Mutations in EMT-Related Genes in ALK Positive Crizotinib Resistant Non-Small Cell Lung Cancers

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
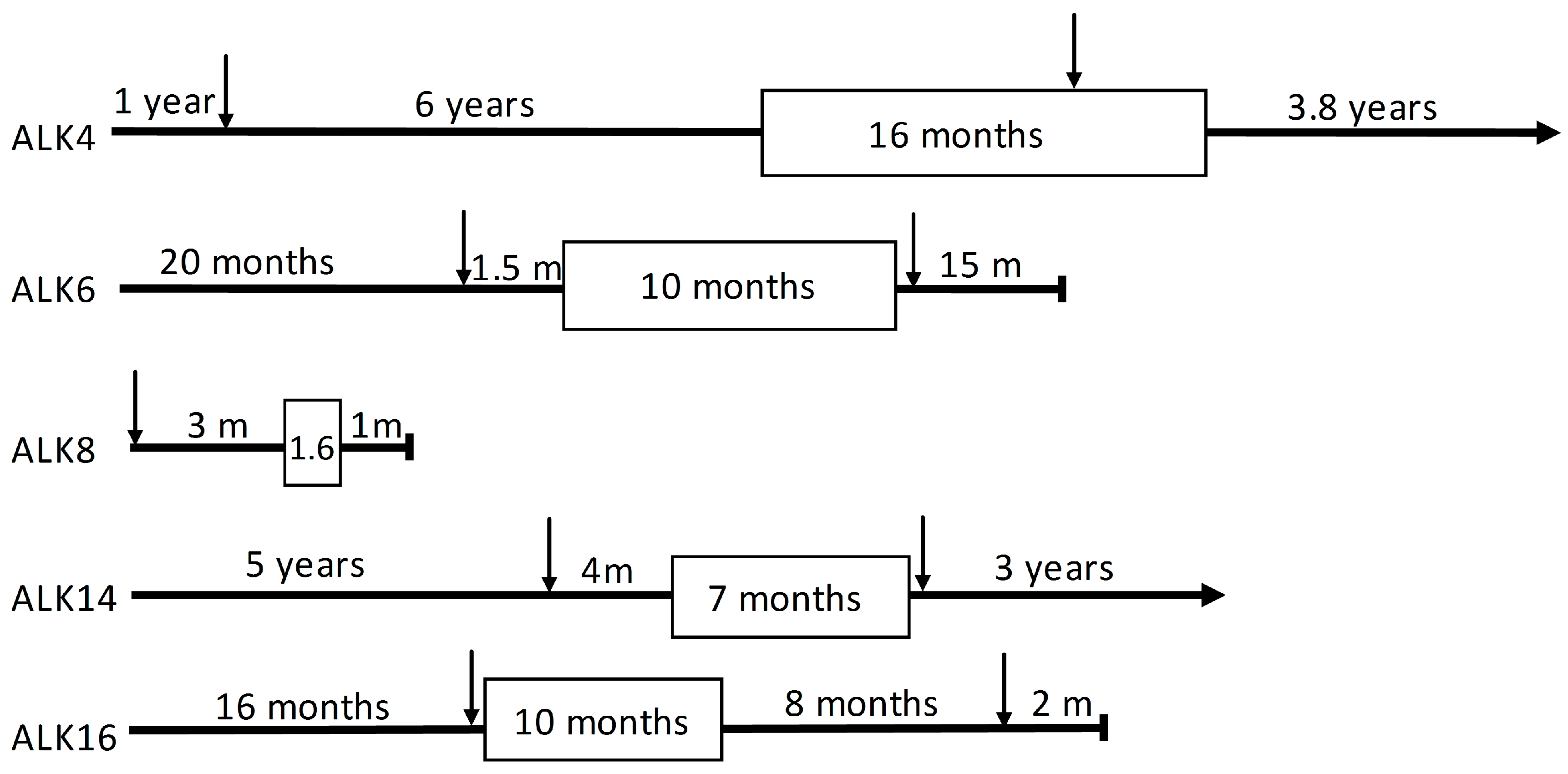
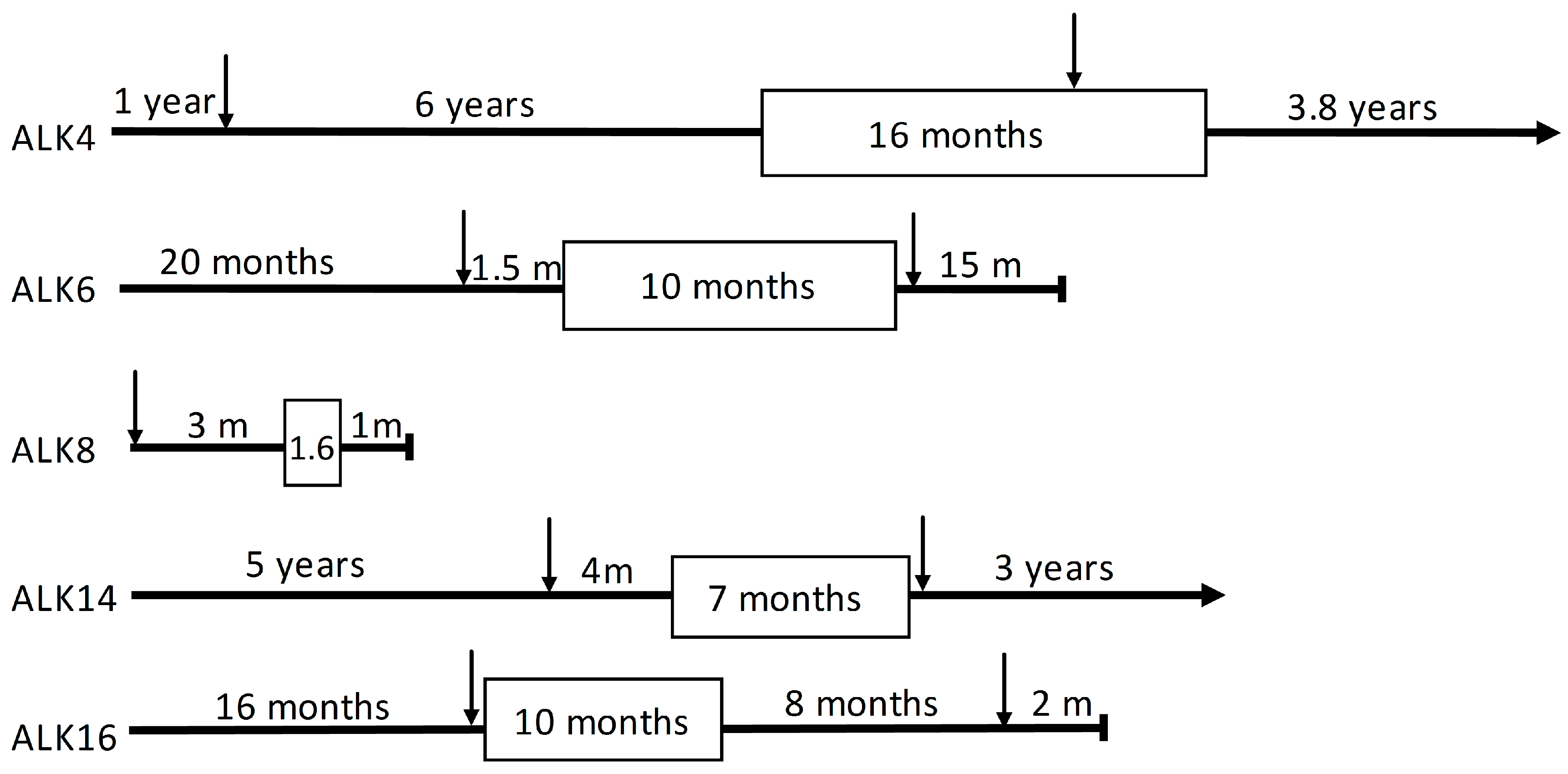
2.1. Patients
2.2. Whole Exome Sequencing
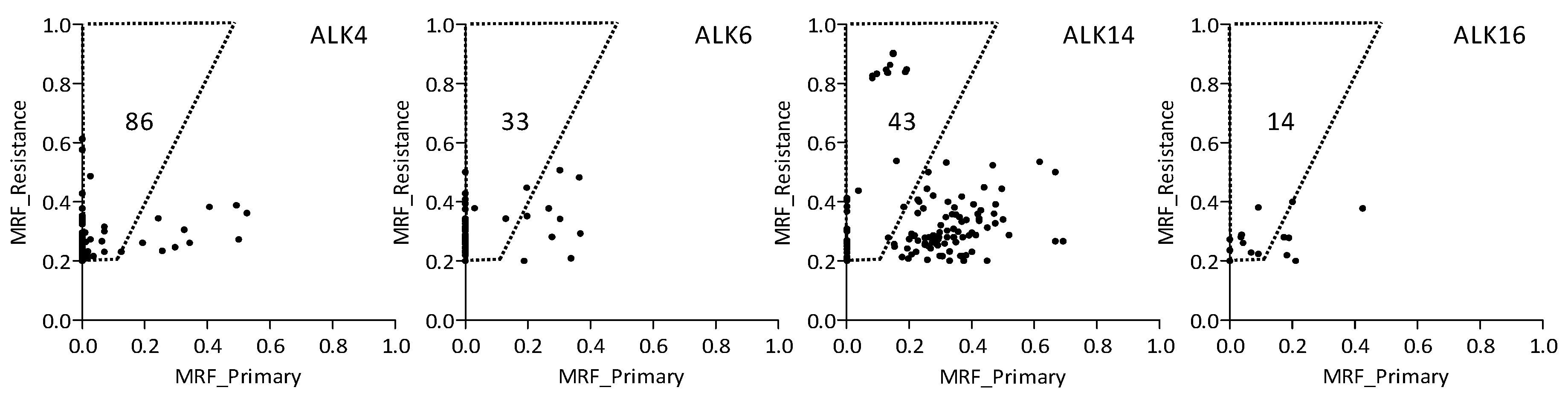
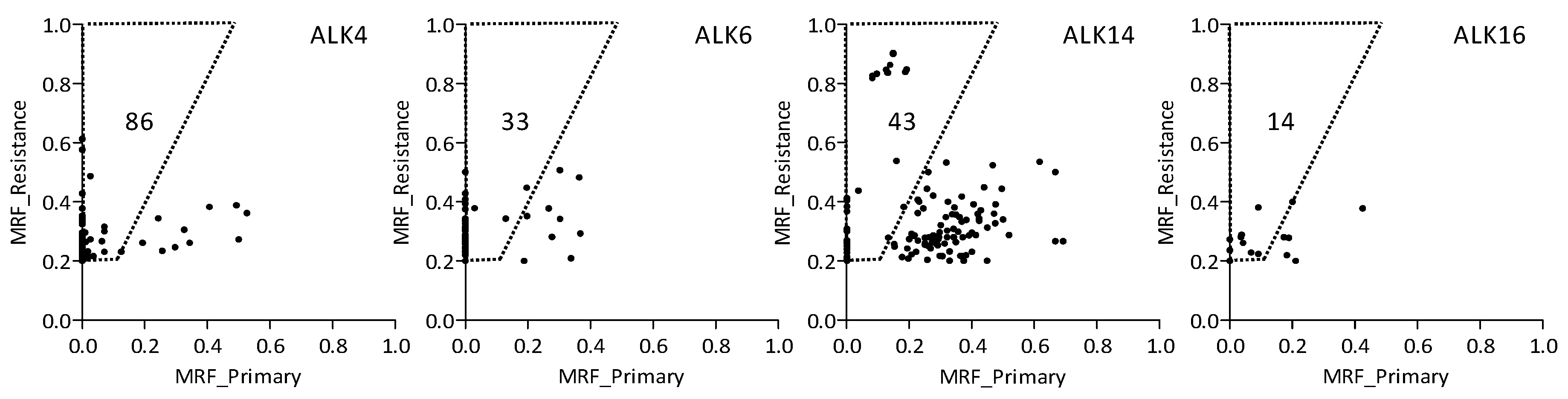
2.3. Variants Related to Crizotinib Treatment
2.4. Treatment-Related Copy Number Alterations
3. Discussion
4. Materials and Methods
4.1. Patient Inclusion
4.2. DNA Isolation
4.3. Whole Exome Sequencing
4.4. Immunohistochemistry
4.5. Data Analysis
4.6. WES-Based Analysis of CNAs
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Malik, P.S.; Raina, V. Lung cancer: Prevalent trends & emerging concepts. Indian J. Med. Res. 2015, 141, 5–7. [Google Scholar] [PubMed]
- Mountain, C.F. Revisions in the international system for staging lung cancer. Chest 1997, 111, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- El-Telbany, A.; Ma, P.C. Cancer genes in lung cancer: Racial disparities: Are there any? Genes Cancer 2012, 3, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.; Varella-Garcia, M.; Camidge, D.R. ALK gene rearrangements a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J. Thorac. Oncol. 2009, 4, 1450–1454. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Tan, D.S.W.; Chiari, R.; Wu, Y.-L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.-J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ascend-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Gandhi, L.; Gadgeel, S.; Riely, G.J.; Cetnar, J.; West, H.; Camidge, D.R.; Socinski, M.A.; Chiappori, A.; Mekhail, T. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: A single-group, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 234–242. