S.; Hung, M.H.; Chen, L.J.; Chen, P.L.; Su, J.C.; Lin, P.Y.; et al. STAT3 mediates regorafenib-induced apoptosis in hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 5768–5776. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Cheng, A.L.; Shiau, C.W.; Liu, C.Y.; Ko, C.H.; Lin, M.W.; Chen, P.J.; Chen, K.F. Dovitinib Induces Apoptosis and Overcomes Sorafenib Resistance in Hepatocellular Carcinoma through SHP-1-Mediated Inhibition of STAT3. Mol. Cancer Ther. 2012, 11, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Su, J.C.; Chiang, H.C.; Tseng, P.H.; Tai, W.T.; Hsu, C.Y.; Li, Y.S.; Huang, J.W.; Ko, C.H.; Lin, M.W.; Chu, P.Y.; et al. RFX-1-dependent activation of SHP-1 inhibits STAT3 signaling in hepatocellular carcinoma cells. Carcinogenesis 2014, 35, 2807–2814. [Google Scholar] [CrossRef] [PubMed]
- Su, J.C.; Tseng, P.H.; Wu, S.H.; Hsu, C.Y.; Tai, W.T.; Li, Y.S.; Chen, I.T.; Liu, C.Y.; Chen, K.F.; Shiau, C.W. SC-2001 overcomes STAT3-mediated sorafenib resistance through RFX-1/SHP-1 activation in hepatocellular carcinoma. Neoplasia 2014, 16, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Su, J.C.; Liu, C.Y.; Huang, J.W.; Chen, K.C.; Chen, W.L.; Tai, W.T.; Shiau, C.W. A novel obatoclax derivative, SC-2001, induces apoptosis in hepatocellular carcinoma cells through SHP-1-dependent STAT3 inactivation. Cancer Lett. 2012, 321, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, A.; Shanmugam, M.K.; Ong, T.H.; Li, F.; Perumal, E.; Chen, L.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br. J. Pharmacol. 2013, 170, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cai, X.; Lu, W.; Hu, C.; Xu, X.; Yu, Q.; Cao, P. Evodiamine inhibits STAT3 signaling by inducing phosphatase shatterproof 1 in hepatocellular carcinoma cells. Cancer Lett. 2013, 328, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Manu, K.A.; Shanmugam, M.K.; Loo, S.Y.; Kumar, A.P.; Sethi, G. gamma-Tocotrienol is a novel inhibitor of constitutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma: Potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. Br. J. Pharmacol. 2011, 163, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Su, H.; Hong, Z.; Yang, T.; Shu, G. Chemical composition of total flavonoids from Polygonum amplexicaule and their pro-apoptotic effect on hepatocellular carcinoma cells: Potential roles of suppressing STAT3 signaling. Food Chem. Toxicol. 