
Mast Cell Targeted Chimeric Toxin Can Be Developed as an Adjunctive Therapy in Colon Cancer Treatment

Abstract
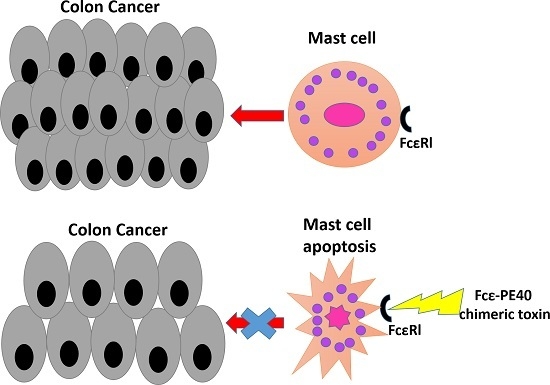
:
1. Introduction
2. Results
2.1. Mast Cell Infiltration Is Associated with Colon Cancer Development and Distant Metastasis
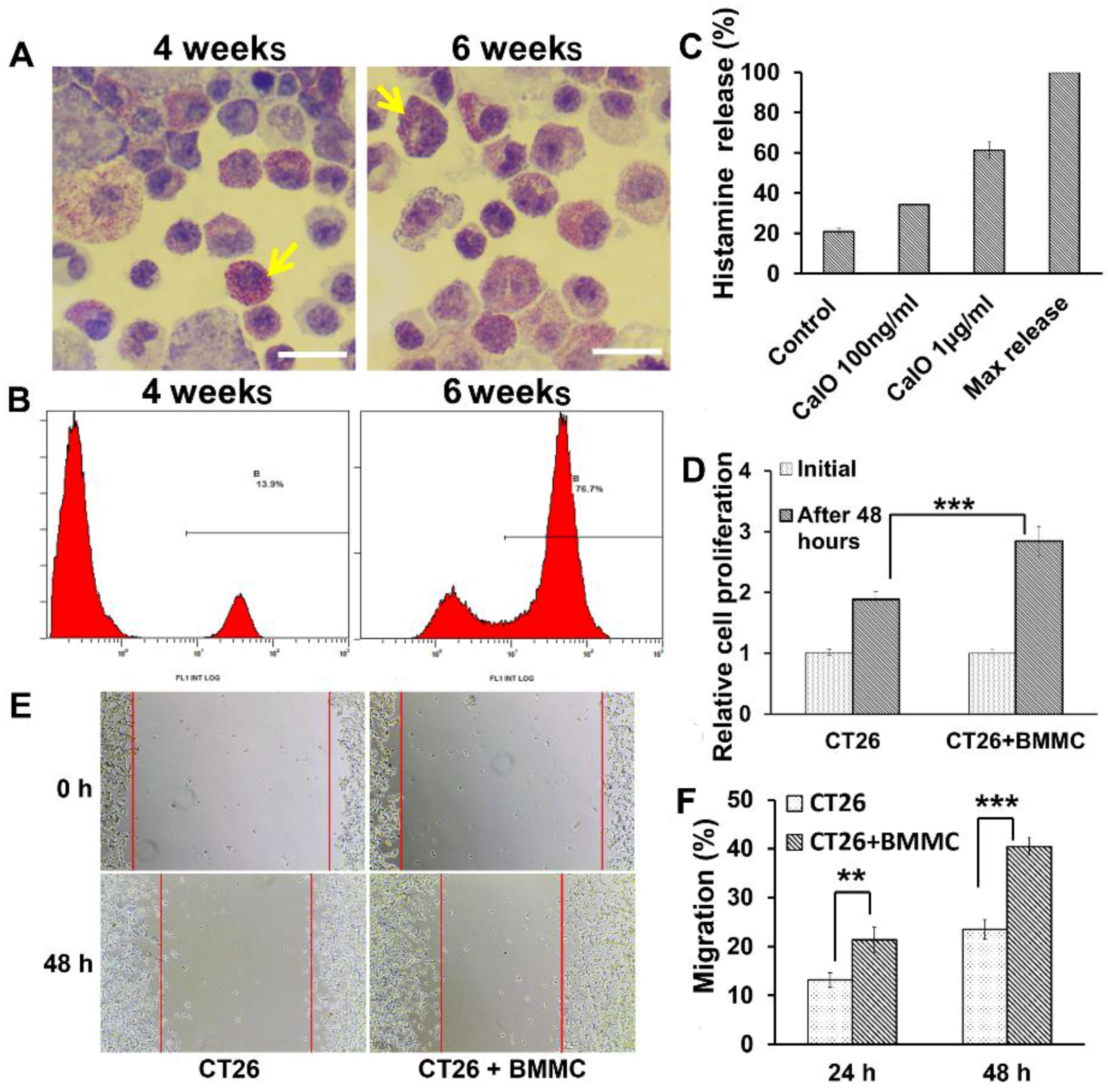
2.2. Mast Cells Induced from Bone Marrow Cells Can Manifest “Piecemeal Degranulation” and Promote Colon Cancer Cell Proliferation and Migration in Vitro
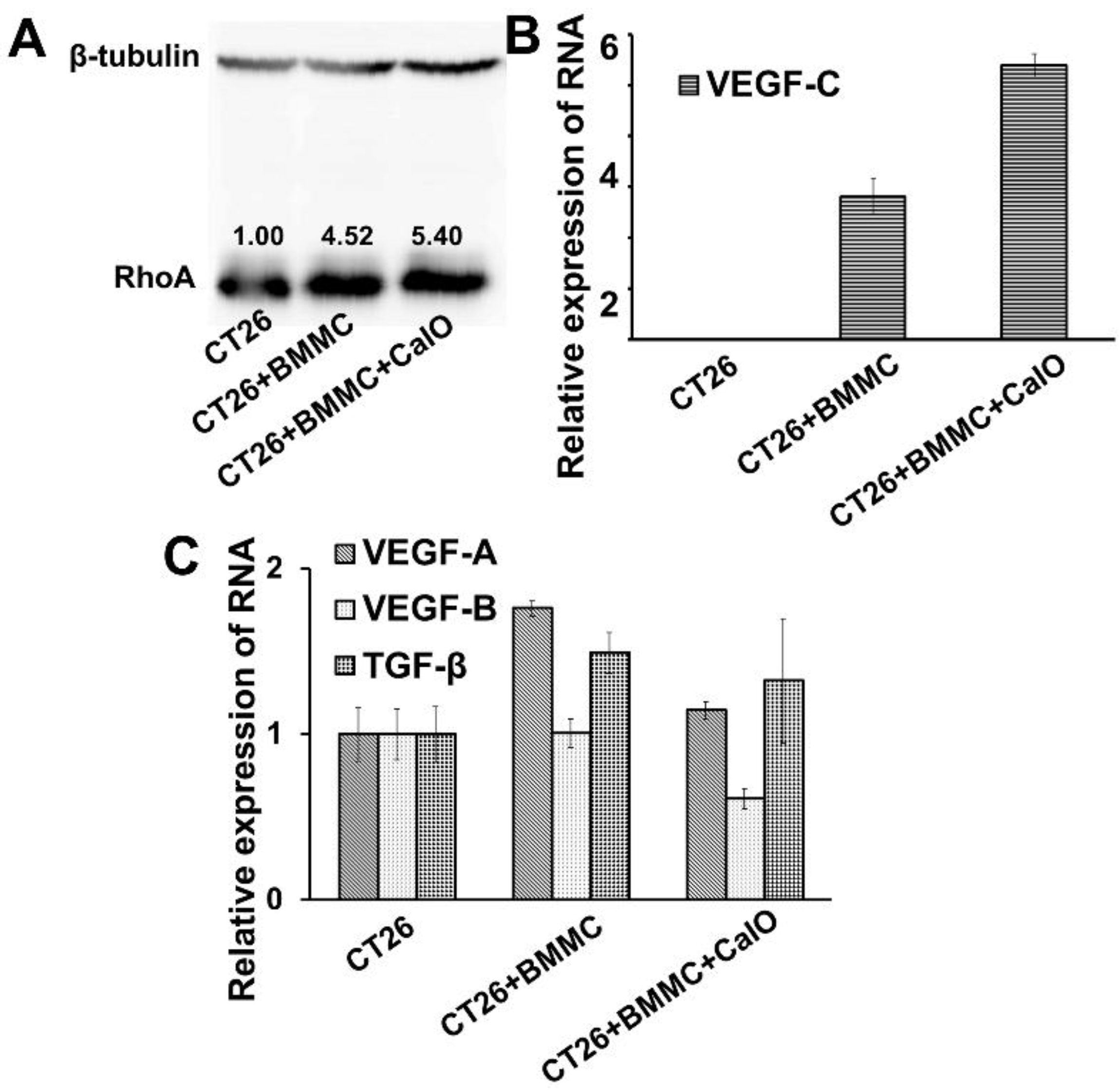
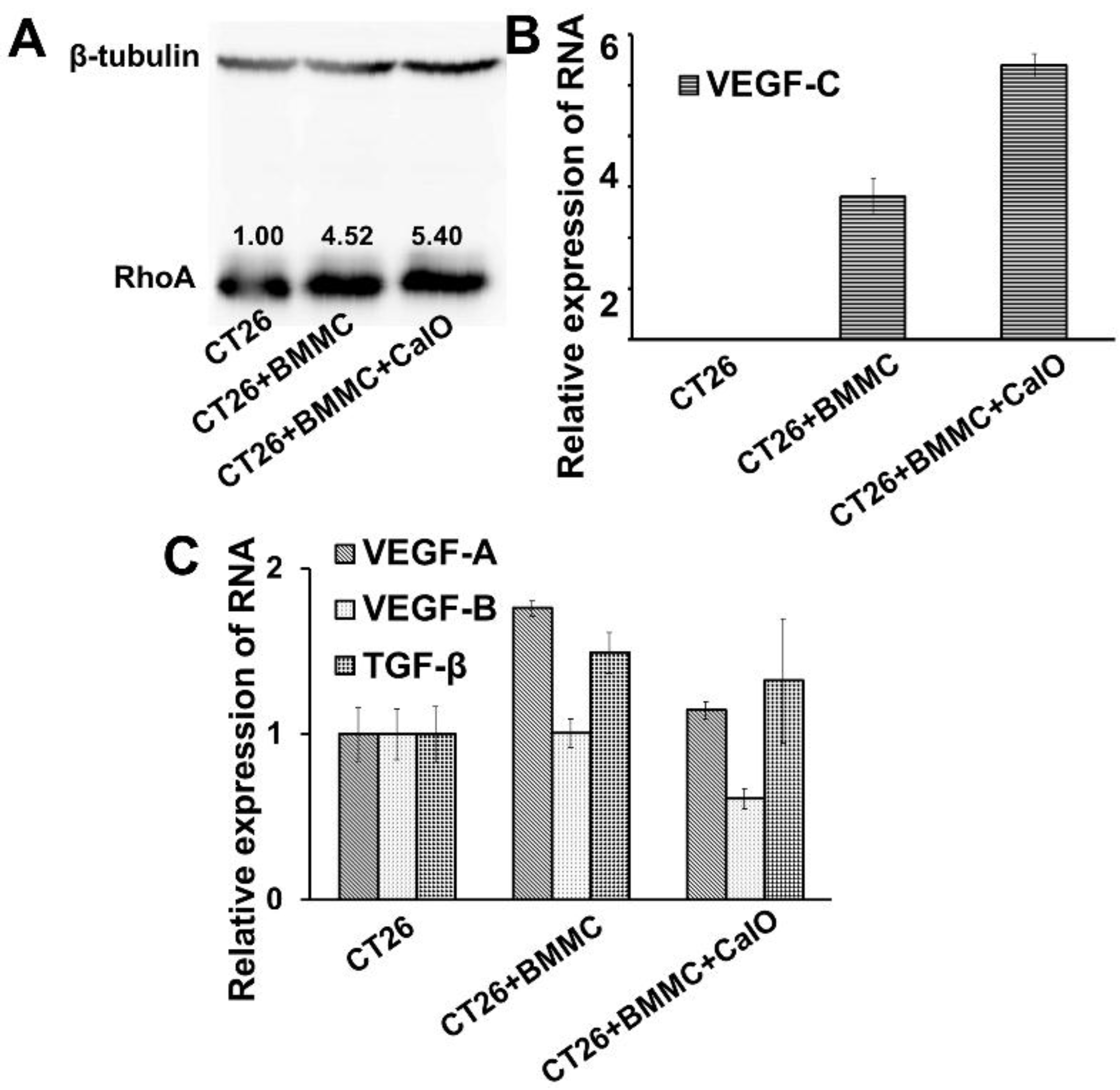
2.3. Mast Cells Can Up-Regulate the Expression of RhoA and VEGFc in Colon Cancer Cells to Promote Cancer Invasion and Angiogenesis
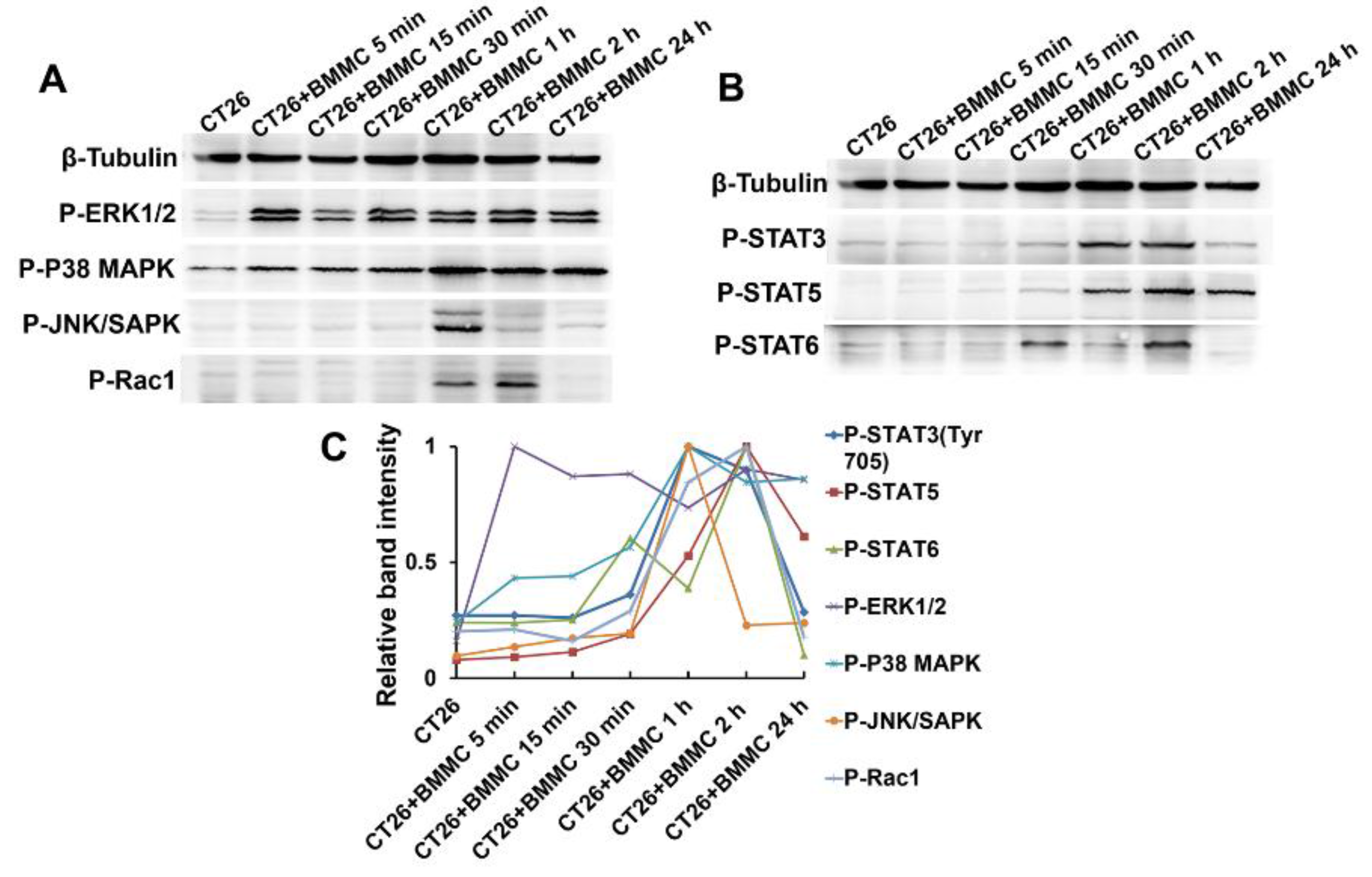
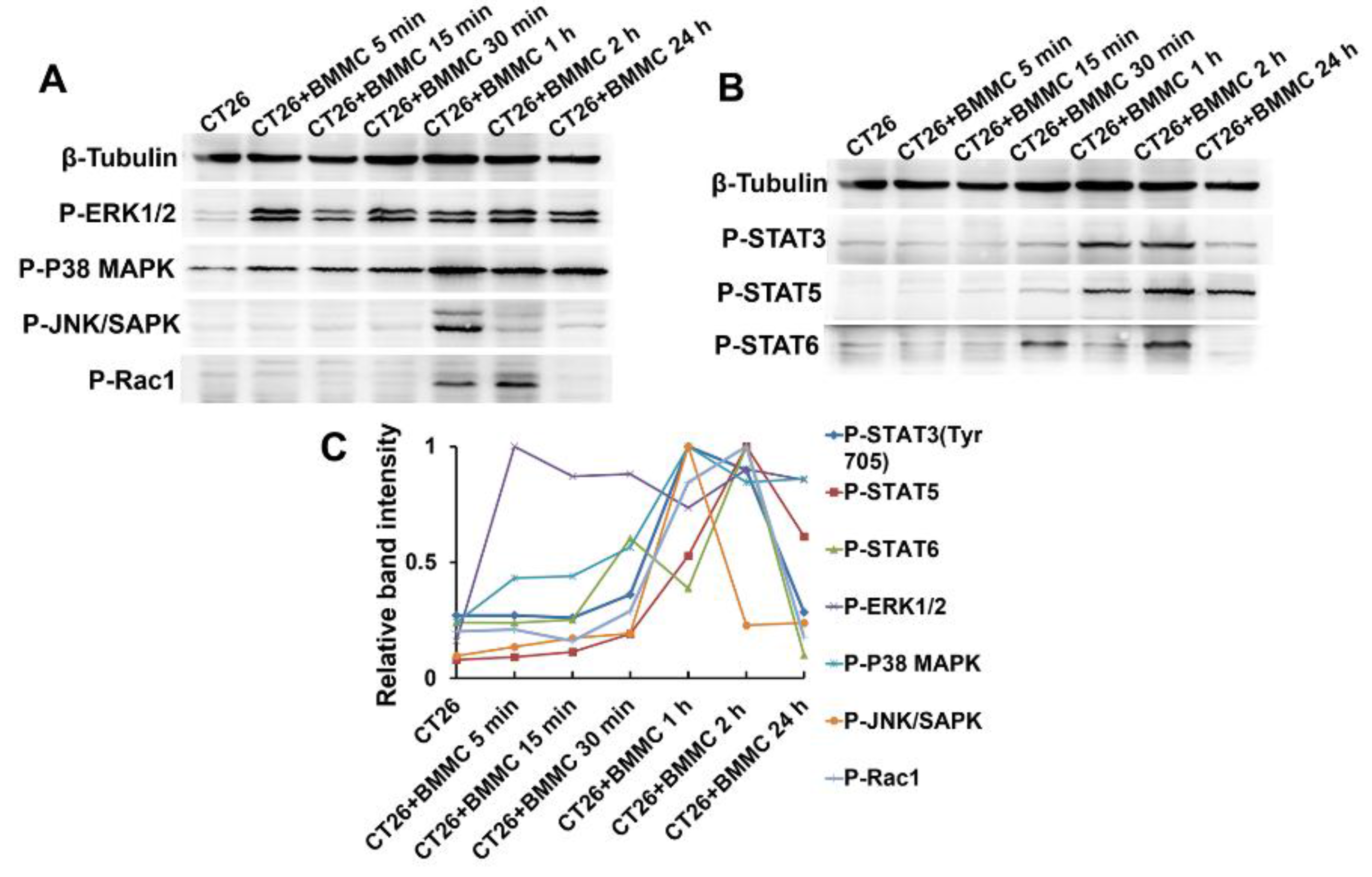
2.4. MAPK/Rho-GTPase/STATs Pathways Participate in the Regulation of Mast Cell Function in Colon Cancer Development
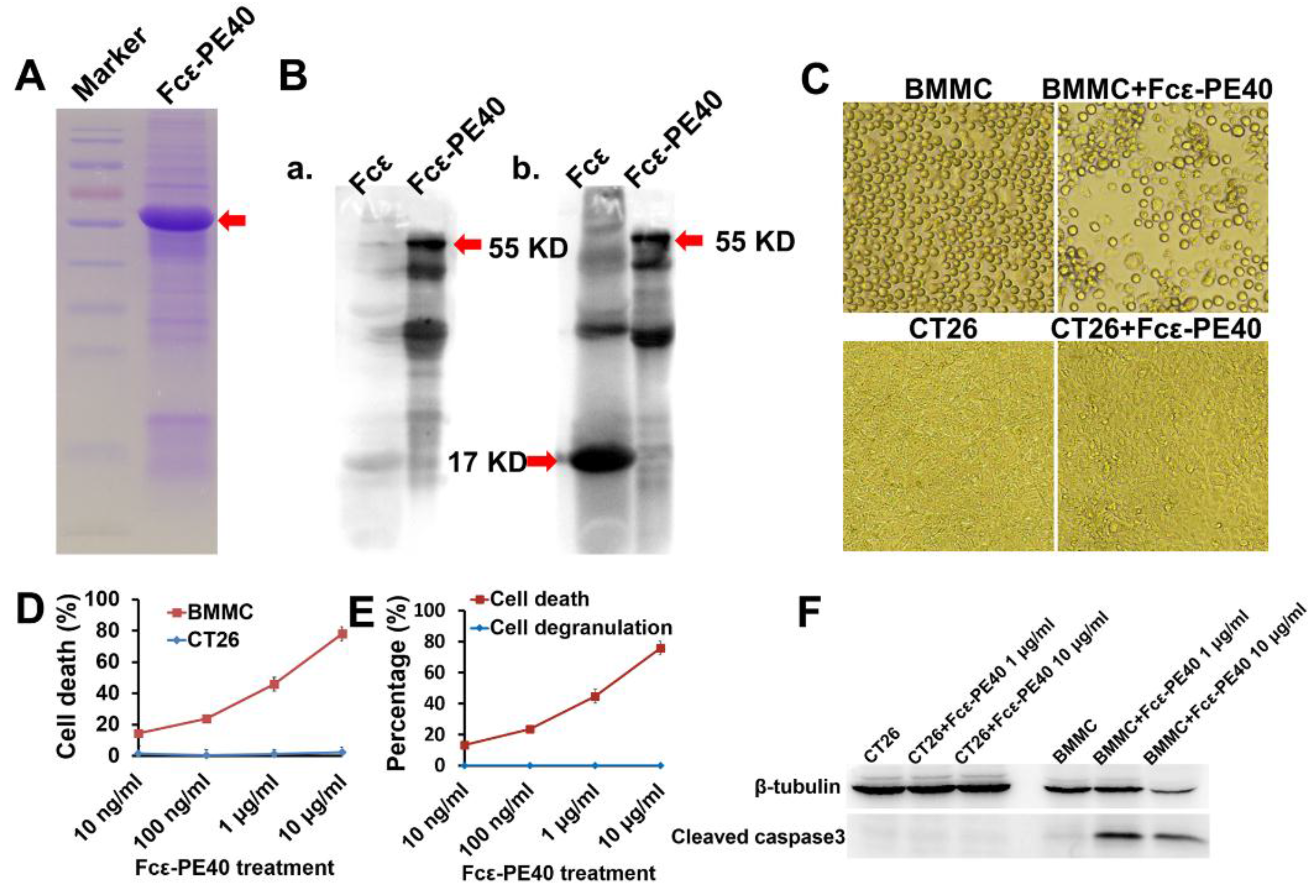
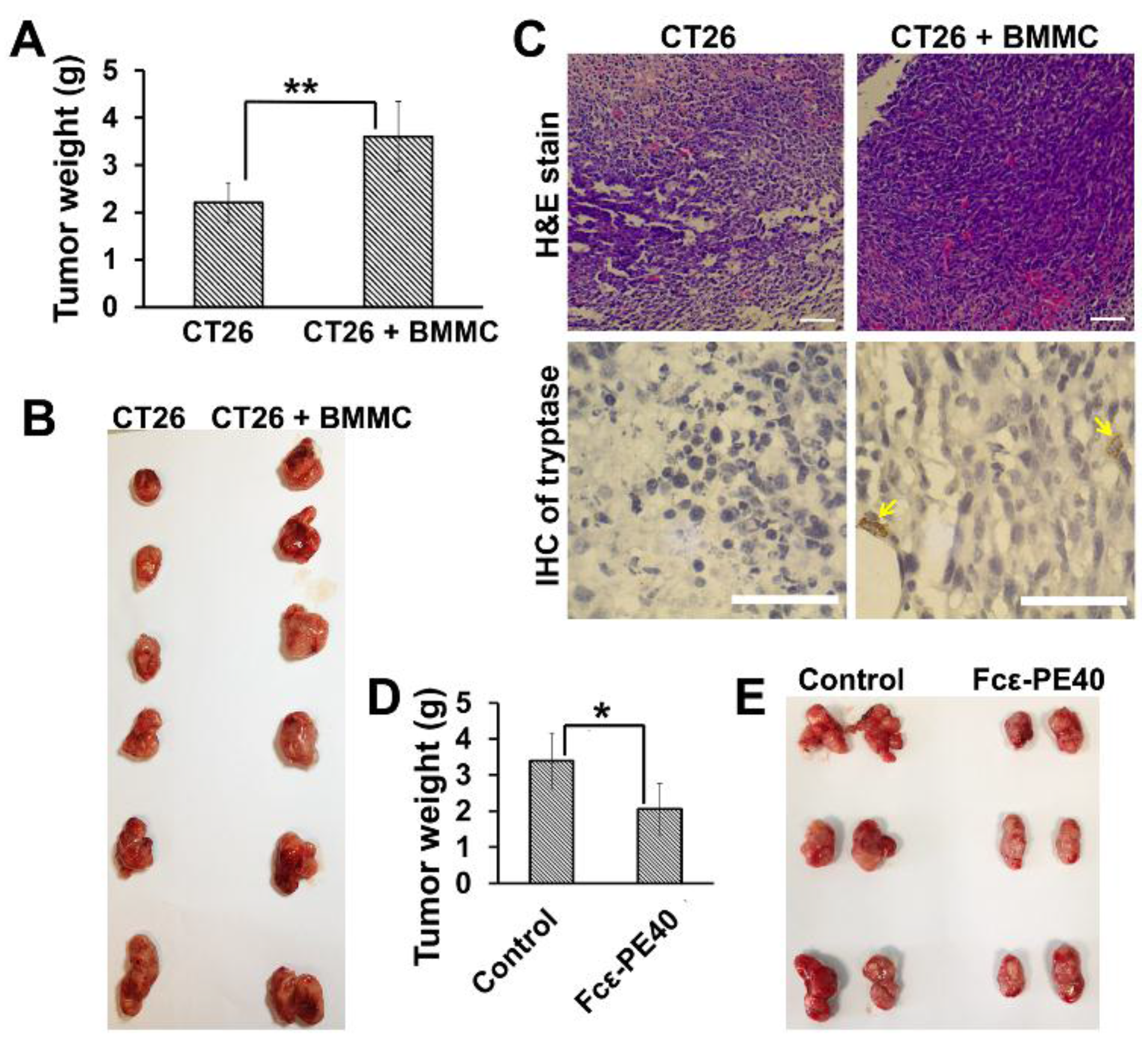
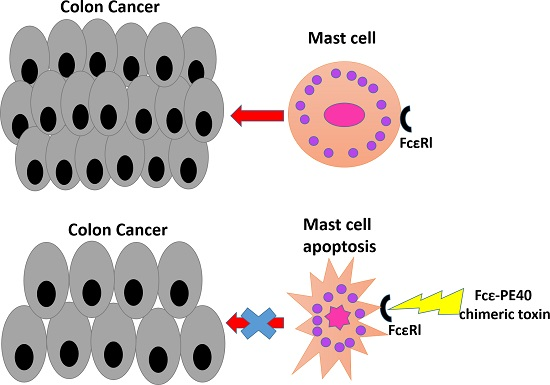
2.5. Targeted Fcε-PE40 Chimeric Toxin Can Specifically Induce BMMCs Apoptosis without Degranulation
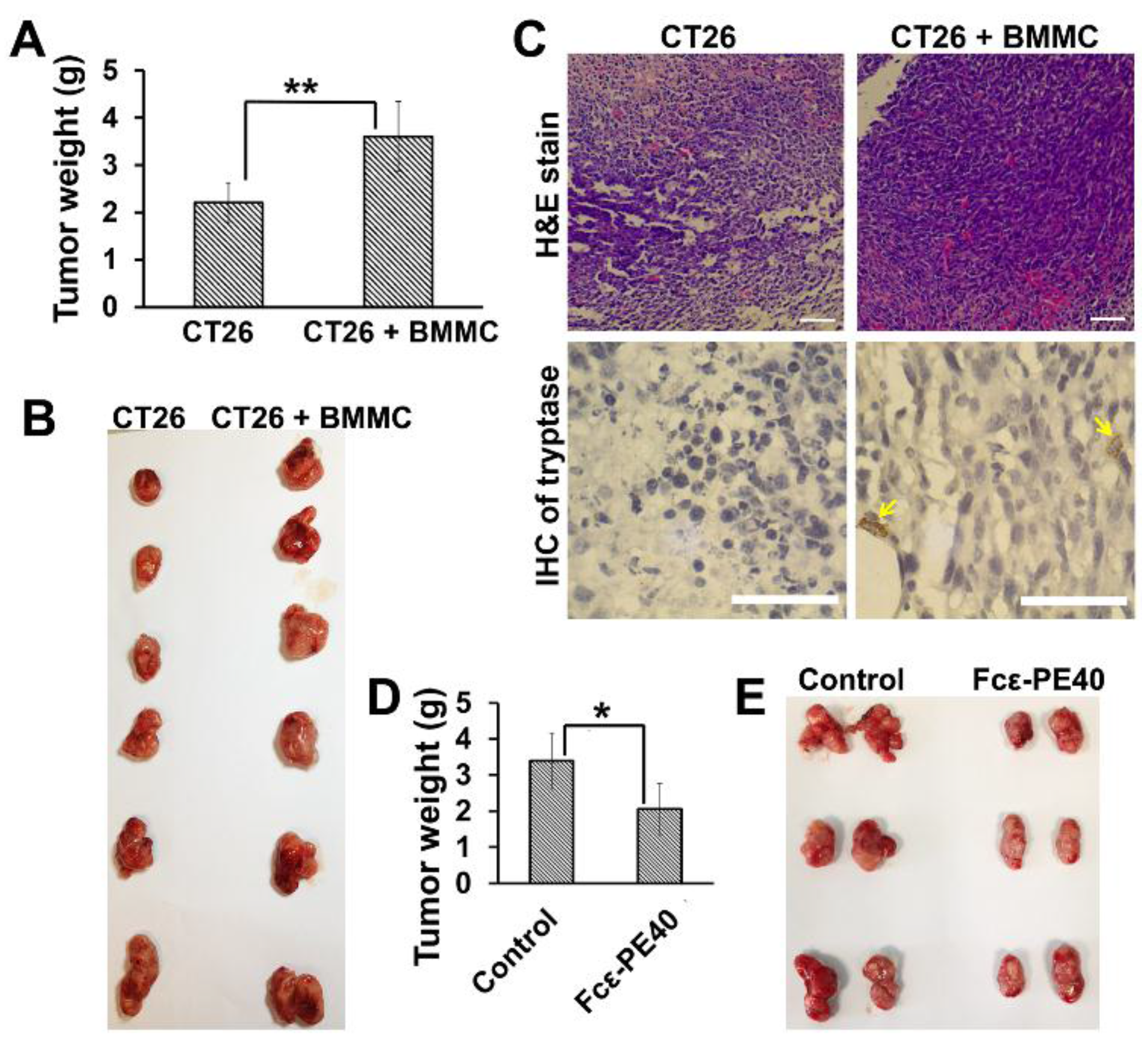
2.6. The Promotion of Mast Cell to Colon Cancer in Vivo Can Be Effectively Controlled by Targeted Fcε-PE40 Chimeric Toxin
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Patients and Tissues
5.2. Mice and Cell Culture
5.3. Immunohistochemistry and MC Counting
5.4. Plasmid Construction, Protein Expression and Partial Purification
5.5. Immunoblotting
5.6. Flow Cytometry
5.7. Histamine Release
5.8. Cell Proliferation Assay
5.9. Colon Cancer Epithelial Cell Invasion Assay
5.10. RNA Extraction and Quantitative Real-Time PCR
5.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PEA | Pseudomonsexotoxin A |
| BMMC | Bone marrow mast cell |
References