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Van der Wekken, A.; Pelgrim, R.; ’t Hart, N.; Werner, N.; Mastik, M.; Hendriks, L.; van der Heijden, E.H.; Looijen-Salamon, M.; de Langen, A.J.; Staal-van den Brekel, J.; et al. Dichotomous ALK-IHC is a better predictor for ALK inhibition outcome than traditional ALK-FISH in advanced non-small cell lung cancer. Clin. Cancer Res. 2017, 23, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Doebele, R.C. Treating ALK-positive lung cancer--early successes and future challenges. Nat. Rev. Clin. Oncol. 2012, 9, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Saber, A.; van der Wekken, A.J.; Kok, K.; Terpstra, M.M.; Bosman, L.J.; Mastik, M.F.; Timens, W.; Schuuring, E.; Hiltermann, T.J.; Groen, H.J.; et al. Genomic aberrations in crizotinib resistant lung adenocarcinoma samples identified by transcriptome sequencing. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Van der Wekken, A.J.; Saber, A.; Hiltermann, T.J.; Kok, K.; van den Berg, A.; Groen, H.J. Resistance mechanisms after tyrosine kinase inhibitors afatinib and crizotinib in non-small cell lung cancer, a review of the literature. Crit. Rev. Oncol. Hematol. 2016, 100, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra117. [Google Scholar] [CrossRef] [PubMed]
- Gouji, T.; Takashi, S.; Mitsuhiro, T.; Yukito, I. Crizotinib can overcome acquired resistance to ch5424802: Is amplification of the MET gene a key factor? J. Thorac. Oncol. 2014, 9, e27–e28. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Khan, T.M.; Benes, C.; Lifshits, E.; Ebi, H.; Rivera, V.M.; Shakespeare, W.C.; Iafrate, A.J.; Engelman, J.A.; Shaw, A.T. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 7535–7540. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, AT. Crizotinib resistance: Implications for therapeutic strategies. Ann.Oncol. 