2015, 80, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Tai, W.T.; Wu, S.Y.; Shih, C.T.; Chen, M.H.; Tsai, M.H.; Kuo, C.W.; Shiau, C.W.; Hung, M.H.; Chen, K.F. Dovitinib Acts As a Novel Radiosensitizer in Hepatocellular Carcinoma by Targeting SHP-1/STAT3 Signaling. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Chen, H.L.; Liu, C.Y.; Tai, W.T.; Ichikawa, K.; Chen, P.J.; Cheng, A.L. Dovitinib sensitizes hepatocellular carcinoma cells to TRAIL and tigatuzumab, a novel anti-DR5 antibody, through SHP-1-dependent inhibition of STAT3. Biochem. Pharmacol. 2012, 83, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.I.; Tai, W.T.; Hung, M.H.; Tsai, M.H.; Chen, M.H.; Chang, M.J.; Shiau, C.W.; Chen, K.F. A combination of sorafenib and SC-43 is a synergistic SHP-1 agonist duo to advance hepatocellular carcinoma therapy. Cancer Lett. 2016, 371, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Tai, W.T.; Hsieh, C.Y.; Hsu, W.M.; Lai, Y.J.; Chen, L.J.; Shiau, C.W.; Chen, K.F. A sorafenib derivative and novel SHP-1 agonist, SC-59, acts synergistically with radiotherapy in hepatocellular carcinoma cells through inhibition of STAT3. Cancer Lett. 2014, 349, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Baburajeev, C.P.; Mohan, C.D.; Ananda, H.; Rangappa, S.; Fuchs, J.E.; Jagadish, S.; Siveen, K.S.; Chinnathambi, A.; Alharbi, S.A.; Zayed, M.E.; et al. Development of Novel Triazolo-Thiadiazoles from Heterogeneous “Green” Catalysis as Protein Tyrosine Phosphatase 1B Inhibitors. Sci. Rep. 2015, 5, 14195. [Google Scholar] [CrossRef] [PubMed]
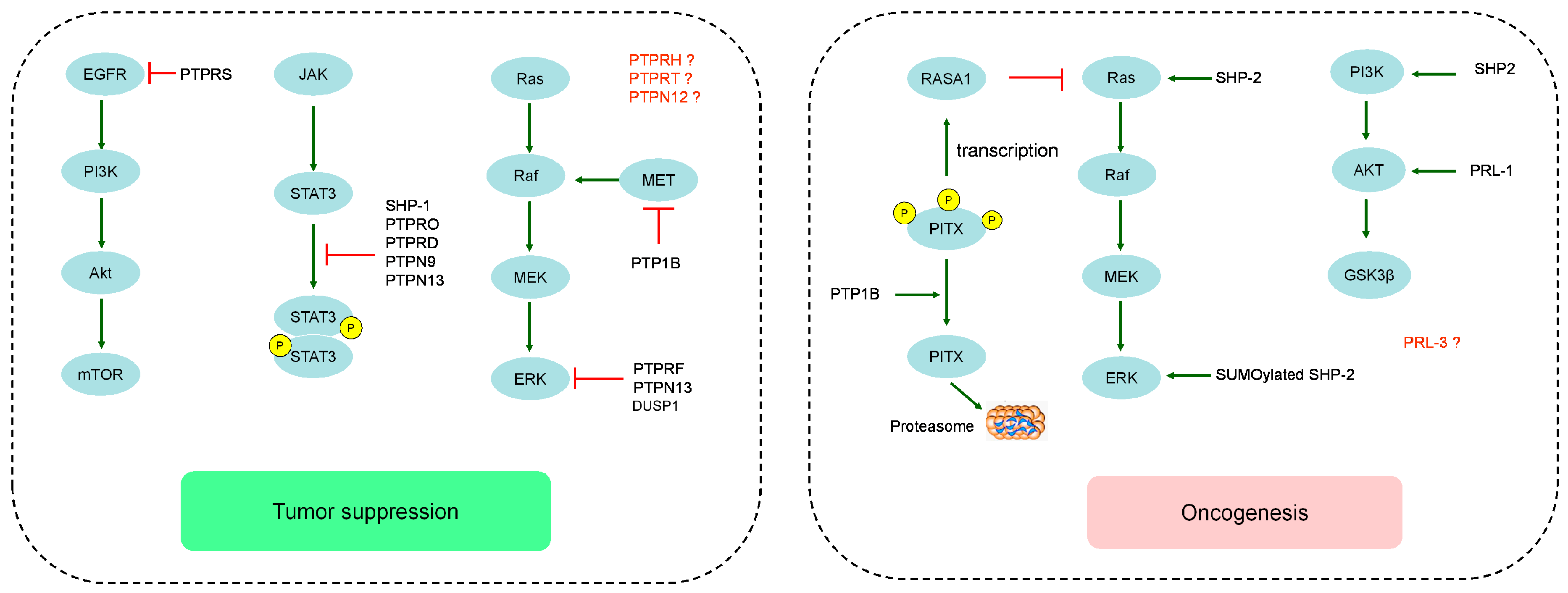

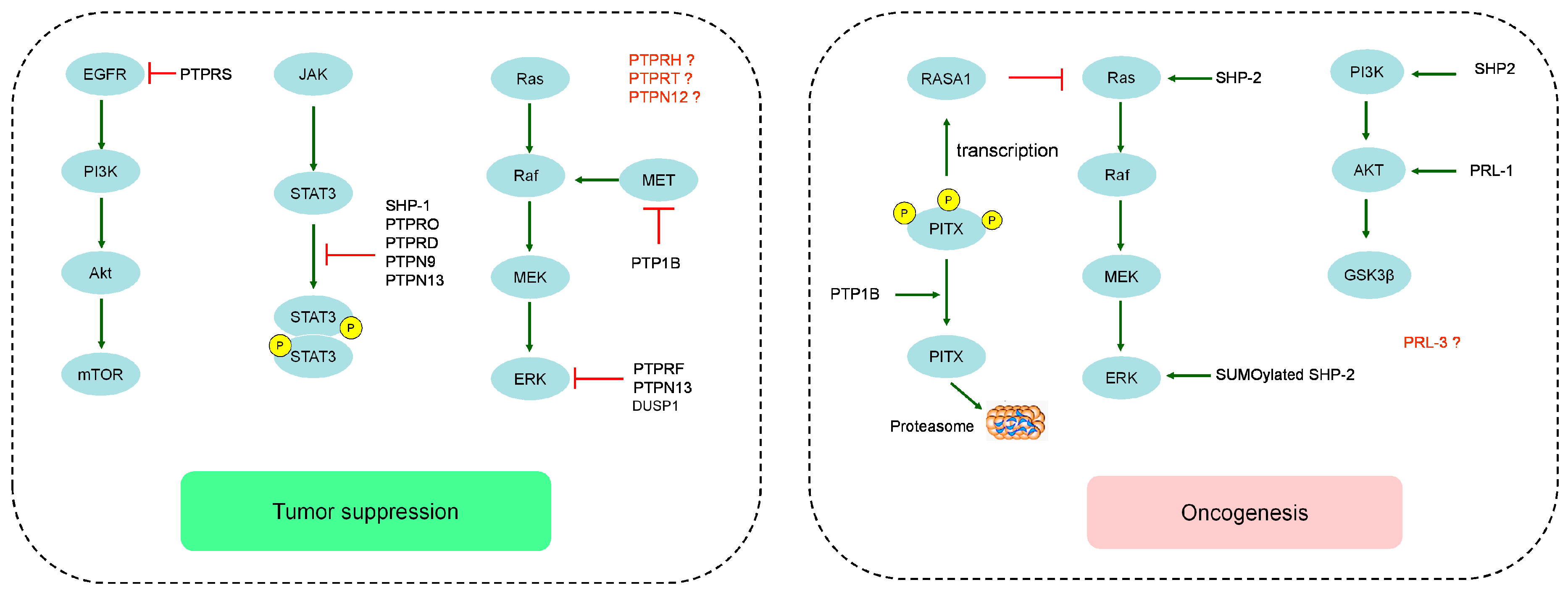{kind=link}
| PTPs | Observations | TSG or Onco | Ref. | |
|---|---|---|---|---|
| Receptor PTPs | PTPRD | PTPRD expression was downregulated or loss; High expression of PTPRD has a long-term survival rate and less chance of recurrent liver cancer. | TSG | [7,8] |
| PTPRF | PTPRF was frequent downregulated in most of HCC patients, and upregulation of PTPRF associated with better prognosis. PTPRF suppressed cell proliferation, colony formation in vitro and inhibited tumor growth in vivo. | TSG | [9] | |
| PTPRH | PTPRH expression in moderately differentiated HCCs and in all poorly differentiated HCCs was greatly reduced or loss. Overexpression of PTPRH reduce both migratory activity and growth rate of cells | TSG | [10] | |
