Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis

Laboratoire de Microbiologie et Génétique Moléculaire (LMGM), Centre National de la Recherche Scientifique (CNRS), Université Paul Sabatier, 118 route de Narbonne, Toulouse 31062, France
*
Author to whom correspondence should be addressed.
Toxins 2014, 6(3), 1002-1020; https://doi.org/10.3390/toxins6031002
Submission received: 19 December 2013
/
Revised: 20 February 2014
/
Accepted: 24 February 2014
/
Published: 6 March 2014
(This article belongs to the Special Issue Toxin-Antitoxin System)
Abstract
:The hallmark of Mycobacterium tuberculosis is its ability to persist for a long-term in host granulomas, in a non-replicating and drug-tolerant state, and later awaken to cause disease. To date, the cellular factors and the molecular mechanisms that mediate entry into the persistence phase are poorly understood. Remarkably, M. tuberculosis possesses a very high number of toxin-antitoxin (TA) systems in its chromosome, 79 in total, regrouping both well-known (68) and novel (11) families, with some of them being strongly induced in drug-tolerant persisters. In agreement with the capacity of stress-responsive TA systems to generate persisters in other bacteria, it has been proposed that activation of TA systems in M. tuberculosis could contribute to its pathogenesis. Herein, we review the current knowledge on the multiple TA families present in this bacterium, their mechanism, and their potential role in physiology and virulence.
1. General Overview of TA Systems in M. tuberculosis
Toxin-antitoxin (TA) systems are small genetic modules originally discovered as plasmid-borne loci that promote plasmid maintenance in bacterial populations by killing daughter cells devoid of the TA encoding plasmid [1]. TA loci were subsequently identified in many bacterial and archeal chromosomes, thus suggesting alternative functions [2]. TA systems are typically composed of a protein toxin and a more labile antagonistic antitoxin, which can be a protein or non-coding RNA [1]. Under certain circumstances, including environmental stress, plasmid loss, or bacteriophage infection, the less stable antitoxin is rapidly degraded and the free active toxin is now capable of targeting essential cellular processes such as DNA replication, cell wall synthesis, cell division or translation, thus leading to growth inhibition and eventually cell death. Growth inhibition by toxins is often reversible when new antitoxins are available and, accordingly, it has been proposed that activation of TA systems could facilitate bacterial survival until conditions become more favorable [3]. To date, five main types of TA families, with respect to the mode of inhibition of the poisonous activity of the protein toxin, have been described: (i) in type I, a small antisense RNA antitoxin specifically inhibits toxin synthesis by interacting with the toxin mRNA; (ii) in type II, the antitoxin is a protein that directly binds and inhibits the toxin; (iii) in type III the antitoxin is an RNA that directly interacts with the protein toxin; (iv) in type IV the antitoxin and the toxin are proteins that have the same target but do not directly interact with each other; and (v) in type V the antitoxin is an endoribonuclease, which specifically degrades the toxin mRNA [1,4,5].
Although the cellular roles of chromosomally-encoded TA systems are not well established, they have been involved in several processes, including stabilization of genomic regions, anti-addiction against similar plasmid-borne toxins, defense against phage infection, biofilm formation, control of the stress response, and, especially, bacterial persistence [1,6]. Persister cells are stochastic phenotypic variants of regular cells, which appear randomly and at low frequency in a population and are tolerant to antibiotic treatments due to slow or arrested growth [7]. Remarkably, it was recently shown that stochastic induction of several chromosomal type II TA systems significantly contributes to such persistence phenotype in E. coli, thus, indicating a major involvement of TAs in this process [8].
Tuberculosis is the deadliest disease due to a single bacterial pathogen, namely Mycobacterium tuberculosis. Besides the synergy with the human immunodeficiency virus (HIV) and the emergence of multi and extensively drug resistant strains, one of the limitations to tuberculosis eradication is its ability to persist in human lungs in a dormant state, which is tolerant to the host immune system and to antibiotic treatments. This phenomenon is thought to be responsible for latent tuberculosis, which affects approximately one third of the human population according to the World Health Organization (WHO), and can reactivate later in case of immune depression. The existence of a persistent subpopulation in the active form of the disease is also believed to be responsible, at least in part, for the exceptionally long duration of the anti-tubercular treatment [9]. M. tuberculosis possesses a remarkably high number of TA systems in its chromosome when compared to other mycobacteria (Figure 1 and Table 1), and it has been proposed that persistence induced by active toxins could contribute to its pathogenesis [10,11]. Interestingly, a transcriptomic analysis of antibiotic-induced persisters of M. tuberculosis revealed that in addition to a general shutdown of metabolic pathways, at least 10 TA systems were significantly up-regulated under these conditions [12], thus, further supporting a potential contribution of TAs in M. tuberculosis persistence.
We have identified a total of 79 TA systems (confirmed or putative) in M. tuberculosis H37Rv: 67 belonging to six well described type II TA families: VapBC (50 systems), MazEF (10 systems), YefM/YoeB (one system), RelBE (two systems), HigBA (two systems), and ParDE (two systems); one tripartite type II TAC (Toxin-Antitoxin-Chaperone) system controlled by a SecB-like chaperone; three potentially type IV systems; and eight uncharacterized putative TA systems. Note that no other type of TA family was identified in M. tuberculosis [13,14]. Most of these systems (63) have been experimentally tested, essentially in E. coli and M. smegmatis, for growth inhibition by the putative toxin and its neutralization by the putative antitoxin. Among these, 37 were shown to be functional in at least one of the conditions tested (Figure 1) [15,16,17,18,19,20]. This review presents the current knowledge on TA systems in M. tuberculosis.
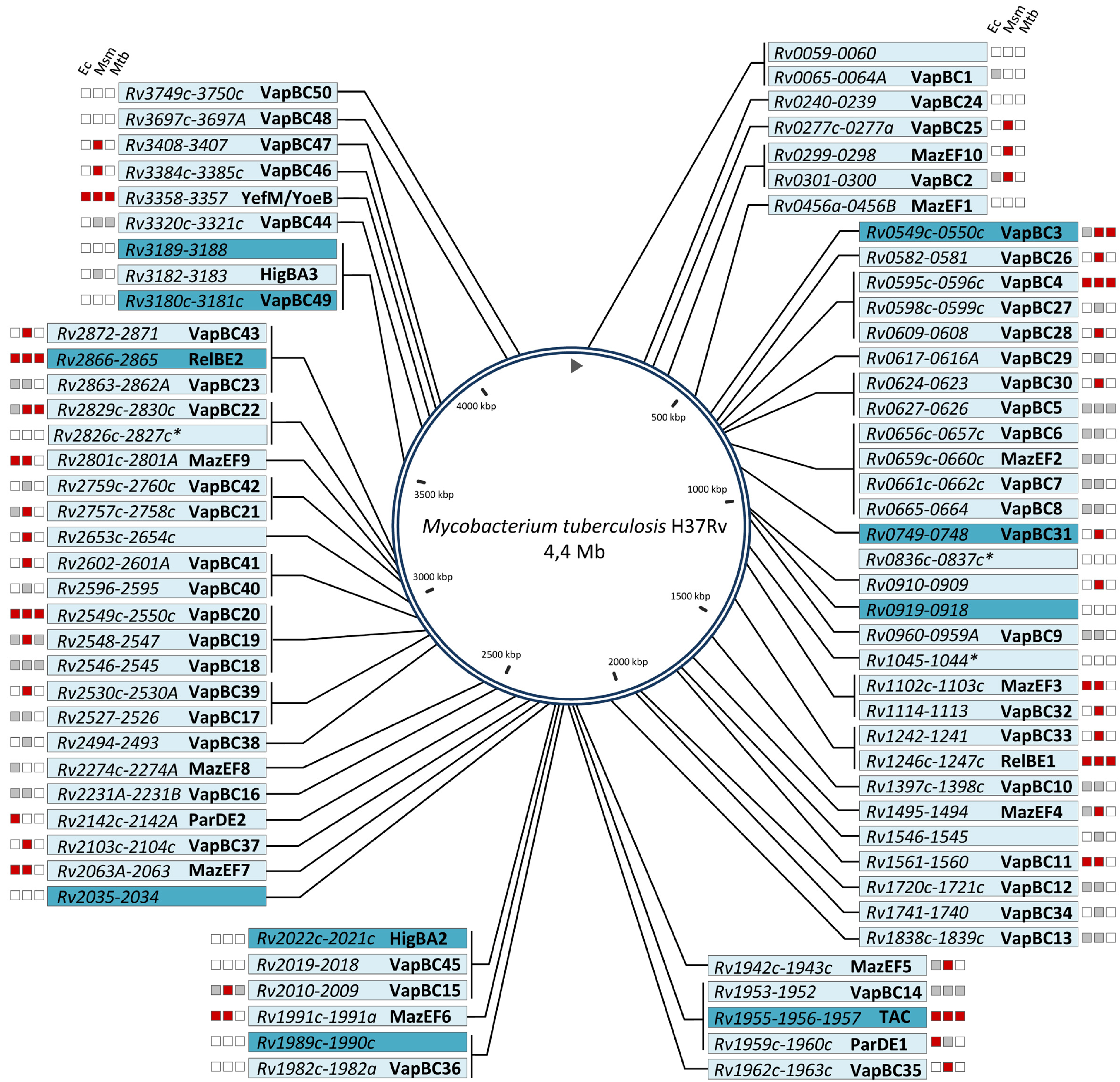
Figure 1.