2016, 27, iii42–iii50. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Seto, T. Updated evidence on the mechanisms of resistance to ALK inhibitors and strategies to overcome such resistance: Clinical and preclinical data. Oncol. Res. Treat. 2015, 38, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, W.S.; Choi, Y.J.; Choi, C.M.; Rho, J.K.; Lee, J.C. Epithelial-mesenchymal transition leads to crizotinib resistance in H-2228 lung cancer cells with EML4-ALK translocation. Mol. Oncol. 2013, 7, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Gower, A.; Hsu, W.H.; Hsu, S.T.; Wang, Y.; Giaccone, G. EMT is associated with, but does not drive resistance to ALK inhibitors among EML4-ALK non-small cell lung cancer. Mol Oncol. 2016, 10, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Endo, H.; Inoue, T.; Nishino, K.; Uchida, J.; Kumagai, T.; Kukita, Y.; Kato, K.; Imamura, F.; Inoue, M. Analysis of ERBB ligand-induced resistance mechanism to crizotinib by primary culture of lung adenocarcinoma with EML4-ALK fusion gene. J. Thorac. Oncol. 2015, 10, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Azzato, E.M.; Deshpande, C.; Aikawa, V.; Aggarwal, C.; Alley, E.; Jacobs, B.; Morrissette, J.; Daber, R. Rare complex mutational profile in an ALK inhibitor-resistant non-small cell lung cancer. Anticancer Res. 2015, 35, 3007–3012. [Google Scholar] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, T.M.; Kim, D.W.; Go, H.; Keam, B.; Lee, S.H.; Ku, J.L.; Chung, D.H.; Heo, D.S. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J. Thorac. Oncol. 2013, 8, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, L.; Zhao, Q.; Liu, Y.; He, L.; Xu, Q.; Sun, X.; Teng, L.; Cheng, H.; Ke, Y. SHP2 positively regulates TGFβ1-induced epithelial-mesenchymal transition modulated by its novel interacting protein Hook1. J. Biol. Chem. 2014, 289, 34152–34160. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liu, W.; Yu, W.-M.; Loh, M.L.; Alter, S.; Guvench, O.; MacKerell, A.D.; Tang, L.-D.; Qu, C.-K. Targeting protein tyrosine phosphatase SHP2 for the treatment of PTPN11-associated malignancies. Mol. Cancer Ther. 2013, 12, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Wanami, L.S.; Chen, H.-Y.; Peiró, S.; de Herreros, A.G.; Bachelder, R.E. Vascular endothelial growth factor—A stimulates Snail expression in breast tumor cells: Implications for tumor progression. Exp. Cell Res. 2008, 314, 2448–2453. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Hasan, M.R. Cancer metabolism and drug resistance. Metabolites 2015, 5, 571–600. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.; Banat, G.; Schmall, A.; Szibor, M.; Pomagruk, D.; Hänze, J.; Kolosionek, E.; Wilhelm, J.; Braun, T.; Grimminger, F. Phosphodiesterase-4 promotes proliferation and angiogenesis of lung cancer by crosstalk with HIF. Oncogene 2013, 32, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Savooji, J.; Liu, D. Second-and third-generation ALK inhibitors for non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Shaw, A.T. ALK inhibitors in non–small cell lung cancer: Crizotinib and beyond. Clin. Adv. Hematol. Oncol. 2014, 12, 429–439. [Google Scholar] [PubMed]
- Ou, S.-H.I.; Ahn, J.S.; Petris, L.D.; Govindan, R.; Yang, J.C.-H.; Hughes, B.; Lena, H.; Moro-Sibilot, D.; Bearz, A.; Ramirez, S.V.; et al. Alectinib in crizotinib-refractory ALK-rearranged non–small-cell lung cancer: A phase II global study. J. Clin. Oncol. 2016, 34, 661–668. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows–wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2.0: A database of human non-synonymous SNVS and their functional predictions and annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S. Cosmic: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.P. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [Green Version]