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Varga, J.; Wang, T.C.; Greten, F.R. The gastrointestinal tumor microenvironment. Gastroenterology 2013, 145, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat. Rev. 2014, 40, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Riese, D.J.; Cullum, R.L. Epiregulin: Roles in normal physiology and cancer. Semin. Cell Dev. Biol. 2014, 28, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Loesch, M.; Chen, G. The p38 mapk stress pathway as a tumor suppressor or more? Front. Biosci. 2008, 13, 3581–3593. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of mapk signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; di Magliano, M.P. Interleukin-6 is required for pancreatic cancer progression by promoting mapk signaling activation and oxidative stress resistance. Cancer Res. 2013, 73, 6359–6374. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.A.; Zheng, Q.; Liu, Z.; Thompson, J.E. Role of p38 and jnk mapk signaling pathways and tumor suppressor p53 on induction of apoptosis in response to ad-eif5a1 in a549 lung cancer cells. Mol. Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.J.; Webb, S.L.; Parr, C.; Mason, M.D.; Jiang, W.G. The type ii transmembrane serine protease, matriptase-2: Possible links to cancer? Anti-Cancer Agents Med. Chem. 2010, 10, 64–69. [Google Scholar] [CrossRef]
- Chia, C.Y.; Kumari, U.; Casey, P.J. Breast cancer cell invasion mediated by galpha12 signaling involves expression of interleukins-6 and -8, and matrix metalloproteinase-2. J. Mol. Signal. 2014, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Lin, B.R.; Chang, C.C.; Chu, C.Y.; Su, H.J.; Chen, S.T.; Jeng, Y.M.; Kuo, M.L. Il-6 induces ags gastric cancer cell invasion via activation of the c-src/rhoa/rock signaling pathway. Int. J. Cancer. 2007, 120, 2600–2608. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Chandrasekaran, S.; Hsu, J.W.; Gidwani, M.; Hughes, A.D.; King, M.R. Phenotypic switch in blood: Effects of pro-inflammatory cytokines on breast cancer cell aggregation and adhesion. PLoS ONE 2013, 8, e54959. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N.; Meshram, R.J. Targeting tumor micro-environment for design and development of novel anti-angiogenic agents arresting tumor growth. Prog. Biophys. Mol. Biol. 2013, 113, 333–354. [Google Scholar] [CrossRef] [PubMed]