Chromosomal map of M. tuberculosis H37Rv TA systems. TA systems are annotated according to the tuberculist database except for VapBC45 (Rv2018-Rv2019), VapBC49 (Rv3181c-Rv3180c), VapBC50 (Rv3750c-Rv3749c), HigBA2 (Rv2022c-Rv2021c), HigBA3 (Rv3182-Rv3183), YefM/YoeB (Rv3357-Rv3358), and MazEF10 (Rv0298-Rv0299). Most of the TA systems depicted here likely belong to type II, expect for those marked with an asterisk, which are putative type IV systems. For each system, the functionality in E. coli (Ec), M. smegmatis (Msm), and M. tuberculosis (Mtb), is depicted: red color stands for “inhibition of growth”, grey for “no inhibition of growth”, and white for “not tested”. The 10 most induced TA systems in drug-tolerant persister cells are highlighted on dark blue background.
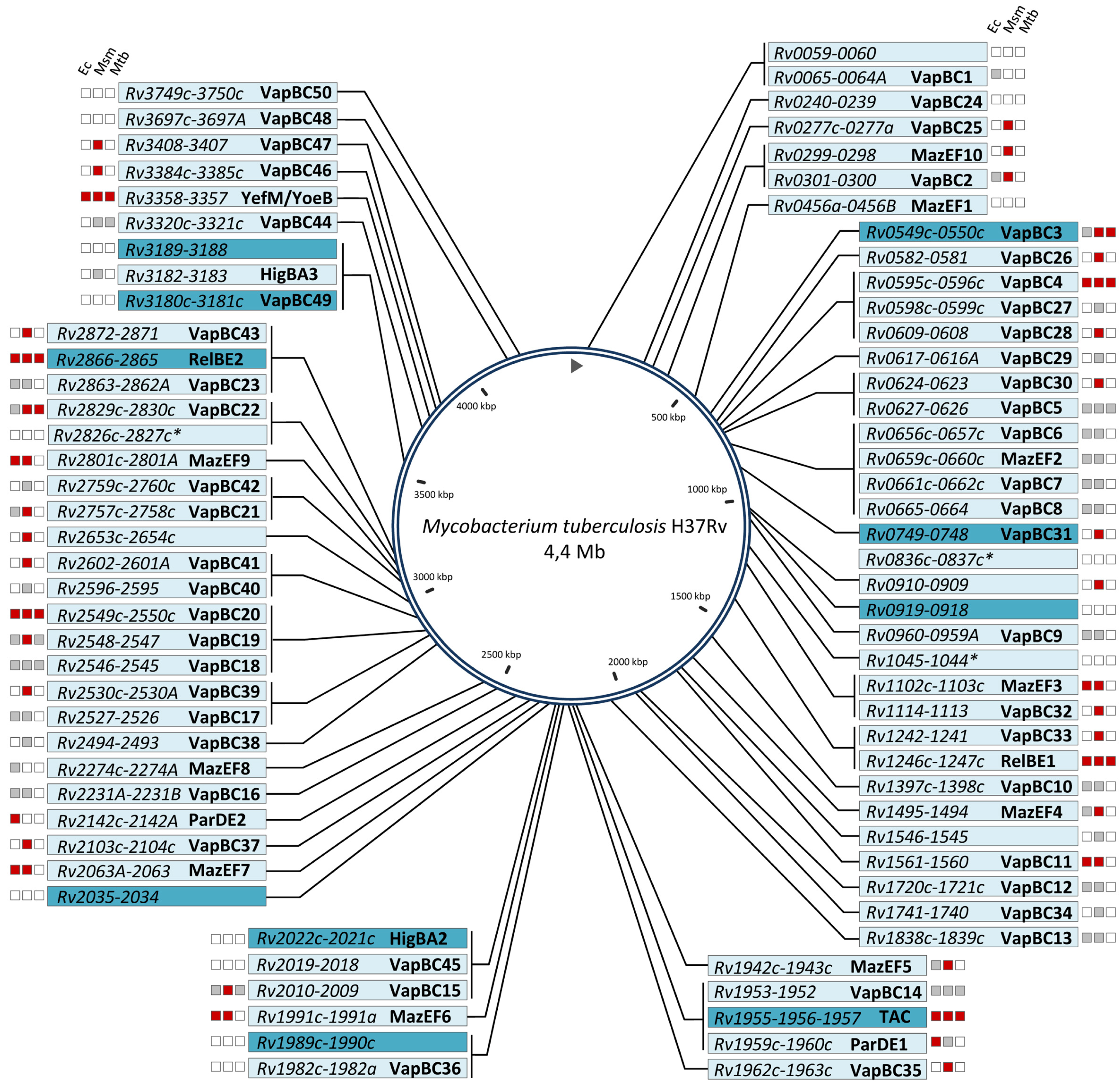
Figure 1.
Chromosomal map of M. tuberculosis H37Rv TA systems. TA systems are annotated according to the tuberculist database except for VapBC45 (Rv2018-Rv2019), VapBC49 (Rv3181c-Rv3180c), VapBC50 (Rv3750c-Rv3749c), HigBA2 (Rv2022c-Rv2021c), HigBA3 (Rv3182-Rv3183), YefM/YoeB (Rv3357-Rv3358), and MazEF10 (Rv0298-Rv0299). Most of the TA systems depicted here likely belong to type II, expect for those marked with an asterisk, which are putative type IV systems. For each system, the functionality in E. coli (Ec), M. smegmatis (Msm), and M. tuberculosis (Mtb), is depicted: red color stands for “inhibition of growth”, grey for “no inhibition of growth”, and white for “not tested”. The 10 most induced TA systems in drug-tolerant persister cells are highlighted on dark blue background.

{kind=link}
{kind=link}
| Genomes | VapBC | MazEF | RelBE | ParDE | YefM/YoeB | HigBA | TAC | Otherb | Total |
|---|---|---|---|---|---|---|---|---|---|
| M. tuberculosis H37Rv | 50 | 10 | 2 | 2 | 1 | 2 | 1 | 11 | 79 |
| M. smegmatis MC2155 | 1 | 1 | 1c | 2 | 4 | ||||
| M. marinum M | 1 | 1 | |||||||
| M. avium 104 | 1 | 2 | 3 | ||||||
| M. avium paratuberculosis K10 | 1 | 1 | 1 | 3 | |||||
| M. abscessus ATCC 19977 | 1 | 7 | 8 | ||||||
| M. ulcerans Agy99 | 1 | 1 | 2 | ||||||
| M. gilvum PYR-GCK | 3 | 1 | 1 | 9 | 14 |
Notes: a Data were collected on TADB (Toxin-Antitoxin Database) or searching for TA homologs using BLASTP on the NCBI server; b Unclassified TA systems: only one couple of conserved domains found in M. abscessus and M. gilvum, namely COG2856-Xre, was not present in M. tuberculosis; DUF1814-COG5340 was found in M. marinum, M. avium and M. gilvum; COG3832-ArsR was found in all the investigated mycobacterial genomes except from M. marinum; one GNAT-RHH and one COG5654-Xre were found in M. gilvum. See Part “Other TA modules” for details; c Indicates that in M. smegmatis the putative TAC system is incomplete due to the absence of toxin gene.
2. The VapBC Family
The VapBC (Virulence associated protein) family of TAs was first discovered as an addiction system of a virulence plasmid of Salmonella dublin [21]. They are the most abundant TA loci in M. tuberculosis (Figure 1), defined by a toxin VapC with a PIN domain (homologous to PilT N-terminal domain) found on proteins from all life kingdoms and generally ribonucleases [22]. Most of the VapC toxins tested so far, including VapC1, VapC2, VapC5, VapC11, VapC20, and VapC29 from M. tuberculosis, exhibit a ribonuclease activity in vitro [16,20,23,24]. However, toxicity of the M. tuberculosis VapC4 seems to be induced by stable RNA binding and not by degradation [25]. Remarkably, Winther and Gerdes [24] showed that two VapC from the entero-pathogenic bacteria Shigella flexneri and Salmonella enterica are specific tRNases that cleave the initiator tRNA. Thus, VapC toxins appear to target RNA by distinct mechanisms. Sequence specificities of both VapC1 and VapC19 from M. tuberculosis were recently investigated by mass spectrometry of mRNA fragments [26]. Both toxins appeared to be redundant, cleaving preferentially GC rich 4mers, with other sequences being cleaved less efficiently. Considering the fact that M. tuberculosis has a GC rich genome, with a GC content of 66%, these findings suggest that VapC cleavage sites are frequent in mRNA, and, thus activation of these toxins is likely to rapidly trigger a general translation inhibition. A recent analysis of VapBC20 revealed that the VapC20 toxin specifically inhibits translation by cleavage of the conserved Sarcin-Ricin loop of the 23S rRNA. In this case, the structure of the loop was essential for recognition and cleavage by VapC20 [27].
Structural analysis of the VapBC5 complex from M. tuberculosis revealed an asymmetric complex with a 1:1 stoichiometry [23]. However, the recently obtained crystal structure of the VapBC3 complex showed a different picture, with a toxin-antitoxin hetero-octamer formed by assembly of two heterotetramers containing two toxins and two antitoxins, similar to VapBC structures from other species, namely Nisseria gonorrhoeae FitAB, Shigella flexneri VapBC, and Rickettsia felis VapBC2 [28]. In spite of such differences, the two mycobacterial VapC toxins present a similar putative catalytic site formed by the conserved acidic residues of their PIN domains that coordinate the Mg2+ ion. In addition, both structures suggest a conserved mechanism of inhibition, in which the VapB antitoxins prevent efficient binding of Mg2+ at the active sites of their cognate VapC [23,28]. The M. tuberculosis VapB antitoxins are generally related to families of transcriptional regulators or DNA binding domains already reported to be associated with VapC toxins, i.e., 33 RHH, 8 Phd, 2 ArbR, and 1 MerR [29]. However, five other VapB do not present any conserved domain.