- Lek, M.; Karczewski, K.; Minikel, E.; Samocha, K.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.; Ware, J.; Hill, A.; Cummings, B. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, R.S.N.; Karjalainen, J.M.; Krajewska, M.; Westra, H.-J.; Maloney, D.; Simeonov, A.; Pers, T.H.; Hirschhorn, J.N.; Jansen, R.C.; Schultes, E.A.; et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 2015, 47, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013; arXiv:1303.3997. [Google Scholar]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. varScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, E.S.; Olshen, A.B. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 2007, 23, 657–663. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Patient | Gender | Age at Diagnosis (Years) | PFS 1 (m) | Smoking Status (py 2) | Tumor Location | |
|---|---|---|---|---|---|---|
| Primary 4 | Resistant 5 | |||||
| ALK4 | female | 34 | 15.9 | non-smoker | ovary | liver |
| ALK6 | male | 55 | 9.5 | past-smoker (15) | cervical lymph node | glenoid |
| ALK8 | male | 76 | 1.6 | past-smoker (NA 3) | lymph node mediastinal 4 right | not done |
| ALK14 | female | 62 | 8.4 | current smoker (20) | brain occipital metastasis | mediastinal lymph node 7 |
| ALK16 | female | 48 | 5.1 | past-smoker (18) | lung | lung |
| Pathway Name | Patient(s) | Genes Mutated | Enrichment Score | Enrichment p-Value 1 |
|---|---|---|---|---|
| Proteoglycans in cancer 2 | ALK6 ALK14 | ANK2, FASLG, HSPG2, PTPN11, STAT3, VEGFA | 8.0 | 0.00 |
| HIF-1 signaling pathway 2 | ALK6 ALK14 | ARNT, STAT3, VEGFA | 4.3 | 0.01 |