- Le Guelte, A.; Dwyer, J.; Gavard, J. Jumping the barrier: Ve-cadherin, vegf and other angiogenic modifiers in cancer. Biol. Cell 2011, 103, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H. Targeting of tak1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 2012, 33, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Vaiopoulos, A.G.; Papachroni, K.K.; Papavassiliou, A.G. Colon carcinogenesis: Learning from nf-kappab and ap-1. Int. J. Biochem. Cell Biol. 2010, 42, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Cardama, A.; Verstovsek, S. Molecular pathways: Jak/stat pathway: Mutations, inhibitors, and resistance. Clin. Cancer Res. 2013, 19, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Laveti, D.; Kumar, M.; Hemalatha, R.; Sistla, R.; Naidu, V.G.; Talla, V.; Verma, V.; Kaur, N.; Nagpal, R. Anti-inflammatory treatments for chronic diseases: A review. Inflamm. Allergy Drug Targets 2013, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Crivellato, E.; Beltrami, C.; Mallardi, F.; Ribatti, D. Paul ehrlich’s doctoral thesis: A milestone in the study of mast cells. Br. J. Haematol. 2003, 123, 19–21. [Google Scholar] [PubMed]
- Wedemeyer, J.; Tsai, M.; Galli, S.J. Roles of mast cells and basophils in innate and acquired immunity. Curr. Opin. Immunol. 2000, 12, 624–631. [Google Scholar] [CrossRef]
- Galli, S.J.; Tsai, M. Mast cells in allergy and infection: Versatile effector and regulatory cells in innate and adaptive immunity. Eur. J. Immunol. 2010, 40, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Ali, H. Regulation of human mast cell and basophil function by anaphylatoxins c3a and c5a. Immunol. Lett. 2010, 128, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Lei, Z.; Zhang, G.M.; Li, D.; Song, C.; Li, B.; Liu, Y.; Yuan, Y.; Unkeless, J.; Xiong, H.; et al. Scf-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood 2008, 112, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Kanbe, N.; Tanaka, A.; Kanbe, M.; Itakura, A.; Kurosawa, M.; Matsuda, H. Human mast cells produce matrix metalloproteinase 9. Eur. J. Immunol. 1999, 29, 2645–2649. [Google Scholar] [CrossRef]