Several VapBC of M. tuberculosis were found to be induced in response to relevant stress conditions encountered during the infection process [16]. This includes hypoxia (VapBC15, VapBC7, and VapBC25) and in IFN-γ-stimulated murine bone marrow-derived macrophages (VapBC11, VapBC3, and VapBC47). In addition, VapBC3, VapBC31, and VapBC49 were shown to be among the TA systems the most up-regulated in drug-tolerant bacteria, suggesting a possible role in persistence [12]. Interestingly, a recent study in the closely related bacterium M. smegmatis showed that its sole VapC toxin is an RNase that couples the rate of glycerol utilization to bacterial growth via post-transcriptional regulation of genes of sugar transport pathways [30]. In M. tuberculosis, it is conceivable that the multiple VapC could act as regulators of translation in response to a plethora of environmental stresses. Intriguingly, the extra cellular concentration of eleven VapC toxins from M. tuberculosis, namely VapC4, VapC5, VapC13, VapC19, VapC22, VapC27, VapC37, VapC39, VapC38, VapC41, and VapC44, was increased in a nutrient starvation model, further suggesting a possible role for these toxins during the establishment of latent infection [31].
3. The MazEF Family
Overexpression of the E. coli sequence-specific endoribonuclease MazF induces a growth arrest via cleavage of almost all cellular mRNAs in a ribosome-independent manner [32]. The E. coli MazEF complex in which the toxin MazF is inactive forms a linear heterohexamer composed of a dimer of the MazE antitoxin symmetrically bound to two MazF dimers [33]. Similar organization and stoichiometry is observed for the MazEF complex of Bacillus subtilis, although the MazE antitoxins are poorly related and present different fold [34]. Several MazF cleavage sites in mRNA have been reported, for example MazF from E. coli cleaves at ACA sequences [35] and the one from B. subtilis at UACAU [36]. Out of the 10 MazF toxin members present in M. tuberculosis, seven were shown to affect E. coli and/or M. smegmatis growth, two exhibited non-noticeable phenotype, and one has not been tested (Figure 1) [15,16,37]. Analysis of cleavage sites in mRNA, which has been performed for several MazF toxins of M. tuberculosis revealed significant differences. Indeed, while MazF9 cleaves at UAC sequences, MazF3 cleaves U rich regions [37], MazF6 at UUCCU sequences [37,38], and MazF4 at UCGCU [39]. Such differences in substrate recognition suggest that MazF toxins may trigger a large panel of responses, going from a global translation inhibition to a fine-tuned regulation of specific mRNAs. For example, in the case of MazF6, 31% of the total mRNA do not present the recognition site and are, thus, predicted to be resistant to toxin cleavage.
In addition to mRNA recognition, certain MazF toxins were shown to target ribosomal RNA. It is indeed the case for the E. coli MazF toxin, which generates specialized ribosomes by cleavage of the 3’ region of the 16S rRNA containing the anti-Shine-Dalgarno sequence, thus, facilitating leaderless mRNAs translation [40]. This mechanism would lead to an alternative translation pathway, which might promote stress adaptation. The MazF6 toxin from M. tuberculosis also acts on ribosomal RNA but by a very distinct mechanism. In this case, the toxin cleaves the 23S rRNA of dissociated ribosomes and subsequently provokes a global inhibition of protein synthesis [38]. Aside from RNA targets, it has been shown that the mycobacterial MazF4 toxin interacts with DNA topoisomerase I and that they mutually inhibit each other [17]. As the DNA topoisomerase I gene is essential in M. tuberculosis [41], this suggests that MazF4 could act by two alternative mechanisms to inhibit bacterial growth.
Expression of mazEF from E. coli is induced under various stress conditions, including DNA damage, heat shock and oxidative stress in a RelA dependent manner [42]. Similarly, it has been shown that M. tuberculosis MazF2 was down-regulated after four hours of nutrient starvation and MazF3 up-regulated during amino acid starvation in a relA deleted strain [43,44], thus, suggesting that MazEF modules from M. tuberculosis could also be regulated by the stringent response. As observed for RelE toxins (see next part), overexpression of either MazF1 (Rv2801c), MazF5 (Rv1942c), or MazF6 (Rv1102c) from M. tuberculosis induced drug-specific effects on the formation of persister cells (data not shown from [18]). Correspondingly, individual deletion of each of these three MazF encoding genes from M. tuberculosis differentially diminished the formation of drug-tolerant persisters [18]. Although not yet studied in detail, these data point towards an implication of MazEF modules in the formation of M. tuberculosis persisters in response to antibiotic treatment.
Partner specificity among TA pairs in M. tuberculosis has been studied as well. The presence of 79 possible TAs in this bacterium represents a unique system to address possible cross-talks, overlaps, or cooperation between TA systems. Remarkably, the M. tuberculosis MazF9 toxin was shown to be neutralized by the non-cognate antitoxins MazE6, VapB27, and VapB40 [19]. Such overlap is in agreement with the very low conservation observed between the different MazE antitoxins in this bacterium (4% to 22% of identity) and with the fact that VapB27 and VapB40 are closely related to the E. coli MazE. Accordingly, the non-cognate antitoxins MazE1 to 7 and MazE9 could neither interact with MazF6 nor alleviate its toxicity [45]. In contrast, M. tuberculosis MazF toxins are well conserved with up to 46% sequence identity. Together, these findings suggest that intricate networks of TA systems may exist in this pathogen.
4. The RelBE and YefM/YoeB Families
Mutations in the relB gene in E. coli were originally identified as conferring a delayed relaxed phenotype of the stringent response to amino acid starvation [46]. Later, it was shown that this phenotype was due to destabilization of the RelB antitoxin, leading to hyperactivation of the toxin RelE [47]. The YefM/YoeB family, named after its E. coli member, was first described as homologous of the Axe-Txe addiction system of the Enterococcus faecium plasmid pRUM [48]. The YefM antitoxins are homologous to Phd, the antitoxin of the Phd-Doc module of phage P1, whereas the YoeB toxins belong to the RelE superfamily [1]. Both RelE and YoeB toxins from E. coli are ribosome-dependent ribonucleases that cleave mRNA at the ribosomal A site [32]. Nevertheless, despite sequence and structural homology, E. coli RelE and YoeB seem to act by distinct mechanisms: RelE binds to the 30S subunit of 70S ribosomes and inhibits translation elongation, whereas YoeB binds to the 50S subunit and inhibits translation initiation [32]. RelE from E. coli was also shown to cleave tmRNA, thus, suggesting delicate interplay between such toxins and quality control mechanisms in response to amino acid starvation [49].
M. tuberculosis encodes for two RelBE systems, namely RelBE1 (Rv1247c-Rv1246c) and RelBE2 (Rv2865-Rv2866), and one YefM/YoeB (Rv3357-Rv3358). Note that the YefM/YoeB module discussed here was previously annotated as RelEB3, due to the fact that the toxin belongs to the RelE superfamily of ribonucleases. Yet, the YefM/YoeB (RelBE3) proteins from M. tuberculosis are very close to those of the E. coli YefM/YoeB system (58% and 68% sequence similarity respectively), with the mycobacterial toxin containing the conserved C-terminal histidine and tyrosine residues of YoeB involved in RNAse activity and not present in RelE [50,51]. Therefore, in contrast with RelBE1 and 2, the RelBE3 system clearly belongs to the YefM/YoeB family.
Previous gel filtration analyses of the M. tuberculosis YefM/YoeB complex suggested that it may form a heterotrimeric complex, as it is the case for the E. coli homologs [52]. However, structural analysis of both RelBE2 and YefM/YoeB revealed very similar heterotetrameric complexes composed of two heterodimers of one toxin bound to one antitoxin, with tetramerization occurring mainly via interaction between the two antitoxins [53]. Interestingly, while both RelE2 and YoeB toxins showed characteristic folds of RelE-like toxins [53], the structure of their respective antitoxins RelB2 and YefM were related the YefM/Phd antitoxins and not to either the E. coli or the archeal RelB [53]. As for YoeB, inactivation of E. coli RelE upon binding to RelB seems to occur via a conformational shift in the catalytic site of the toxin [50,54].
The two RelBE and the YefM/YoeB systems of M. tuberculosis were shown to be functional TA systems in E. coli, M. smegmatis, and in M. tuberculosis [18,55]. In agreement with their different mechanisms of action, the toxin YoeB was less toxic than RelE1 and RelE2 when expressed in M. smegmatis [53]. Similar results were obtained in E. coli, where YoeB expression only weakly affected colony-forming [47]. Intriguingly, Yang and colleagues [56] proposed that there might be complex interplays between these three systems with different patterns of cross interaction and promoter regulation. For example, they showed that YefM could interact only with its cognate toxin YoeB, whereas RelB1 and RelB2 were capable of interacting with any of the RelE1, RelE2, or YoeB toxins. Moreover, YefM is able to bind its promoter on its own, whereas RelB1 and RelB2 need to be part of the TA complex, but not necessarily with their cognate toxin [56]. Such multiple ways to inactivate toxins or to regulate expression of TA genes further emphasizes the complexity of toxin-antitoxin networks in this bacterium.
