The Activation Effect of Hainantoxin-I, a Peptide Toxin from the Chinese Spider, Ornithoctonus hainana, on Intermediate-Conductance Ca2+-Activated K+ Channels

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
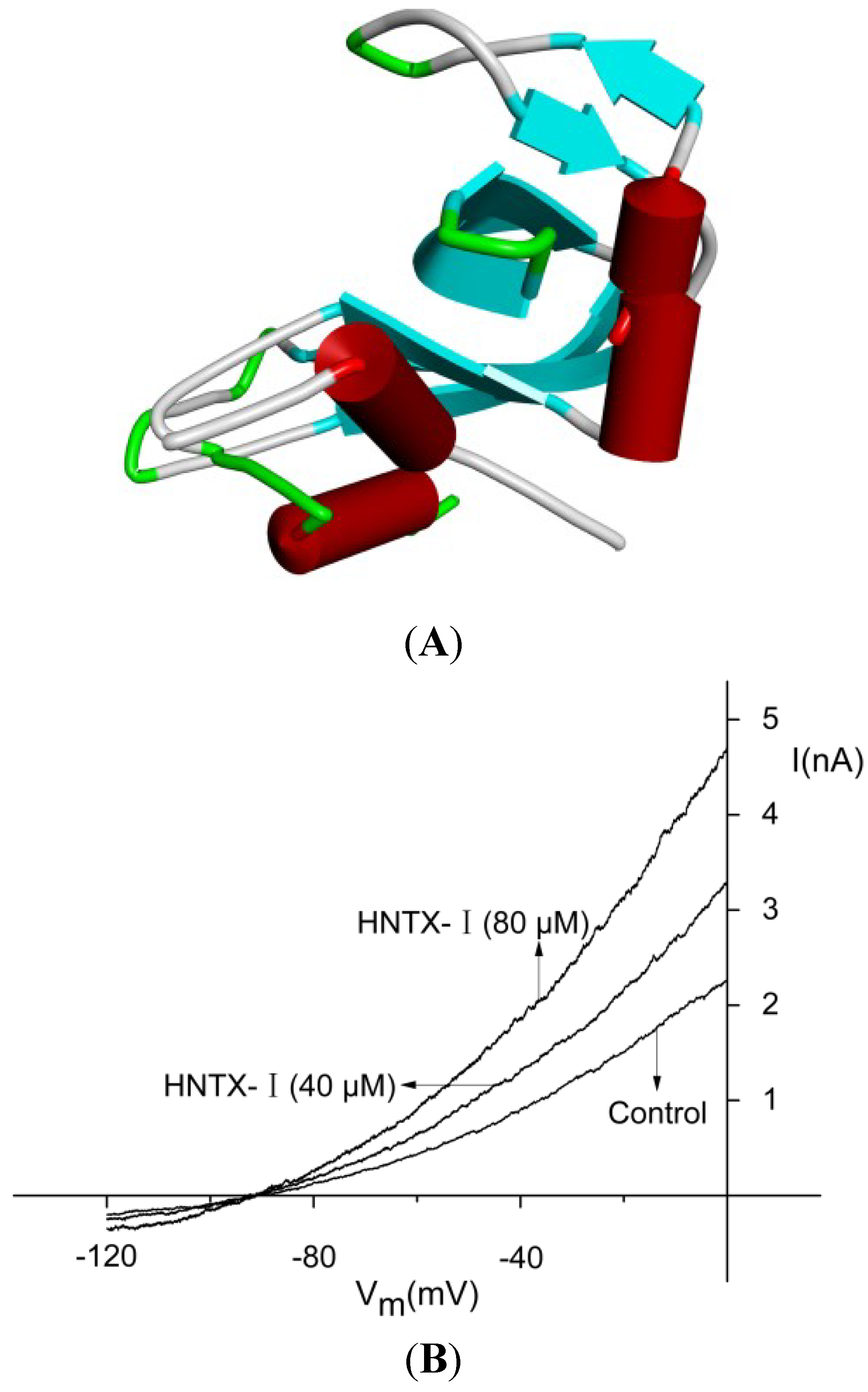
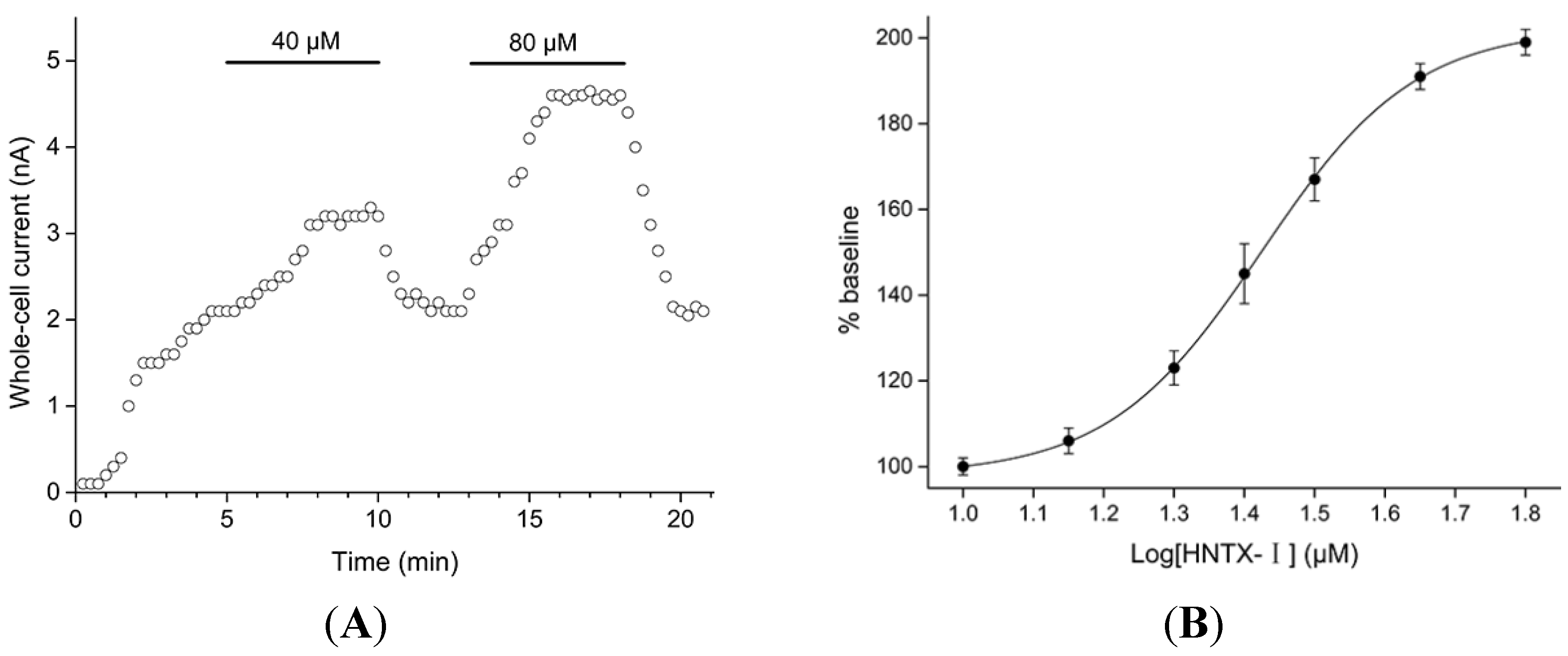
2.1. Defining the HNTX-I for hIK1 Activate
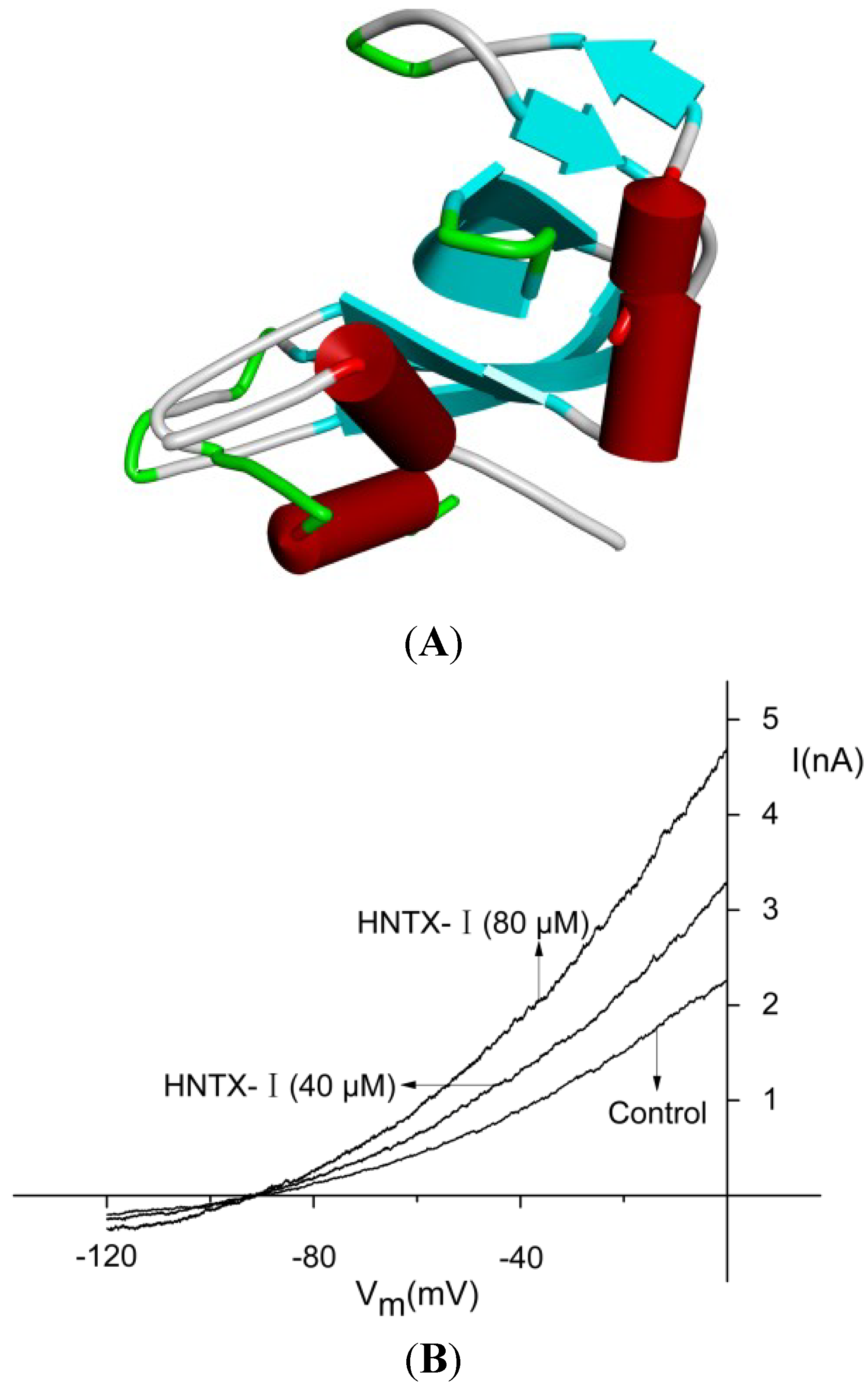
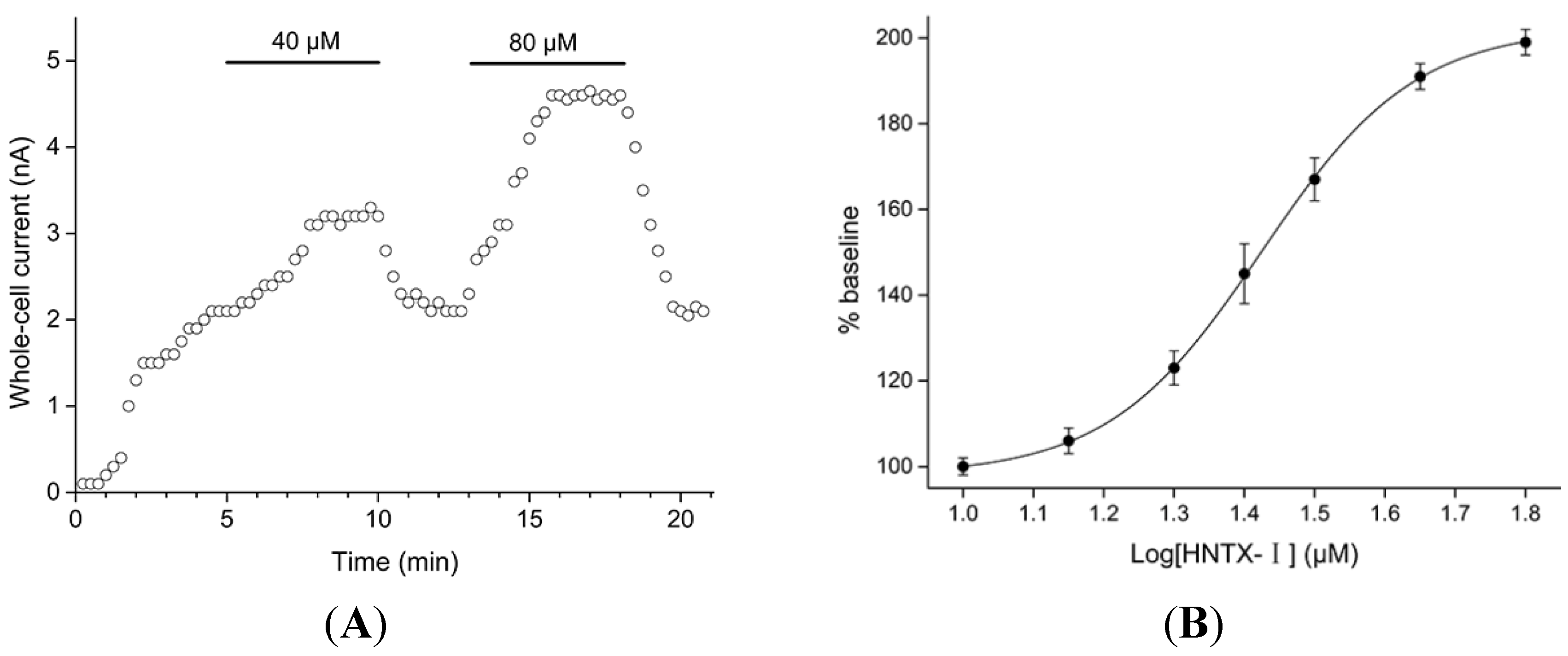

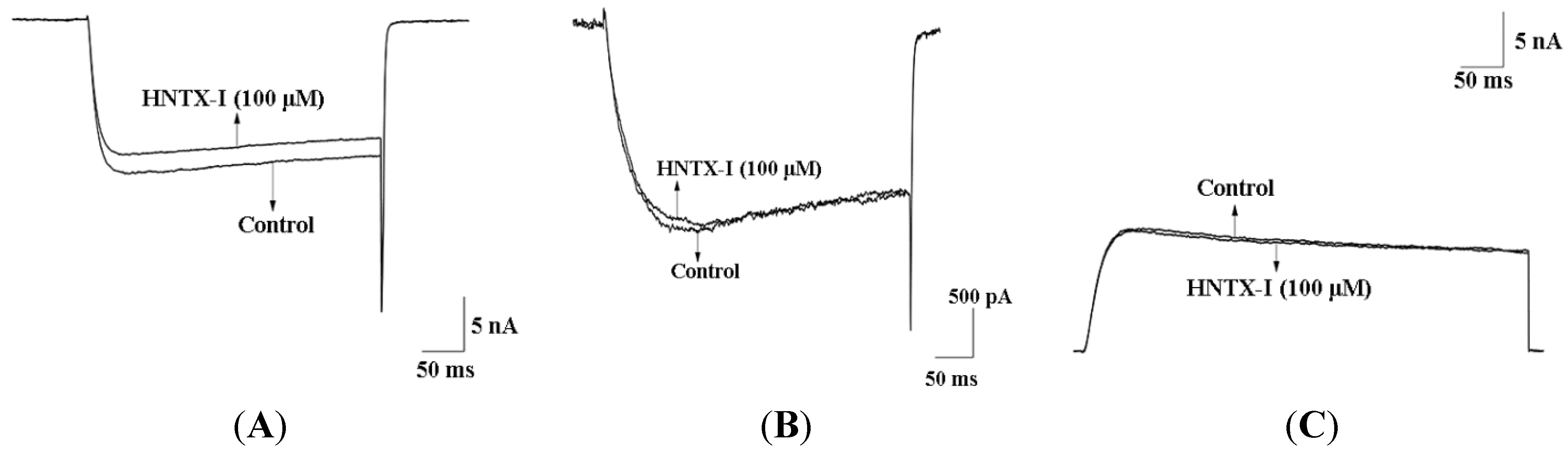
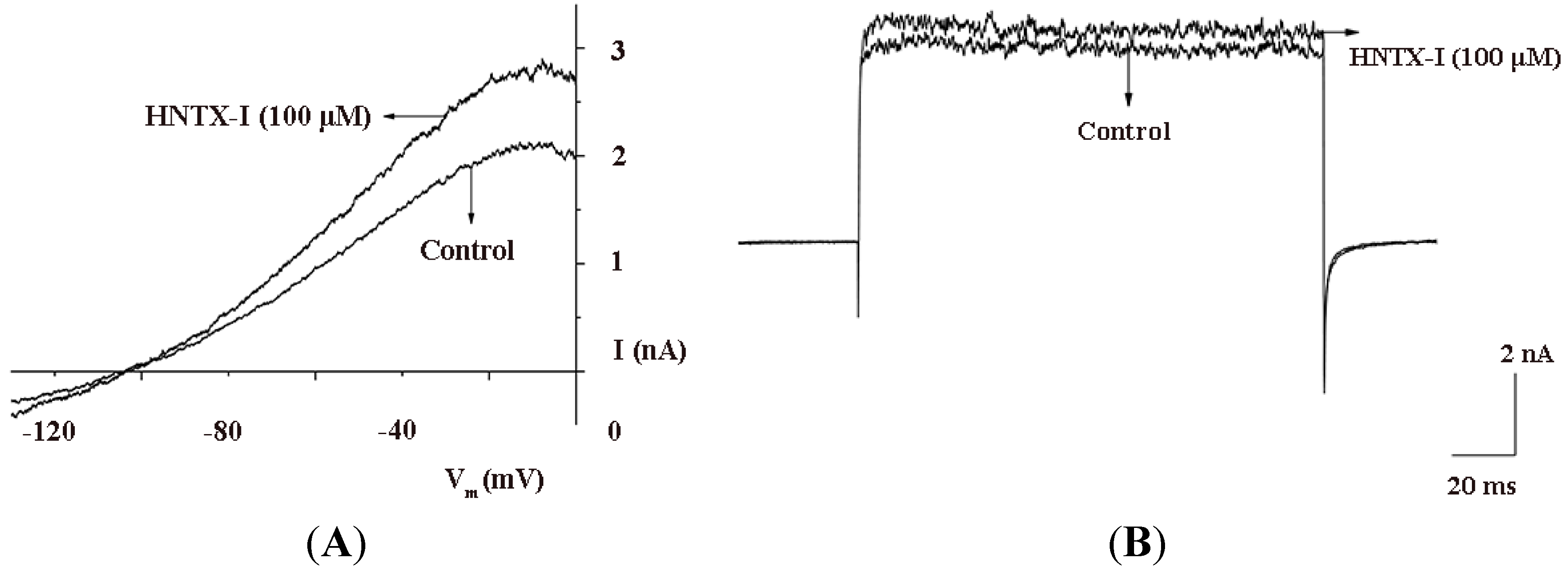
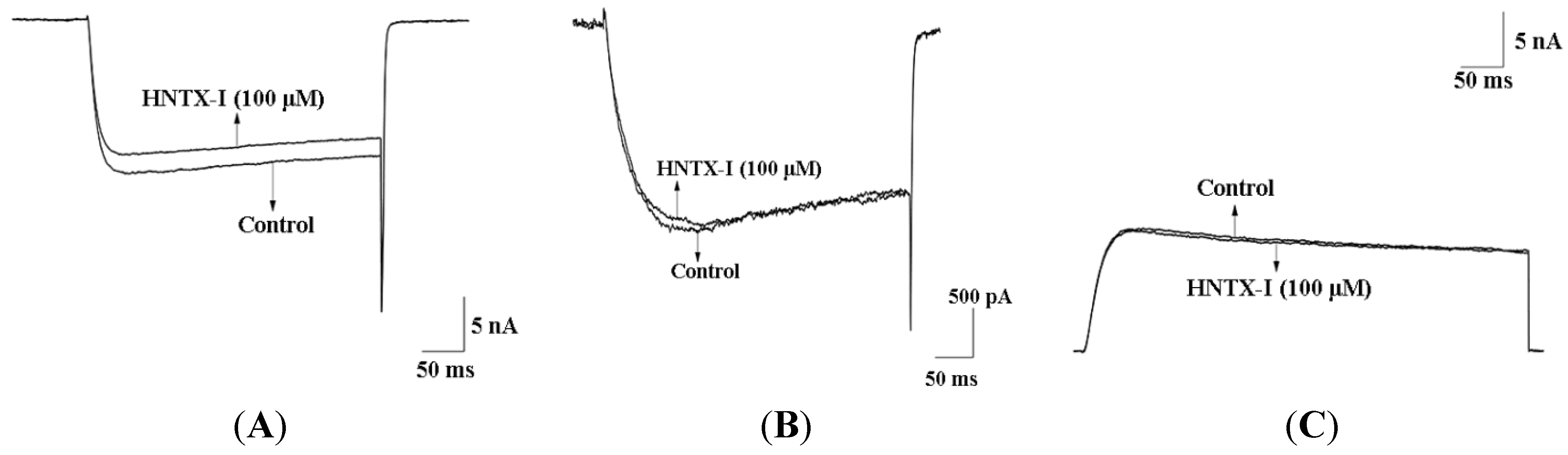
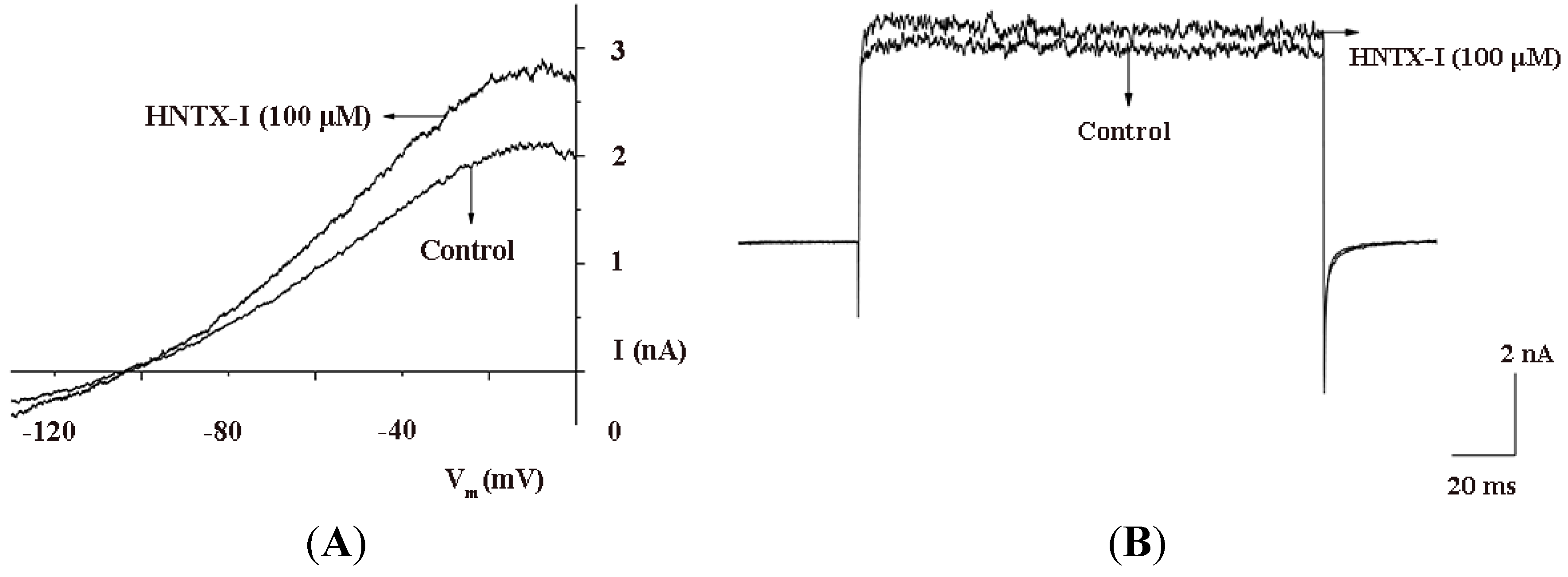
2.2. HNTX-I Is a Highly Selective Activator of hIK1 Current
2.3. HNTX-I Has No Obvious Block on Phrenic Nerve Conduction in Mice
2.4. HNTX-I Is Nontoxic in an in vivo Toxicity Test
3. Experimental Section
3.1. Toxins
3.2. Cells
3.3. Electrophysiology
3.4. Blocked Studies of Phrenic Nerve Conduction
3.5. Acute in vivo Toxicity Determinations