- Crivellato, E.; Finato, N.; Ribatti, D.; Beltrami, C.A. Piecemeal degranulation in human tumour pheochromocytes. J. Anat. 2005, 206, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.A.; Ieni, A.; Fedele, F.; Zuccala, V.; Riccardo, M.; Parisi, E.; Parisi, A. Degranulation patterns of eosinophils in advanced gastric carcinoma: An electron microscopic study. Ultrastruct. Pathol. 2005, 29, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Conti, P. Mast cells: The jekyll and hyde of tumor growth. Trends Immunol. 2004, 25, 235–241. [Google Scholar] [CrossRef] [PubMed]
- McNeil, H.P.; Adachi, R.; Stevens, R.L. Mast cell-restricted tryptases: Structure and function in inflammation and pathogen defense. J. Biol. Chem. 2007, 282, 20785–20789. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Tinkle, C.L.; Hanahan, D.; Werb, Z. Mmp-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 2000, 103, 481–490. [Google Scholar] [CrossRef]
- Maltby, S.; Khazaie, K.; McNagny, K.M. Mast cells in tumor growth: Angiogenesis, tissue remodelling and immune-modulation. Biochim. Biophys. Acta 2009, 1796, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Crivellato, E.; Nico, B.; Ribatti, D. Mast cells and tumour angiogenesis: New insight from experimental carcinogenesis. Cancer Lett. 2008, 269, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Mast cells and macrophages exert beneficial and detrimental effects on tumor progression and angiogenesis. Immunol. Lett. 2013, 152, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Adams, W.J.; Morris, D.L. Short-course cimetidine and survival with colorectal cancer. Lancet 1994, 344, 1768–1769. [Google Scholar] [CrossRef]
- Matsumoto, S. Cimetidine and survival with colorectal cancer. Lancet 1995, 346, 115. [Google Scholar] [CrossRef]