| FoxO signaling pathway 2 | ALK6 ALK14 | FASLG, SMAD4, STAT3 | 3.7 | 0.03 |
| ECM-receptor interaction 3 | ALK6 ALK14 | HSPG2, LAMA2 | 3.3 | 0.04 |
| Adherens junction 3 | ALK6 ALK14 | CTNNA3, SMAD4 | 3.0 | 0.05 |
| Ras signaling pathway | ALK14 | FASLG, PTPN11, VEGFA | 2.7 | 0.07 |
| Jak-STAT signaling pathway | ALK6 ALK14 | PTPN11, STAT3 | 1.9 | ns |
| PI3K-AKT signaling pathway | ALK14 | FASLG, LAMA2, VEGFA | 1.8 | ns |
| Focal adhesion 3 | ALK14 | LAMA2, VEGFA | 1.6 | ns |
| VEGF signaling pathway 3 | ALK14 | VEGFA | 1.3 | ns |
| TGF-beta signaling pathway | ALK6 | SMAD4 | 1.2 | ns |
| MAPK signaling pathway | ALK14 | FASLG, MAP3K20 | 1.1 | ns |
| Wnt signaling pathway 3 | ALK6 | SMAD4 | 1.0 | ns |
| Regulation of actin cytoskeleton | ALK6 | SCIN | 0.5 | ns |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, J.; Van der Wekken, A.J.; Saber, A.; Terpstra, M.M.; Schuuring, E.; Timens, W.; Hiltermann, T.J.N.; Groen, H.J.M.; Van den Berg, A.; Kok, K. Mutations in EMT-Related Genes in ALK Positive Crizotinib Resistant Non-Small Cell Lung Cancers. Cancers 2018, 10, 10. https://doi.org/10.3390/cancers10010010
Wei J, Van der Wekken AJ, Saber A, Terpstra MM, Schuuring E, Timens W, Hiltermann TJN, Groen HJM, Van den Berg A, Kok K. Mutations in EMT-Related Genes in ALK Positive Crizotinib Resistant Non-Small Cell Lung Cancers. Cancers. 2018; 10(1):10. https://doi.org/10.3390/cancers10010010
Chicago/Turabian StyleWei, Jiacong, Anthonie J. Van der Wekken, Ali Saber, Miente M. Terpstra, Ed Schuuring, Wim Timens, T. Jeroen N. Hiltermann, Harry J. M. Groen, Anke Van den Berg, and Klaas Kok. 2018. "Mutations in EMT-Related Genes in ALK Positive Crizotinib Resistant Non-Small Cell Lung Cancers" Cancers 10, no. 1: 10. https://doi.org/10.3390/cancers10010010