Transcriptional analyses revealed that RelBE2 is among the 10 most induced TA systems in M. tuberculosis drug-tolerant persisters [12]. Furthermore, while the yefM/yoeB, relBE1, and relBE2 genes were all expressed during bacterial growth in Luria-Bertani medium, only relE1, relB2, and yoeB transcripts could be detected in human macrophages at late stages of infection [55]. All three relE1, relE2, and yoeB toxin-encoding genes were also up-regulated in response to antibiotic treatment, as well as in lung tissues of infected mice [18], thus, suggesting that such toxins could contribute to persistence. Accordingly, overexpression of each of the three toxins also led to an increased level of drug-tolerant persisters and the deletion of either relE2 or yoeB, but not relE1, reduced the formation of persisters by four- to nine-fold [18]. However, none of the toxin mutants was affected for survival in mice model, indicating that the toxins may not individually contribute to infection in vivo [18].
5. The HigBA Family
The first identified higBA locus (host inhibition of growth) originates from the Rts1 plasmid of Proteus vulgaris where it functions as an addiction module [57]. TA members of this family encode- for a RelE-like toxin directly followed by an antitoxin that contains a HTH Xre-domain [29,58]. The HigB toxin from Rts1 plasmid was shown to act as a ribosome-dependent ribonuclease with a mechanism distinct from RelE and YoeB, in which the toxin binds the ribosome 50S subunit and cleaves preferentially at AAA sequences on the processing mRNAs [59]. HigB1 and HigB2 from V. cholerae were also studied and showed different, less specific cleavage patterns [60]. The recently solved crystal structure of HigBA from P. vulgaris revealed a heterotetrameric HigB-(HigA)2-HigB complex in which two HigA-HigB heterodimers are assembled via additional HigA-HigA interactions [61]. The structure of HigA in such complex was very similar to that of previously identified HigA-like antitoxin structures obtained in the absence of toxin (i.e.,: HigA from Coxiella burnetii, PDB entry 3trb and YddM from E. coli CFT073, PDB entries 2ICT and 2ICP; [62]), thus, indicating that HigA may not undergo major conformational changes upon toxin binding. Remarkably, the structure also shows that inactivation of HigB may not occur by direct occlusion of its active site by HigA, thus suggesting a novel mechanism of inhibition in which the large HigB-(HigA)2-HigB complex might sterically inhibit interaction of the toxin with ribosome-bound mRNA, as proposed by the authors [61].
M. tuberculosis H37Rv possesses three TA systems homologous to HigBA, i.e., two classical two-component TA pairs HigBA2 (Rv2022c-Rv2021c) and HigBA3 (Rv3182-Rv3183) and one atypical tripartite system, named TAC (Toxin-Antitoxin-Chaperone), composed of a HigBA1 pair (Rv1955-Rv1956) coupled to a molecular chaperone Rv1957 (Figure 1). To date, nothing is known about the involvement of HigBAs in M. tuberculosis pathogenesis. However, it has been shown that both higBA1 and higBA2 locus, which are located on the same genomic island comprising other genes potentially involved in dormancy, are among the 10 most up-regulated TA systems in M. tuberculosis drug-tolerant persisters [12,63]. While the effect of HigB2 toxin on bacterial growth was not investigated yet, conditional expression of HigB3 did not significantly affect M. smegmatis growth (Figure 1) [16]. In contrast, HigB1 exhibits a robust toxicity both in E. coli and in mycobacteria, and has been more extensively studied as part of the TAC system.
6. The Tripartite TAC System
The TAC (toxin-antitoxin-chaperone) system from M. tuberculosis is encoded by three genes organized in operon, Rv1955-Rv1956-Rv1957, respectively encoding the HigBA1 TA pair (Rv1955-Rv1956) and a chaperone Rv1957 related to the generic export chaperone SecB [64]. The operon also contains the upstream less conserved Rv1954a gene of unknown function. It has been shown that HigBA1 of TAC is specifically controlled by Rv1957 through a direct interaction between the chaperone and the antitoxin, allowing antitoxin folding and protection from degradation (Figure 2) [64]. Accordingly, expression of the HigA1 antitoxin does not counteract the severe toxicity induced by HigB1 in the absence of the chaperone, both in E. coli and mycobacteria [64]. In addition, single deletion of higA1 in M. tuberculosis is lethal [65], while disruption of Rv1957 alone exhibits a slow growth phenotype most likely due to a reduced antitoxin activity [41]. Therefore, it is very likely that the HigB1 activation cascade is triggered either by a decrease in expression or by direct hijacking of the chaperone at the post-translational level (Figure 2).
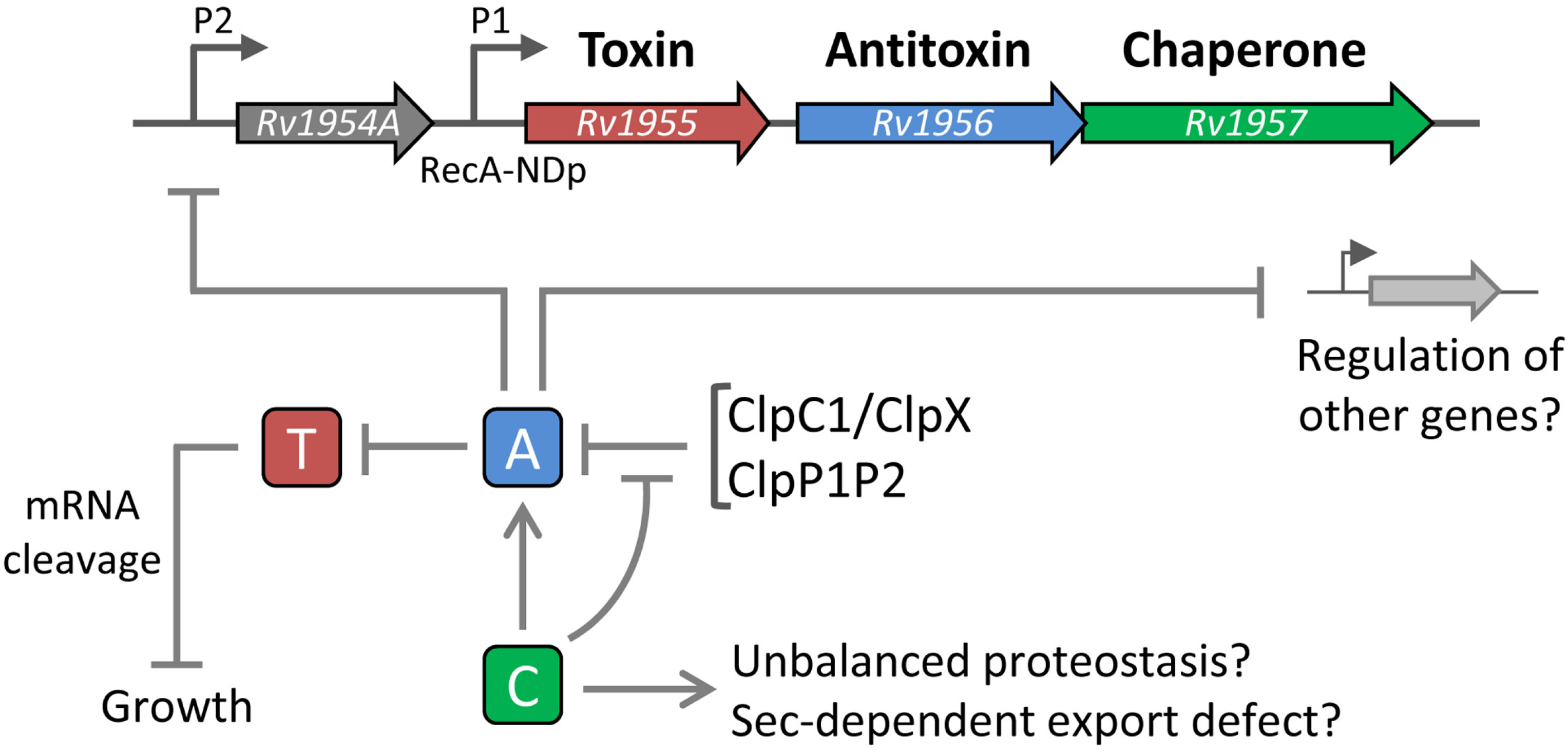
Figure 2.
Proposed mechanism for TAC. In M. tuberculosis, the TAC genes Rv1955-Rv1956-Rv1957, respectively, encode the toxin HigB1 (T), the antitoxin HigA1 (A) and the chaperone Rv1957 (C). They are located within an operon also containing the less conserved upstream Rv1954A gene of unknown function. Expression of TAC is regulated by two promoters: the promoter P1, which contains a RecA-NDp motif characteristic of LexA/RecA-independent genes in M. tuberculosis and the promoter P2, recognized by the HigA1 antitoxin. Repression of the TAC operon by HigA1 could occur in complex with its HigB1 and/or Rv1957 partners and HigA1 could regulate other genes as well. The SecB-like chaperone Rv1957 facilitates the folding of HigA1 and prevents its degradation by proteases (potentially ClpC1 or ClpX together with the ClpP1ClpP2 proteolytic subunit). Although interaction with the chaperone renders HigA1 competent to neutralize the toxin, it is not known whether Rv1957 is part of the final inactive complex. Activation of the HigB1 toxin in response to stress, which induces growth inhibition through mRNA cleavage, mostly likely requires chaperone unavailability and subsequent degradation of the antitoxin, perhaps triggered by recruitment of the chaperone for Sec-dependent functions or by unbalanced proteostasis.