- Matsumoto, S.; Imaeda, Y.; Umemoto, S.; Kobayashi, K.; Suzuki, H.; Okamoto, T. Cimetidine increases survival of colorectal cancer patients with high levels of sialyl lewis-x and sialyl lewis-a epitope expression on tumour cells. Br. J. Cancer 2002, 86, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Khazaie, K.; Blatner, N.R.; Khan, M.W.; Gounari, F.; Gounaris, E.; Dennis, K.; Bonertz, A.; Tsai, F.N.; Strouch, M.J.; Cheon, E.; et al. The significant role of mast cells in cancer. Cancer Metastasis Rev. 2011, 30, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, D.S.; Nechushtan, H. Mast cells and cancer—No longer just basic science. Crit. Rev. Oncol. Hematol. 2008, 68, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, N.; Takayama, H.; Nishimura, K.; Oka, D.; Nakai, Y.; Shiba, M.; Tsujimura, A.; Nakayama, M.; Aozasa, K.; Okuyama, A. Decreased number of mast cells infiltrating into needle biopsy specimens leads to a better prognosis of prostate cancer. Br. J. Cancer 2007, 97, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Strouch, M.J.; Cheon, E.C.; Salabat, M.R.; Krantz, S.B.; Gounaris, E.; Melstrom, L.G.; Dangi-Garimella, S.; Wang, E.; Munshi, H.G.; Khazaie, K.; et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin. Cancer Res. 2010, 16, 2257–2265. [Google Scholar] [CrossRef] [PubMed]
- Goffredo, V.; Gadaleta, C.D.; Laterza, A.; Vacca, A.; Ranieri, G. Tryptase serum levels in patients suffering from hepatocellular carcinoma undergoing intra-arterial chemoembolization: Possible predictive role of response to treatment. Mol. Clin. Oncol. 2013, 1, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.H.; Jayabaskar, T.; Yip, G.; Tan, Y.; Hilmy, M.; Selvarajan, S.; Bay, B.H. P53 and c-kit (cd117) protein expression as prognostic indicators in breast phyllodes tumors: A tissue microarray study. Mod. Pathol. 2005, 18, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Gulubova, M.; Vlaykova, T. Prognostic significance of mast cell number and microvascular density for the survival of patients with primary colorectal cancer. J. Gastroenterol. Hepatol. 