3.6. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wei, A.D.; Gutman, G.A.; Aldrich, R.; Chandy, K.G.; Grissmer, S.; Wulff, H. International union of pharmacology. Lii. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol. Rev. 2005, 57, 463–472. [Google Scholar]
- Ishii, T.M.; Silvia, C.; Hirschberg, B.; Bond, C.T.; Adelman, J.P.; Maylie, J. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA 1997, 94, 11651–11656. [Google Scholar] [PubMed]
- Strobaek, D.; Teuber, L.; Jorgensen, T.D.; Ahring, P.K.; Kjaer, K.; Hansen, R.S.; Olesen, S.P.; Christophersen, P.; Skaaning-Jensen, B. Activation of human IK and SK Ca2+-activated K+ channels by NS309 (6,7-dichloro-1h-indole-2,3-dione 3-oxime). Biochim. Biophys. Acta 2004, 1665, 1–5. [Google Scholar]
- Singh, S.; Syme, C.A.; Singh, A.K.; Devor, D.C.; Bridges, R.J. Benzimidazolone activators of chloride secretion: Potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther. 2001, 296, 600–611. [Google Scholar] [PubMed]
- Dalsgaard, T.; Kroigaard, C.; Simonsen, U. Calcium-activated potassium channels—A therapeutic target for modulating nitric oxide in cardiovascular disease? Expert Opin. Ther. Targets 2010, 14, 825–837. [Google Scholar]
- Wulff, H.; Zhorov, B.S. K+ channel modulators for the treatment of neurological disorders and autoimmune diseases. Chem. Rev. 2008, 108, 1744–1773. [Google Scholar] [CrossRef] [PubMed]
- Morimura, K.; Yamamura, H.; Ohya, S.; Imaizumi, Y. Voltage-dependent Ca2+-channel block by openers of intermediate and small conductance Ca2+-activated K+ channels in urinary bladder smooth muscle cells. J. Pharmacol. Sci. 2006, 100, 237–241. [Google Scholar] [PubMed]
- Grgic, I.; Kaistha, B.P.; Hoyer, J.; Kohler, R. Endothelial Ca+-activated K+ channels in normal and impaired edhf-dilator responses—Relevance to cardiovascular pathologies and drug discovery. Br. J. Pharmacol. 2009, 157, 509–526. [Google Scholar] [PubMed]