Figure 2.
Proposed mechanism for TAC. In M. tuberculosis, the TAC genes Rv1955-Rv1956-Rv1957, respectively, encode the toxin HigB1 (T), the antitoxin HigA1 (A) and the chaperone Rv1957 (C). They are located within an operon also containing the less conserved upstream Rv1954A gene of unknown function. Expression of TAC is regulated by two promoters: the promoter P1, which contains a RecA-NDp motif characteristic of LexA/RecA-independent genes in M. tuberculosis and the promoter P2, recognized by the HigA1 antitoxin. Repression of the TAC operon by HigA1 could occur in complex with its HigB1 and/or Rv1957 partners and HigA1 could regulate other genes as well. The SecB-like chaperone Rv1957 facilitates the folding of HigA1 and prevents its degradation by proteases (potentially ClpC1 or ClpX together with the ClpP1ClpP2 proteolytic subunit). Although interaction with the chaperone renders HigA1 competent to neutralize the toxin, it is not known whether Rv1957 is part of the final inactive complex. Activation of the HigB1 toxin in response to stress, which induces growth inhibition through mRNA cleavage, mostly likely requires chaperone unavailability and subsequent degradation of the antitoxin, perhaps triggered by recruitment of the chaperone for Sec-dependent functions or by unbalanced proteostasis.
TAC is conserved in all members of the M. tuberculosis complex and a recent evolutionarily study showed that SecB-like chaperones associated with toxin-antitoxin systems can be found in many bacterial phyla [66]. Remarkably, this analysis also revealed that the presence of a chaperone was not restricted to HigBA TA pairs [66]. In addition, some bacteria, including M. smegmatis, only possess an antitoxin/chaperone pair, possibly due to the loss of the upstream toxin gene [66].
As stated above, expression of the HigB1 toxin in the absence of HigA1 and Rv1957 severely inhibits E. coli, M. smegmatis, M. marinum, and M. tuberculosis growth (Figure 2) [15,16,67]. A recent study performed in M. tuberculosis ΔTAC mutant revealed a general decrease of over 30 transcripts upon HigB1 overexpression, whereas only a limited number of genes were found induced in this context [67]. Strikingly, most of the dysregulated mRNA transcripts were putative targets of the iron- and zinc-transcriptional regulators IdeR/Rv2711 and/or Zur/Rv2359, known to limit the expression of iron and zinc uptake systems in conditions of high iron and zinc amounts, respectively. Therefore, these results suggest that HigB1 could respond to iron and/or zinc overload, and allow the rapid degradation of metal uptake systems, once the bacterial cell is replenished in iron and/or zinc, in order to avoid intoxication [68].
Transcription of the TAC operon is induced by several relevant stress conditions including DNA damage [69], heat shock [70], nutrient starvation [43], hypoxia [16], drug-persistence [12], and in host phagocytes [71]. The TAC operon is under the control of two known promoters: the higB1P1 located 51 nucleotides upstream of the start codon of higB1, thus, controlling the expression of higB1-higA1-Rv1957, and the more distal promoter higB1P2 located 29 nucleotides upstream of the start codon of the upstream Rv1954a [69]. Noticeably, the DNA-damage inducible P1 promoter possesses a RecA-NDp motif characteristic of LexA/RecA-independent genes in M. tuberculosis [69]. As observed for most TA loci, TAC is autorepressed by HigA1 binding to a palindromic motif overlapping the -35 element of the higB1P2 promoter [65]. Although very likely, it is not known whether HigB1 and/or Rv1957 also participate in this process (Figure 2).
A role for HigA1 as a global transcriptional regulator has been proposed as well. Indeed, microarray analysis of the ΔTAC mutant revealed that the fadB, Rv3173c and Rv3662c genes might be regulated by HigA1 [65]. Moreover, a one-hybrid reporter system showed that HigA1 is capable of interacting specifically with the promoter region of gene clusters containing stress response genes whiB7, rubA, rubB, and fatty acid metabolism genes echA20, fadE28, fadE29, ufaA2, and fad26 [72]. Finally, ChIP-Seq analysis using FLAG-tagged HigA1 expressed in M. tuberculosis H37Rv revealed at least 30 potential HigA1 binding sites in intergenic regions, including the higBA3 locus described above (TB Database sever). These data suggest that HigA could be part of a network of stress-associated regulons involved in M. tuberculosis pathogenesis.
As stated above, Rv1957 shares sequence similarities with the export chaperone SecB known to facilitate protein export in most Gram-negative bacteria. In E. coli, the homotetrameric SecB chaperone binds unfolded presecretory proteins, maintains them in a protected yet nonnative state and passes them to the SecA subunit of the Sec translocase, thus facilitating their export [73,74,75]. Remarkably, it has been shown that Rv1957 can functionally replace SecB during protein export in vivo in E. coli and prevent aggregation of the outer membrane protein precursor proOmpC in vitro [64]. Similarly, the SecB-like protein SmegB from M. smegmatis was also capable of performing such SecB-like function in E. coli [66]. The presence of a well-defined outer membrane and a remarkably large number of putative outer membrane proteins in M. tuberculosis and M. smegmatis [76,77,78] suggests that these bacteria could make use of a functional export chaperone under certain circumstances (Figure 2). Although nothing is known yet about such a role in protein export, an attractive possibility is that SecB-like chaperones specifically connect the activation of stress-responsive TA systems to the export process. Under certain stress affecting the Sec translocon, the cytoplasmic accumulation of certain presecretory proteins could thus compete with HigA1 for binding to the Rv1957 chaperone, facilitating degradation of the free antitoxin and the subsequent activation of the toxin (Figure 2). The relatively well-conserved association of SecB-like chaperones with stress-responsive TA systems, including members of the HigBA, HicAB, and MqsRA families [66] further suggests that the control of TA activation by molecular chaperones might represent a more commonly used mechanism to control bacterial growth in response to environmental changes affecting protein homeostasis. In agreement with such a hypothesis, it was recently shown that the major E. coli stress chaperone DnaK (HSP70) interacts with at least four antitoxins in vivo, namely MazE, RelB, MqsA and DinJ, whose respective toxin partners MazF, RelE MqsR, and YafQ were shown to affect translation [79]. Therefore, DnaK could control the activation of several TA systems in a manner comparable to that of Rv1957 from TAC. Further reinforcing the potential link between molecular chaperones and toxin activation, an independent study showed that overexpression of the HipA toxin of the type II TA system HipAB facilitates E. coli adaptation and survival in response to a severe proteotoxic stress induced by the lack of both major chaperones Trigger Factor and DnaK [80]. More work is warranted to elucidate such fundamental links between networks of stress-induced molecular chaperones and TA systems in bacteria.
7. The ParDE Family
M. tuberculosis H37Rv possesses two ParDE systems (Figure 1). ParDE was originally discovered on the broad-host-range plasmid RK2 as a stabilization system [81]. The ParE toxin does not target RNA but acts by inhibiting the DNA gyrase, thereby blocking DNA replication [82]. The crystal structure of the ParDE1 complex from Caulobacter crescentus revealed a heterotetrameric complex composed of two homodimers of toxin and antitoxin [54]. As C. crescentus ParD1, the ParD1 antitoxin from M. tuberculosis is predicted to have an RHH DNA binding domain, whereas ParD2 does not seem to contain any conserved domain. Ectopic expression of the ParE2 toxin from M. tuberculosis inhibited both E. coli and M. smegmatis growth, whereas, under the same conditions, ParE1 only affected E. coli growth [15,16]. Thus far, none of these systems were tested in M. tuberculosis.
8. Other TA Modules
M. tuberculosis H37Rv genome potentially encodes 11 TA systems that do not yet belong to canonic well-characterized TA families (Figure 1). Among these systems, three were experimentally tested and two were functional in M. smegmatis (Figure 1) [16]. Four of these TA modules were identified as part of the 10 most induced TA systems in drug-tolerant persister cells, however, none of them have been tested for TA functions (Figure 1) [12]. Except for the Rv2653c-Rv2654c system, all these TA pairs contain a conserved domain in the toxin and/or in the antitoxin. These modules are paired together as follows, with the toxin domain designated first (NCD stands for No Conserved Domain): PF10604-PF14013 (Rv0910-Rv0909), PF10604-NCD (Rv1546-Rv1545), GNAT-RHH (Rv0919-Rv0918), COG5654-Xre (Rv1989c-Rv1990, Rv3189-Rv3188), COG3832-ArsR (Rv2035-Rv2034), NCD-COG2110 (Rv0059-Rv0060), DUF1814-COG5340 (Rv1045-Rv1044, Rv2826c-Rv2827c), DUF1814-COG4861 (Rv0836c-Rv0837c). Note that all of these TA domain pairs have been predicted either by Makarova et al. [29], Sberro et al. [83], or Dy et al. [84].
In the case of the DUF1814-COG5340 (Rv1045-Rv1044, Rv2826c-Rv2827c) TA pairs, homologous systems from diverse bacteria were experimentally tested and indeed behaved as bona fide TAs [83]. This includes the MosAT TA system involved in the maintenance of the SXT conjugative element in Vibrio cholerae [85] and the recently discovered lactococcal AbiE bacteriophage abortive infection system [84]. DUF1814 toxins belong to the polymerase β nucleotidyltransferase (polβ NTase) superfamily and, accordingly, it has been shown that the toxin of AbiE specifically binds GTP in vitro [84]. Remarkably, no interaction between the toxin and the antitoxin of AbiE could be detected by co-immunoprecipitation experiments performed in E. coli, thus, suggesting that conserved DUF1814-COG5340 pairs might belong to type IV TA systems [84]. This study also revealed that the DUF1814-COG4861 (Rv0836c-Rv0837c) pair might belong to the same AbiE family and thus potentially represents a third type IV member in M. tuberculosis.