2009, 24, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Yodavudh, S.; Tangjitgamol, S.; Puangsa-art, S. Prognostic significance of microvessel density and mast cell density for the survival of thai patients with primary colorectal cancer. J. Med. Assoc. Thail. 2008, 91, 723–732. [Google Scholar]
- Acikalin, M.F.; Oner, U.; Topcu, I.; Yasar, B.; Kiper, H.; Colak, E. Tumour angiogenesis and mast cell density in the prognostic assessment of colorectal carcinomas. Dig. Liver Dis. 2005, 37, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Fishman, A.; Lorberboum-Galski, H. Targeted elimination of cells expressing the high-affinity receptor for ige (fc epsilon ri) by a pseudomonas exotoxin-based chimeric protein. Eur. J. Immunol. 1997, 27, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Fishman, A.; Prus, D.; Belostotsky, R.; Lorberboum-Galski, H. Targeted fc2′-3-pe40 chimeric protein abolishes passive cutaneous anaphylaxis in mice. Clin. Exp. Immunol. 2000, 119, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.G.; Bloom, G.D. Structural features of histamine release in rat peritoneal mast cells. A study with toluidine blue. Int. Arch. Allergy Appl. Immunol. 1974, 46, 740–752. [Google Scholar] [PubMed]
- Nitta, N.; Aoki, Y.; Isogawa, Y.; Tsuchiya, T.; Kanegasaki, S. Image analysis of mast cell degranulation in a concentration gradient of stimuli formed in the channel between a glass plate and a silicon substrate. Eur. J. Cell Biol. 2009, 88, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Kovarikova, P.; Michalova, E.; Knopfova, L.; Bouchal, P. Methods for studying tumor cell migration and invasiveness. Klin. Onkol. 2014, 27 (Suppl. 1), S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.B.; MacDonald, A.J.; Thornton, E.M.; Newlands, G.F.; Galli, S.J.; Miller, H.R. Stem cell factor enhances immunoglobulin e-dependent mediator release from cultured rat bone marrow-derived mast cells: Activation of previously unresponsive cells demonstrated by a novel elispot assay. Immunology 1996, 87, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.H.; Taub, D.D.; Nagel, J.; Smith, R.; Hogaboam, C.M.; Berlin, A.; Lukacs, N.W. Stem cell factor induces eosinophil activation and degranulation: Mediator release and gene array analysis. Blood 2002, 100, 4291–4297. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Grimbaldeston, M.A.; Pearce, A.L.; Robertson, B.O.; Coventry, B.J.; Marshman, G.; Finlay-Jones, J.J.; Hart, P.H. Association between melanoma and dermal mast cell prevalence in sun-unexposed skin. Br. J. Dermatol. 2004, 150, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Anuradha, A.; Kiran Kumar Naik, B.; Vijay Srinivas, G.; Devi, R.S.; Puneet, H.K. Incidence of mast cells in oral squamous cell carcinoma: A short study. J. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rabenhorst, A.; Schlaak, M.; Heukamp, L.C.; Forster, A.; Theurich, S.; von Bergwelt-Baildon, M.; Buttner, R.; Kurschat, P.; Mauch, C.; Roers, A.; et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood 2012, 120, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Crivellato, E. The controversial role of mast cells in tumor growth. Int. Rev. Cell Mol. Biol. 2009, 275, 89–131. [Google Scholar] [PubMed]
- Marichal, T.; Tsai, M.; Galli, S.J. Mast cells: Potential positive and negative roles in tumor biology. Cancer Immunol. Res. 2013, 1, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi Narendra, B.; Eshvendar Reddy, K.; Shantikumar, S.; Ramakrishna, S. Immune system: A double-edged sword in cancer. Inflamm. Res. 2013, 62, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Ritter, G.; Nishikawa, H. Antibody-based therapy in colorectal cancer. Immunotherapy 2013, 5, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Keppler-Hafkemeyer, A.; Brinkmann, U.; Pastan, I. Role of caspases in immunotoxin-induced apoptosis of cancer cells. Biochemistry 1998, 37, 16934–16942. [Google Scholar] [CrossRef] [PubMed]