- Li, D.; Xiao, Y.; Hu, W.; Xie, J.; Bosmans, F.; Tytgat, J.; Liang, S. Function and solution structure of hainantoxin-I, a novel insect sodium channel inhibitor from the chinese bird spider Selenocosmia hainana. FEBS Lett. 2003, 555, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Cestele, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Aglieco, F.; Dib-Hajj, S.D. Critical molecular determinants of voltage-gated sodium channel sensitivity to mu-conotoxins GIIIA/B. Mol. Pharmacol. 2002, 61, 1192–1201. [Google Scholar] [PubMed]
- Hui, K.; Lipkind, G.; Fozzard, H.A.; French, R.J. Electrostatic and steric contributions to block of the skeletal muscle sodium channel by mu-conotoxin. J. Gen. Physiol. 2002, 119, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Li, R.A.; Sato, K.; Kodama, K.; Kohno, T.; Xue, T.; Tomaselli, G.F.; Marban, E. Charge conversion enables quantification of the proximity between a normally-neutral mu-conotoxin (GIIIA) site and the Na+ channel pore. FEBS Lett. 2002, 511, 159–164. [Google Scholar] [PubMed]
- Terlau, H.; Heinemann, S.H.; Stuhmer, W.; Pusch, M.; Conti, F.; Imoto, K.; Numa, S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991, 293, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Kohda, D.; Hatanaka, H.; Lancelin, J.M.; Ishida, Y.; Oya, M.; Nakamura, H.; Inagaki, F.; Sato, K. Structure-activity relationships of mu-conotoxin GIIIA: Structure determination of active and inactive sodium channel blocker peptides by nmr and simulated annealing calculations. Biochemistry 1992, 31, 12577–12584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pascal, J.M.; Schumann, M.; Armen, R.S.; Zhang, J.F. Identification of the functional binding pocket for compounds targeting small-conductance Ca2+-activated potassium channels. Nat. Commun. 2012, 3, 1021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pascal, J.M.; Zhang, J.F. Unstructured to structured transition of an intrinsically disordered protein peptide in coupling Ca2+-sensing and SK channel activation. Proc. Natl. Acad. Sci. USA 2013, 110, 4828–4833. [Google Scholar] [CrossRef] [PubMed]