| PTPRO | PTPRO expression was significantly reduced in human HCC specimens. Overexpression of PTPRO promoted apoptosis i and nhibited proliferation in vitro, and tumor size and number were increased in PTPRO knockout mice. | TSG | [11] | |
| PTPRS | Downregulation of PTPRS was observed in HCC cell lines and samples, and significantly associated with decreased overall survival and high risk of recurrence. PTPRS silencing promoted cell proliferation, migration and invasion both in vitro and in vivo. | TSG | [12] | |
| PTPRT | Increased expression of PTPRT in HepG2-xΔ127 cells treated decreased the tumor weight and volume in vivo | TSG | [13] | |
| non-receptor PTPs | PTP1B | PTP1B prime protein degradation of PITX1 to reduce p120RasGAP transcription. | Onco | [14] |
| Patients with low PTP1B expression had either shorter disease-free survival or worse overall survival. PTP1B could reduce phosphorylation of MET receptor to block vascular invasion and metastasis. | TSG | [15,16] | ||
| PTPN9 | PTPN9 expression was significantly reduced in tumor tissues, and low expression of PTPN9 predicted poor survival. PTPN9 silencing reduced apoptosis and promoted proliferation in HepG2 cells. | TSG | [17] | |
| PTPN12 | PTPN12 expression is frequently decreased or loss in HCC tissues. down-regulation of PTPN12 also significantly increased the migration of HCC cell lines. | TSG | [18] | |
| PTPN13 | PTPN13 expression was often downregulated or loss in HCC tissues and HCC cell lines, and positively correlated with overall survival but negatively correlated with the recurrence rate. Overexpression of PTPN13 inhibit EMT in HCC progression. | TSG | [19,20] | |
| SHP-1 | SHP-1 expression has a negative correlation with p-STAT3 Tyr705 in HCC, and SHP-1 overexpression abolished TGF-β1-induced p-STAT3 Tyr705. | TSG | [21] | |
| SHP-2 | SHP2 knockout mice result in development of tumors and enhance diethylenenitrite (DEN)-induced HCC development. | Onco | [22] | |
| low Shp2 expression was significantly associated with short overall survival time. SHP2 could promote HCC cell growth and metastasis by coordinately activating the Ras/Raf/Erk pathway and the PI3-K/Akt/mTOR cascade. | TSG | [23] | ||
| DUSPs | PRL1 | High PRL-1 expression was associated with more aggressive phenotype and poorer prognosis in HCC patients. Overpression of PRL-1 markedly enhanced migration and invasion of HCC cells. | Onco | [24,25] |