- Perentesis, J.P.; Waddick, K.G.; Bendel, A.E.; Shao, Y.; Warman, B.E.; Chandan-Langlie, M.; Uckun, F.M. Induction of apoptosis in multidrug-resistant and radiation-resistant acute myeloid leukemia cells by a recombinant fusion toxin directed against the human granulocyte macrophage colony-stimulating factor receptor. Clin. Cancer Res. 1997, 3, 347–355. [Google Scholar] [PubMed]
- Turcotte, S.; Rosenberg, S.A. Immunotherapy for metastatic solid cancers. Adv. Surg. 2011, 45, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Tartour, E.; Sandoval, F.; Bonnefoy, J.Y.; Fridman, W.H. Cancer immunotherapy: Recent breakthroughs and perspectives. Med. Sci. 2011, 27, 833–841. [Google Scholar]
- Dalton, D.K.; Noelle, R.J. The roles of mast cells in anticancer immunity. Cancer immunology, immunotherapy. Cancer Immunol. Immunother. 2012, 61, 1511–1520. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | Sequences (5′ → 3′) | Amplified Fragment Length |
|---|---|---|
| VEGF-a | GAGAGCAGAAGTCCCATGAAGTGA | 185 bp |
| GCACTCCAGGGCTTCATCGTTA | ||
| VEGF-b | CAGAAGAAAGTGGTGCCATGGAT | 177 bp |
| CACACATTCCAGGCCATCGTC | ||
| VEGF-c | CTCACAAGGCCCCAAACCAG | 145 bp |
| GTTAGCTGCCTGACACTGTGGTAAT | ||
| TGF-β | GGCTGAACCAAGGAGACGGAAT | 145 bp |
| CGGTTCATGTCATGGATGGTGC | ||
| GAPDH | GTGGAAGGGCTCATGACCACA | 188 bp |
| AAGGCCATGCCAGTGAGCTTC |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Li, L.; Shi, R.; Liu, X.; Zhang, J.; Zou, Z.; Hao, Z.; Tao, A. Mast Cell Targeted Chimeric Toxin Can Be Developed as an Adjunctive Therapy in Colon Cancer Treatment. Toxins 2016, 8, 71. https://doi.org/10.3390/toxins8030071
Wang S, Li L, Shi R, Liu X, Zhang J, Zou Z, Hao Z, Tao A. Mast Cell Targeted Chimeric Toxin Can Be Developed as an Adjunctive Therapy in Colon Cancer Treatment. Toxins. 2016; 8(3):71. https://doi.org/10.3390/toxins8030071
Chicago/Turabian StyleWang, Shan, Linmei Li, Renren Shi, Xueting Liu, Junyan Zhang, Zehong Zou, Zhuofang Hao, and Ailin Tao. 2016. "Mast Cell Targeted Chimeric Toxin Can Be Developed as an Adjunctive Therapy in Colon Cancer Treatment" Toxins 8, no. 3: 71. https://doi.org/10.3390/toxins8030071