9. Role of M. tuberculosis Proteases in TA Activation
Toxins from type II TA systems are generally activated following degradation of their cognate antitoxins. The major stress proteases Lon and/or ClpP together with its AAA+ subunit ClpX, ClpA or ClpC, were identified as key players in this process [86,87,88,89,90,91]. Although it clearly appears that certain antitoxins possess intrinsically flexible domains sensitive to proteolysis that are protected when bound to the toxin, very little is known about specific degradation signals within antitoxins that determine selective proteolysis [92,93,94]. Likewise, the cellular cascades that trigger activation of proteases towards TA systems are not well understood. In several cases, it has been shown that TA systems are induced in response to nutritional stress [86,87]. Such conditions are known to induce the stringent response characterized by the synthesis of the small alarmone molecule (p)ppGpp, which causes growth arrest, activation of the stress response, and bacterial persistence [95]. Remarkably, increased persistence in E. coli strongly relies on both the activation of Lon by inorganic polyphosphates known to accumulate in response to (p)ppGpp synthesis, and the presence of TA systems, as shown recently [8]. Although it has been shown that the stringent response is important for M. tuberculosis persistence, it is not known whether activation of TA systems by specific stress proteases contributes to such phenomenon [44].
In contrast with E. coli, M. tuberculosis and closely related species do not have the proteases Lon. However, they do have two clpP genes, clpP1 and clpP2, encoding the proteolytic subunits and three predicted ClpP-associated regulatory subunits encoding genes, clpX, clpC1, and clpC2, involved in substrate recognition, unfolding and translocation into the proteolytic chamber [96]. Note that in contrast with ClpC1 and ClpX, the ClpC2 sequence does not contain any AAA+ domain and it is thus not clear whether it can cooperate with ClpP proteases. Yet, the most striking difference with E. coli is that M. tuberculosis possesses a 20S proteasome-like degradation machinery to which proteins are addressed by addition of an ubiquitine-like tag called Pup (Prokaryotic ubiquitin-like protein), giving the name of “Pupylation” to this pathway [97]. Analysis of the Pup proteome of M. tuberculosis revealed that at least 4 toxins, namely Rv0059 (unclassified), Rv0749 (VapC31), Rv2035 (Unclassified) and Rv2527 (VapC17) are pupylated, while no pupylated antitoxin could be detected [98]. Although there is no evidence for a role of the proteasome in TA activation, the observation that pupylation seems to concern only toxins suggest that an additional, yet unexplored level of regulation of TA systems by proteolysis could exist.
10. Concluding Remarks
In contrast with closely related mycobacteria, M. tuberculosis encodes a remarkably high number of TA systems, with at least 37 being functional in vivo. The existence of such intricate networks of TAs, often induced during persistence or under stress conditions encountered during the infection process, raises the question of their implication in the virulence of this dangerous pathogen. While ectopic expression of certain toxins inhibits M. tuberculosis growth and results in increased persistence, mutation of distinct TA genes did not yet reveal a direct involvement of these systems in the infection process in vivo. The fact that decreased persistence in E. coli could only be observed upon successive deletions of several TA systems strongly suggests that TA functions are redundant and that similar situation might occur in M. tuberculosis. Finally, even though the signals that trigger activation of toxins in M. tuberculosis are unknown, the observation that a single protease can induce persistence in E. coli via stochastic activation of several TA systems indicates that a search for specific stress proteases that target antitoxins in M. tuberculosis might reveal important links between TA activation and persistence in this bacterium.
Acknowledgements
We thank Claude Gutierrez and Olivier Neyrolles for insightful discussions. This work was supported by a French MENRT fellowship to Ambre Sala and a mycoTAC ANR grant to Pierre Genevaux.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yamaguchi, Y.; Park, J.H.; Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acid Res. 2005, 33, 966–976. [Google Scholar] [CrossRef]
- Gerdes, K.; Maisonneuve, E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012, 66, 103–123. [Google Scholar] [CrossRef]
- Masuda, H.; Tan, Q.; Awano, N.; Wu, K.P.; Inouye, M. Yeeu enhances the bundling of cytoskeletal polymers of Mreb and Ftsz, antagonizing the Cbta (Yeev) toxicity in Escherichia coli. Mol. Microbiol. 2012, 84, 979–989. [Google Scholar] [CrossRef]
- Wang, X.; Lord, D.M.; Cheng, H.Y.; Osbourne, D.O.; Hong, S.H.; Sanchez-Torres, V.; Quiroga, C.; Zheng, K.; Herrmann, T.; Peti, W.; et al. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 2012, 8, 855–861. [Google Scholar] [CrossRef]
- Van Melderen, L. Toxin-antitoxin systems: Why so many, what for? Curr. Opin. Microbiol. 2010, 13, 781–785. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells. Ann. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Maisonneuve, E.; Castro-Camargo, M.; Gerdes, K. (p)ppgpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 2013, 154, 1140–1150. [Google Scholar] [CrossRef]
- Barry, C.E., 3rd.; Boshoff, H.I.; Dartois, V.; Dick, T.; Ehrt, S.; Flynn, J.; Schnappinger, D.; Wilkinson, R.J.; Young, D. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009, 7, 845–855. [Google Scholar]
- Georgiades, K.; Raoult, D. Genomes of the most dangerous epidemic bacteria have a virulence repertoire characterized by fewer genes but more toxin-antitoxin modules. PLoS ONE 2011, 6, e17962. [Google Scholar] [CrossRef]
- Sala, A.; Bordes, P.; Fichant, G.; Genevaux, P. Toxin-antitoxin loci in Mycobacterium tuberculosis. In Prokaryotic Toxin-Antitoxin; Gerdes, K., Ed.; Springer: Berlin, Heidelberg, Germany, 2013; pp. 295–314. [Google Scholar]
- Keren, I.; Minami, S.; Rubin, E.; Lewis, K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2011, 2, e00100–e00111. [Google Scholar]
- Fozo, E.M.; Makarova, K.S.; Shabalina, S.A.; Yutin, N.; Koonin, E.V.; Storz, G. Abundance of type I toxin-antitoxin systems in bacteria: Searches for new candidates and discovery of novel families. Nucleic Acid Res. 2010, 38, 3743–3759. [Google Scholar] [CrossRef]
- Blower, T.R.; Short, F.L.; Rao, F.; Mizuguchi, K.; Pei, X.Y.; Fineran, P.C.; Luisi, B.F.; Salmond, G.P. Identification and classification of bacterial type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012, 40, 6158–6173. [Google Scholar]
- Gupta, A. Killing activity and rescue function of genome-wide toxin-antitoxin loci of Mycobacterium tuberculosis. FEMS Microbiol. Lett. 2009, 290, 45–53. [Google Scholar] [CrossRef]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009, 5, e1000767. [Google Scholar] [CrossRef]
- Huang, F.; He, Z.G. Characterization of an interplay between a Mycobacterium tuberculosis MazF homolog, Rv1495 and its sole DNA topoisomerase I. Nucleic Acids Res. 2010, 38, 8219–8230. [Google Scholar] [CrossRef]
- Singh, R.; Barry, C.E., 3rd.; Boshoff, H.I. The three RelE homologs of Mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. J. Bacteriol. 2010, 192, 1279–1291. [Google Scholar] [CrossRef]
- Zhu, L.; Sharp, J.D.; Kobayashi, H.; Woychik, N.A.; Inouye, M. Noncognate Mycobacterium tuberculosis toxin-antitoxins can physically and functionally interact. J. Biol. Chem. 2010, 285, 39732–39738. [Google Scholar] [CrossRef]
- Ahidjo, B.A.; Kuhnert, D.; McKenzie, J.L.; Machowski, E.E.; Gordhan, B.G.; Arcus, V.; Abrahams, G.L.; Mizrahi, V. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS ONE 2011, 6, e21738. [Google Scholar] [CrossRef]
- Pullinger, G.D.; Lax, A.J. A Salmonella dublin virulence plasmid locus that affects bacterial growth under nutrient-limited conditions. Mol. Microbiol. 1992, 6, 1631–1643. [Google Scholar] [CrossRef]
- Arcus, V.L.; McKenzie, J.L.; Robson, J.; Cook, G.M. The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng. Des. Sel. 2011, 24, 33–40. [Google Scholar] [CrossRef]