| PRL-3 | PRL-3 expression (both mRNA and protein) was significantly associated with poor differentiation and prognosis, and positive correlation with matrix metalloproteinase MMP1, MMP9, MMP10 and MMP12. | Onco | [26] | |
| DUSP1 | High DUSP1 expression was associated with better prognosis. In rat models, low DUSP1 expression was more susceptible to develop HCC. | TSG | [27,28] | |
| PTPs | Mechanisms for PTPs Regulation in HCC |
|---|---|
| PTPRD | Epigenetic silencing is partly responsible for the reduction of PTPRD expression [8]. Deletion and mutation of PTPRD gene are also identified in HCC cell lines and tumor tissues [30,36], miR-135a-5p targeting PTPRD mRNA impairs PTPRD expression [7]. |
| PTPRF | NA |
| PTPRH | NA |
| PTPRO | Epigenetic silencing is responsible for the reduction of PTPRO expression [43]. |
| PTPRS | Epigenetic silencing is responsible for the reduction of PTPRS expression [12]. |
| PTPRT | miR-215 targets PTPRT and down-regulates its protein expression in HepG2 cells [13] |
| PTP1B | NA |
| SHP-1 | NA |
| PTPN9 | NA |
| SHP-2 | SHP-2 activation may be due to protein SUMOylation at lysine residue 590 [106]. |
| PTPN12 | NA |
| PTPN13 | Epigenetic silencing is responsible for the reduction of PTPN13 expression [20]. |
| PRL-1 | Copy number amplification may be responsible for the increase expression of PRL-1 [25] |
| PRL-3 | NA |
| DUSP1 | DUSP1 inactivation is due to ubiquitination or promoter hypermethylation associated with loss of heterozygosity at the DUSP1 locus [27]. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Zhang, Y.; Ge, L.; Lin, Y.; Kwok, H.F. The Roles of Protein Tyrosine Phosphatases in Hepatocellular Carcinoma. Cancers 2018, 10, 82. https://doi.org/10.3390/cancers10030082
Huang Y, Zhang Y, Ge L, Lin Y, Kwok HF. The Roles of Protein Tyrosine Phosphatases in Hepatocellular Carcinoma. Cancers. 2018; 10(3):82. https://doi.org/10.3390/cancers10030082
Chicago/Turabian StyleHuang, Yide, Yafei Zhang, Lilin Ge, Yao Lin, and Hang Fai Kwok. 2018. "The Roles of Protein Tyrosine Phosphatases in Hepatocellular Carcinoma" Cancers 10, no. 3: 82. https://doi.org/10.3390/cancers10030082
APA StyleHuang, Y., Zhang, Y., Ge, L., Lin, Y., & Kwok, H. F. (2018). The Roles of Protein Tyrosine Phosphatases in Hepatocellular Carcinoma. Cancers, 10(3), 82. https://doi.org/10.3390/cancers10030082