- Hancox, J.C.; McPate, M.J.; El Harchi, A.; Zhang, Y.H. The herg potassium channel and herg screening for drug-induced torsades de pointes. Pharmacol. Ther. 2008, 119, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Raschi, E.; Vasina, V.; Poluzzi, E.; De Ponti, F. The herg K+ channel: Target and antitarget strategies in drug development. Pharmacol. Res. 2008, 57, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dai, J.; Chen, Z.; Hu, W.; Xiao, Y.; Liang, S. Isolation and characterization of hainantoxin-IV, a novel antagonist of tetrodotoxin-sensitive sodium channels from the chinese bird spider Selenocosmia hainana. Cell. Mol. Life Sci. 2003, 60, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Rash, L.; Zhu, S.; Diochot, S.; Lazdunski, M.; Escoubas, P.; Tytgat, J. Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol. Pharmacol. 2006, 69, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Struyk, A.F.; Scoggan, K.A.; Bulman, D.E.; Cannon, S.C. The human skeletal muscle Na channel mutation R669H associated with hypokalemic periodic paralysis enhances slow inactivation. J. Neurosci. 2000, 20, 8610–8617. [Google Scholar] [PubMed]
- Rowe, A.H.; Xiao, Y.; Rowe, M.P.; Cummins, T.R.; Zakon, H.H. Voltage-gated sodium channel in grasshopper mice defends against bark scorpion toxin. Science 2013, 342, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.P.; Zhang, D.Y.; Pan, X.; Chen, Q.; Zhou, P.A. Properties and amino acid sequence of huwentoxin-I, a neurotoxin purified from the venom of the chinese bird spider Selenocosmia huwena. Toxicon 1993, 31, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.A.; Xie, X.J.; Li, M.; Yang, D.M.; Xie, Z.P.; Zong, X.; Liang, S.P. Blockade of neuromuscular transmission by huwentoxin-i, purified from the venom of the chinese bird spider Selenocosmia huwena. Toxicon 1997, 35, 39–45. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, P.; Zhang, Y.; Chen, X.; Zhu, L.; Yin, D.; Zeng, X.; Liang, S. The Activation Effect of Hainantoxin-I, a Peptide Toxin from the Chinese Spider, Ornithoctonus hainana, on Intermediate-Conductance Ca2+-Activated K+ Channels. Toxins 2014, 6, 2568-2579. https://doi.org/10.3390/toxins6082568
Huang P, Zhang Y, Chen X, Zhu L, Yin D, Zeng X, Liang S. The Activation Effect of Hainantoxin-I, a Peptide Toxin from the Chinese Spider, Ornithoctonus hainana, on Intermediate-Conductance Ca2+-Activated K+ Channels. Toxins. 2014; 6(8):2568-2579. https://doi.org/10.3390/toxins6082568
Chicago/Turabian StyleHuang, Pengfei, Yiya Zhang, Xinyi Chen, Li Zhu, Dazhong Yin, Xiongzhi Zeng, and Songping Liang. 2014. "The Activation Effect of Hainantoxin-I, a Peptide Toxin from the Chinese Spider, Ornithoctonus hainana, on Intermediate-Conductance Ca2+-Activated K+ Channels" Toxins 6, no. 8: 2568-2579. https://doi.org/10.3390/toxins6082568