- Miallau, L.; Faller, M.; Chiang, J.; Arbing, M.; Guo, F.; Cascio, D.; Eisenberg, D. Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J. Biol. Chem. 2009, 284, 276–283. [Google Scholar] [CrossRef]
- Winther, K.S.; Gerdes, K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. USA 2011, 108, 7403–7407. [Google Scholar] [CrossRef]
- Sharp, J.D.; Cruz, J.W.; Raman, S.; Inouye, M.; Husson, R.N.; Woychik, N.A. Growth and translation inhibition through sequence specific RNA binding by a Mycobacterium tuberculosis VapC toxin. J. Biol. Chem. 2012. [Google Scholar] [CrossRef]
- McKenzie, J.L.; Duyvestyn, J.M.; Smith, T.; Bendak, K.; Mackay, J.; Cursons, R.; Cook, G.M.; Arcus, V.L. Determination of ribonuclease sequence-specificity using pentaprobes and mass spectrometry. RNA 2012, 18, 1267–1278. [Google Scholar] [CrossRef]
- Winther, K.S.; Brodersen, D.E.; Brown, A.K.; Gerdes, K. VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nat. Commun. 2013, 4, 2796. [Google Scholar]
- Min, A.B.; Miallau, L.; Sawaya, M.R.; Habel, J.; Cascio, D.; Eisenberg, D. The crystal structure of the Rv0301-Rv0300 VapBC-3 toxin-antitoxin complex from M. tuberculosis reveals a Mg2+ ion in the active site and a putative RNA-binding site. Protein Sci. 2012, 21, 1754–1767. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of type-2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct. 2009, 4, 19. [Google Scholar] [CrossRef]
- McKenzie, J.L.; Robson, J.; Berney, M.; Smith, T.C.; Ruthe, A.; Gardner, P.P.; Arcus, V.L.; Cook, G.M. A VapBC toxin-antitoxin module is a post-transcriptional regulator of metabolic flux in Mycobacteria. J. Bacteriol. 2012. [Google Scholar] [CrossRef]
- Albrethsen, J.; Agner, J.; Piersma, S.R.; Hojrup, P.; Pham, T.V.; Weldingh, K.; Jimenez, C.R.; Andersen, P.; Rosenkrands, I. Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol. Cell. Proteomics 2013, 12, 1180–1191. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Inouye, M. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol. 2011, 9, 779–790. [Google Scholar] [CrossRef]
- Kamada, K.; Hanaoka, F.; Burley, S.K. Crystal structure of the MazE/MazF complex: Molecular bases of antidote-toxin recognition. Mol. Cell. 2003, 11, 875–884. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Yamaguchi, Y.; Park, J.H.; Inouye, M.; Patel, D.J. Structural basis of mRNA recognition and cleavage by toxin MazF and its regulation by antitoxin MazE in Bacillus subtilis. Mol. Cell. 2013, 52, 447–458. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, L.; Zhang, J.; Inouye, M. Characterization of ChpbK, an mRNA interferase from Escherichia coli. J. Biol. Chem. 2005, 280, 26080–26088. [Google Scholar] [CrossRef]
- Park, J.H.; Yamaguchi, Y.; Inouye, M. Bacillus subtilis MazF-bs (EndoA) is a UACAU-specific mRNA interferase. FEBS Lett. 2011, 585, 2526–2532. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, Y.; Teh, J.S.; Zhang, J.; Connell, N.; Rubin, H.; Inouye, M. Characterization of mRNA interferases from Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 18638–18643. [Google Scholar]
- Schifano, J.M.; Edifor, R.; Sharp, J.D.; Ouyang, M.; Konkimalla, A.; Husson, R.N.; Woychik, N.A. Mycobacterial toxin MazF-mt6 inhibits translation through cleavage of 23S rRNA at the ribosomal A site. Proc. Natl. Acad. Sci. USA 2013, 110, 8501–8506. [Google Scholar]
- Zhu, L.; Phadtare, S.; Nariya, H.; Ouyang, M.; Husson, R.N.; Inouye, M. The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Mol. Microbiol. 2008, 69, 559–569. [Google Scholar] [CrossRef]
- Vesper, O.; Amitai, S.; Belitsky, M.; Byrgazov, K.; Kaberdina, A.C.; Engelberg-Kulka, H.; Moll, I. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell 2011, 147, 147–157. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef]
- Hazan, R.; Sat, B.; Engelberg-Kulka, H. Escherichia coli MazEF-mediated cell death is triggered by various stressful conditions. J. Bacteriol. 2004, 186, 3663–3669. [Google Scholar] [CrossRef]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef]
- Dahl, J.L.; Kraus, C.N.; Boshoff, H.I.; Doan, B.; Foley, K.; Avarbock, D.; Kaplan, G.; Mizrahi, V.; Rubin, H.; Barry, C.E., 3rd. The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10026–10031. [Google Scholar] [CrossRef]
- Ramirez, M.V.; Dawson, C.C.; Crew, R.; England, K.; Slayden, R.A. MazF6 toxin of Mycobacterium tuberculosis demonstrates antitoxin specificity and is coupled to regulation of cell growth by a Soj-like protein. BMC Microbiol. 2013, 13, 240. [Google Scholar] [CrossRef]
- Diderichsen, B.; Fiil, N.P.; Lavalle, R. Genetics of the RelB locus in Escherichia coli. J. Bacteriol. 1977, 131, 30–33. [Google Scholar]
- Christensen, S.K.; Gerdes, K. Delayed-relaxed response explained by hyperactivation of RelE. Mol. Microbiol. 2004, 53, 587–597. [Google Scholar] [CrossRef]
- Grady, R.; Hayes, F. Axe-txe, a broad-spectrum proteic toxin-antitoxin system specified by a multidrug-resistant, clinical isolate of Enterococcus faecium. Mol. Microbiol. 2003, 47, 1419–1432. [Google Scholar] [CrossRef]
- Christensen, S.K.; Gerdes, K. RelE toxins from bacteria and archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 2003, 48, 1389–1400. [Google Scholar] [CrossRef]
- Kamada, K.; Hanaoka, F. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol. Cell. 2005, 19, 497–509. [Google Scholar] [CrossRef]
- Cherny, I.; Gazit, E. The YefM antitoxin defines a family of natively unfolded proteins: Implications as a novel antibacterial target. J. Biol. Chem. 2004, 279, 8252–8261. [Google Scholar] [CrossRef]
- Kumar, P.; Issac, B.; Dodson, E.J.; Turkenburg, J.P.; Mande, S.C. Crystal structure of Mycobacterium tuberculosis YefM antitoxin reveals that it is not an intrinsically unstructured protein. J. Mol. Biol. 2008, 383, 482–493. [Google Scholar] [CrossRef]
- Miallau, L.; Jain, P.; Arbing, M.A.; Cascio, D.; Phan, T.; Ahn, C.J.; Chan, S.; Chernishof, I.; Maxson, M.; Chiang, J.;et al. Comparative proteomics identifies the cell-associated lethality of M. tuberculosis RelBE-like toxin-antitoxin complexes. Structure 2013, 21, 627–637. [Google Scholar] [CrossRef]
- Blower, T.R.; Salmond, G.P.; Luisi, B.F. Balancing at survival's edge: The structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr. Opin. Struct. Biol. 2011, 21, 109–118. [Google Scholar] [CrossRef]
- Korch, S.B.; Contreras, H.; Clark-Curtiss, J.E. Three Mycobacterium tuberculosis Rel toxin-antitoxin modules inhibit mycobacterial growth and are expressed in infected human macrophages. J. Bacteriol. 2009, 191, 1618–1630. [Google Scholar] [CrossRef]
- Yang, M.; Gao, C.; Wang, Y.; Zhang, H.; He, Z.G. Characterization of the interaction and cross-regulation of three Mycobacterium tuberculosis RelBE modules. PLoS ONE 2010, 5, e10672. [Google Scholar] [CrossRef]
- Tian, Q.B.; Ohnishi, M.; Tabuchi, A.; Terawaki, Y. A new plasmid-encoded proteic killer gene system: Cloning, sequencing, and analyzing hig locus of plasmid rts1. Biochem. Biophys. Res. Commun. 1996, 220, 280–284. [Google Scholar] [CrossRef]
- Gerdes, K.; Christensen, S.K.; Lobner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef]
- Hurley, J.M.; Woychik, N.A. Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. J. Biol. Chem. 2009, 284, 18605–18613. [Google Scholar] [CrossRef]
- Christensen-Dalsgaard, M.; Gerdes, K. Two HigBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol. Microbiol. 2006, 62, 397–411. [Google Scholar] [CrossRef]
- Schureck, M.A.; Maehigashi, T.; Miles, S.J.; Marquez, J.; Ei Cho, S.; Erdman, R.; Dunham, C.M. Structure of the P. vulgaris HigB-(HigA)2-HigB toxin-antitoxin complex. J. Biol. Chem. 2013. [Google Scholar] [CrossRef]
- Arbing, M.A.; Handelman, S.K.; Kuzin, A.P.; Verdon, G.; Wang, C.; Su, M.; Rothenbacher, F.P.; Abashidze, M.; Liu, M.; Hurley, J.M.; et al. Crystal structures of Phd-Doc, HigA, and YeeU establish multiple evolutionary links between microbial growth-regulating toxin-antitoxin systems. Structure 2010, 18, 996–1010. [Google Scholar] [CrossRef]
- Stinear, T.P.; Seemann, T.; Harrison, P.F.; Jenkin, G.A.; Davies, J.K.; Johnson, P.D.; Abdellah, Z.; Arrowsmith, C.; Chillingworth, T.; Churcher, C.; et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008, 18, 729–741. [Google Scholar] [CrossRef]
- Bordes, P.; Cirinesi, A.M.; Ummels, R.; Sala, A.; Sakr, S.; Bitter, W.; Genevaux, P. SecB-like chaperone controls a toxin-antitoxin stress-responsive system in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2011, 108, 8438–8443. [Google Scholar] [CrossRef]
- Fivian-Hughes, A.S.; Davis, E.O. Analyzing the regulatory role of the HigA antitoxin within Mycobacterium tuberculosis. J. Bacteriol. 2010, 192, 4348–4356. [Google Scholar] [CrossRef]
- Sala, A.; Calderon, V.; Bordes, P.; Genevaux, P. TAC from Mycobacterium tuberculosis: A paradigm for stress-responsive toxin-antitoxin systems controlled by SecB-like chaperones. Cell Stress Chap. 2013, 18, 129–135. [Google Scholar] [CrossRef]
- Schuessler, D.L.; Cortes, T.; Fivian-Hughes, A.S.; Lougheed, K.E.; Harvey, E.; Buxton, R.S.; Davis, E.O.; Young, D.B. Induced ectopic expression of HigB toxin in Mycobacterium tuberculosis results in growth inhibition, reduced abundance of a subset of mRNAs and cleavage of tmRNA. Mol. Microbiol. 2013, 90, 195–207. [Google Scholar]
- Botella, H.; Peyron, P.; Levillain, F.; Poincloux, R.; Poquet, Y.; Brandli, I.; Wang, C.; Tailleux, L.; Tilleul, S.; Charriere, G.M.; et al. Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell. Host Microbe 2011, 10, 248–259. [Google Scholar] [CrossRef]
- Smollett, K.L.; Fivian-Hughes, A.S.; Smith, J.E.; Chang, A.; Rao, T.; Davis, E.O. Experimental determination of translational start sites resolves uncertainties in genomic open reading frame predictions - application to Mycobacterium tuberculosis. Microbiology 2009, 155, 186–197. [Google Scholar] [CrossRef]
- Stewart, G.R.; Wernisch, L.; Stabler, R.; Mangan, J.A.; Hinds, J.; Laing, K.G.; Young, D.B.; Butcher, P.D. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology 2002, 148, 3129–3138. [Google Scholar]
- Tailleux, L.; Waddell, S.J.; Pelizzola, M.; Mortellaro, A.; Withers, M.; Tanne, A.; Castagnoli, P.R.; Gicquel, B.; Stoker, N.G.; Butcher, P.D.; et al. Probing host pathogen cross-talk by transcriptional profiling of both Mycobacterium tuberculosis and infected human dendritic cells and macrophages. PLoS ONE 2008, 3, e1403. [Google Scholar] [CrossRef]
- Guo, M.; Feng, H.; Zhang, J.; Wang, W.; Wang, Y.; Li, Y.; Gao, C.; Chen, H.; Feng, Y.; He, Z.G. Dissecting transcription regulatory pathways through a new bacterial one-hybrid reporter system. Genome Res. 2009, 19, 1301–1308. [Google Scholar] [CrossRef]
- Driessen, A.J.; Nouwen, N. Protein translocation across the bacterial cytoplasmic membrane. Annu. Rev. Biochem. 2008, 77, 643–667. [Google Scholar] [CrossRef]
- Randall, L.L.; Hardy, S.J. SecB, one small chaperone in the complex milieu of the cell. Cell. Mol. Life Sci. 2002, 59, 1617–1623. [Google Scholar] [CrossRef]
- Castanie-Cornet, M.P.; Bruel, N.; Genevaux, P. Chaperone networking facilitates protein targeting to the bacterial cytoplasmic membrane. Biochim. Biophys. Acta 2013. [Google Scholar] [CrossRef]
- Zuber, B.; Chami, M.; Houssin, C.; Dubochet, J.; Griffiths, G.; Daffe, M. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 2008, 190, 5672–5680. [Google Scholar] [CrossRef]
- Niederweis, M.; Danilchanka, O.; Huff, J.; Hoffmann, C.; Engelhardt, H. Mycobacterial outer membranes: In search of proteins. Trends Microbiol. 2010, 18, 109–116. [Google Scholar] [CrossRef]
- Sani, M.; Houben, E.N.; Geurtsen, J.; Pierson, J.; de Punder, K.; van Zon, M.; Wever, B.; Piersma, S.R.; Jimenez, C.R.; Daffe, M.; et al. Direct visualization by cryo-EM of the mycobacterial capsular layer: A labile structure containing ESX-1-secreted proteins. PLoS Path. 2010, 6, e1000794. [Google Scholar] [CrossRef]
- Calloni, G.; Chen, T.; Schermann, S.M.; Chang, H.C.; Genevaux, P.; Agostini, F.; Tartaglia, G.G.; Hayer-Hartl, M.; Hartl, F.U. DnaK functions as a central hub in the E. coli chaperone network. Cell. Rep. 2012, 1, 251–264. [Google Scholar] [CrossRef]
- Bruel, N.; Castanie-Cornet, M.P.; Cirinesi, A.M.; Koningstein, G.; Georgopoulos, C.; Luirink, J.; Genevaux, P. Hsp33 controls elongation factor-Tu stability and allows Escherichia coli growth in the absence of the major DnaK and trigger factor chaperones. J. Biol Chem 2012, 287, 44435–44446. [Google Scholar] [CrossRef]
- Roberts, R.C.; Helinski, D.R. Definition of a minimal plasmid stabilization system from the broad-host-range plasmid rk2. J. Bacteriol. 1992, 174, 8119–8132. [Google Scholar]
- Jiang, Y.; Pogliano, J.; Helinski, D.R.; Konieczny, I. ParE toxin encoded by the broad-host-range plasmid rk2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 2002, 44, 971–979. [Google Scholar] [CrossRef]
- Sberro, H.; Leavitt, A.; Kiro, R.; Koh, E.; Peleg, Y.; Qimron, U.; Sorek, R. Discovery of functional toxin/antitoxin systems in bacteria by shotgun cloning. Mol. Cell. 2013, 50, 136–148. [Google Scholar] [CrossRef]
- Dy, R.L.; Przybilski, R.; Semeijn, K.; Salmond, G.P.; Fineran, P.C. A widespread bacteriophage abortive infection system functions through a type IV toxin-antitoxin mechanism. Nucleic Acid Res. 2014. [Google Scholar] [CrossRef]
- Wozniak, R.A.; Waldor, M.K. A toxin-antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet. 2009, 5, e1000439. [Google Scholar] [CrossRef]
- Christensen, S.K.; Mikkelsen, M.; Pedersen, K.; Gerdes, K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. USA 2001, 98, 14328–14333. [Google Scholar] [CrossRef]
- Aizenman, E.; Engelberg-Kulka, H.; Glaser, G. An Escherichia coli chromosomal “addiction module” regulated by guanosine [corrected] 3',5'-bispyrophosphate: A model for programmed bacterial cell death. Proc. Natl. Acad. Sci. USA 1996, 93, 6059–6063. [Google Scholar] [CrossRef]
- Christensen-Dalsgaard, M.; Jorgensen, M.G.; Gerdes, K. Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol. Microbiol. 2010, 75, 333–348. [Google Scholar] [CrossRef]
- Ning, D.; Liu, S.; Xu, W.; Zhuang, Q.; Wen, C.; Tang, X. Transcriptional and proteolytic regulation of the toxin-antitoxin locus VapBC10 (ssr2962/slr1767) on the chromosome of Synechocystis sp. Pcc 6803. PLoS ONE 2013, 8, e80716. [Google Scholar]
- Donegan, N.P.; Thompson, E.T.; Fu, Z.; Cheung, A.L. Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 2010, 192, 1416–1422. [Google Scholar] [CrossRef]
- Van Melderen, L.; Bernard, P.; Couturier, M. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol. Microbiol. 1994, 11, 1151–1157. [Google Scholar] [CrossRef]
- Van Melderen, L.; Thi, M.H.; Lecchi, P.; Gottesman, S.; Couturier, M.; Maurizi, M.R. ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J. Biol. Chem. 1996, 271, 27730–27738. [Google Scholar]
- Loris, R.; Marianovsky, I.; Lah, J.; Laeremans, T.; Engelberg-Kulka, H.; Glaser, G.; Muyldermans, S.; Wyns, L. Crystal structure of the intrinsically flexible addiction antidote MazE. J. Biol. Chem. 2003, 278, 28252–28257. [Google Scholar] [CrossRef]
- Hansen, S.; Vulic, M.; Min, J.; Yen, T.J.; Schumacher, M.A.; Brennan, R.G.; Lewis, K. Regulation of the Escherichia coli HipBA toxin-antitoxin system by proteolysis. PLoS ONE 2012, 7, e39185. [Google Scholar]
- Boutte, C.C.; Crosson, S. Bacterial lifestyle shapes stringent response activation. Trends Microbiol. 2013, 21, 174–180. [Google Scholar] [CrossRef]
- Ribeiro-Guimaraes, M.L.; Pessolani, M.C. Comparative genomics of mycobacterial proteases. Microb. Pathogen. 2007, 43, 173–178. [Google Scholar] [CrossRef]
- Pearce, M.J.; Mintseris, J.; Ferreyra, J.; Gygi, S.P.; Darwin, K.H. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 2008, 322, 1104–1107. [Google Scholar] [CrossRef]
- Festa, R.A.; McAllister, F.; Pearce, M.J.; Mintseris, J.; Burns, K.E.; Gygi, S.P.; Darwin, K.H. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis [corrected]. PLoS ONE 2010, 5, e8589. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Sala, A.; Bordes, P.; Genevaux, P. Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis. Toxins 2014, 6, 1002-1020. https://doi.org/10.3390/toxins6031002
AMA Style
Sala A, Bordes P, Genevaux P. Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis. Toxins. 2014; 6(3):1002-1020. https://doi.org/10.3390/toxins6031002
Chicago/Turabian StyleSala, Ambre, Patricia Bordes, and Pierre Genevaux. 2014. "Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis" Toxins 6, no. 3: 1002-1020. https://doi.org/10.3390/toxins6031002