Deficient Glutathione in the Pathophysiology of Mycotoxin-Related Illness

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress
3. Glutathione Sources in the Body
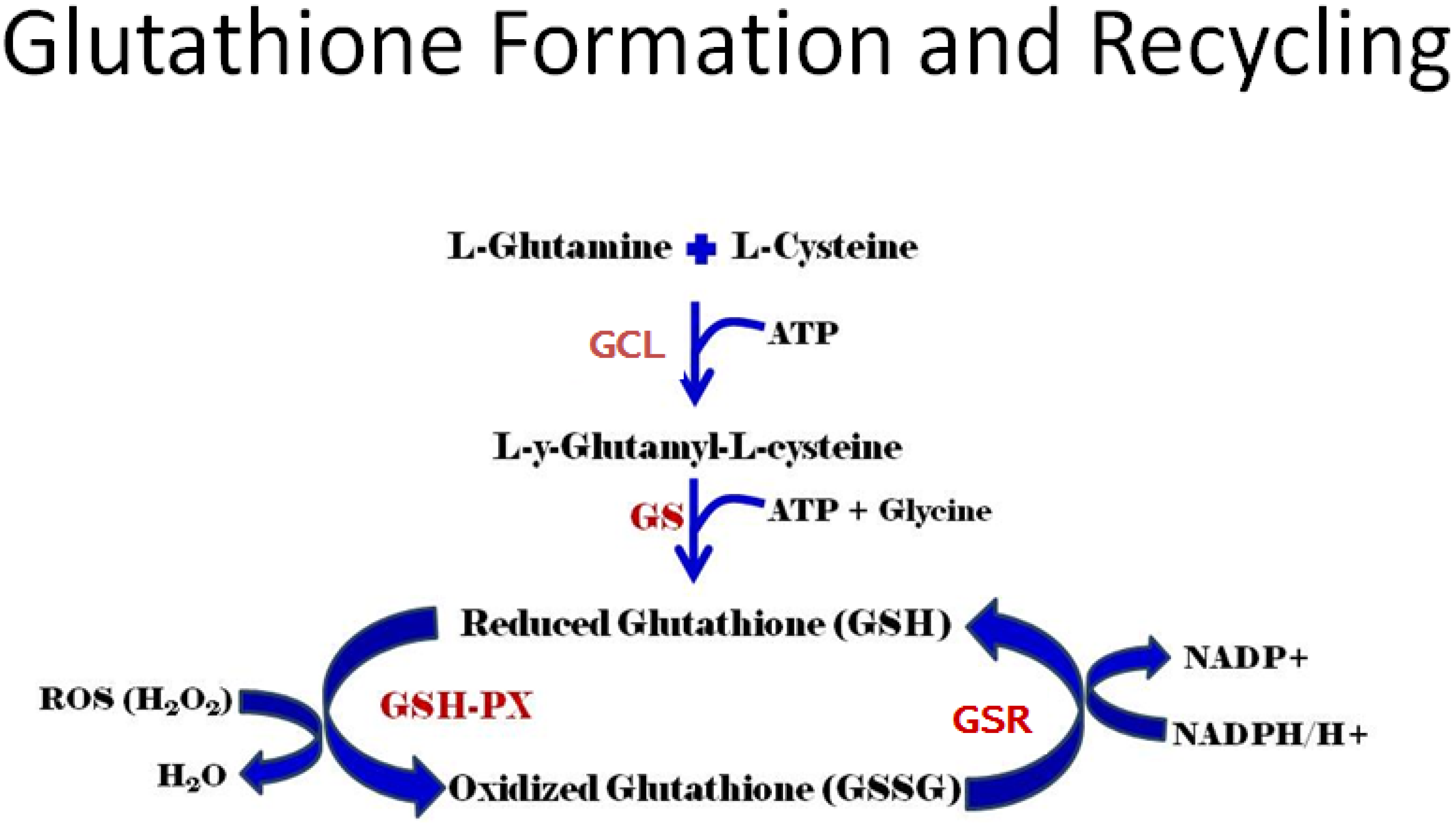
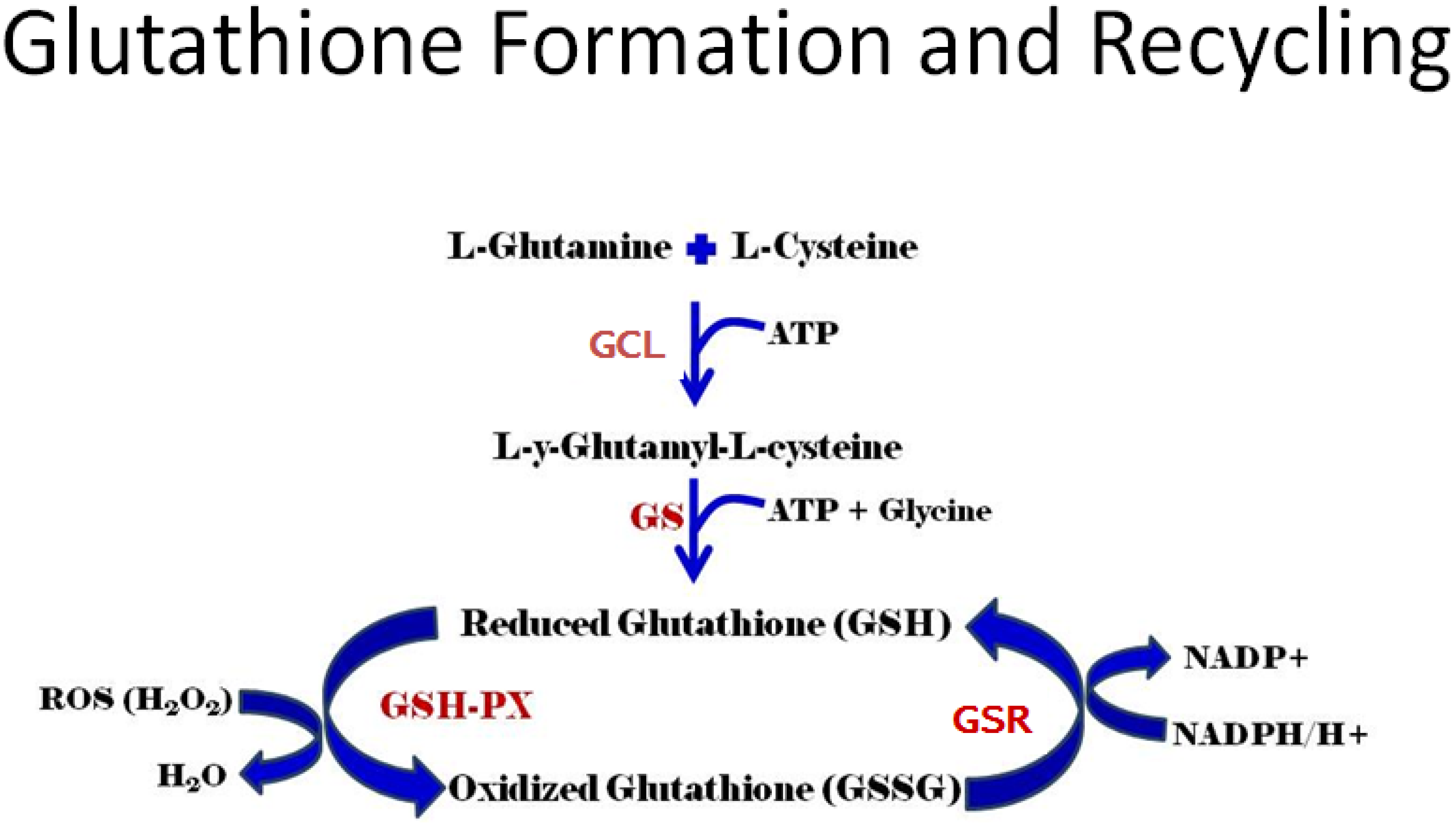
3.1. Gene Control of GSH Production
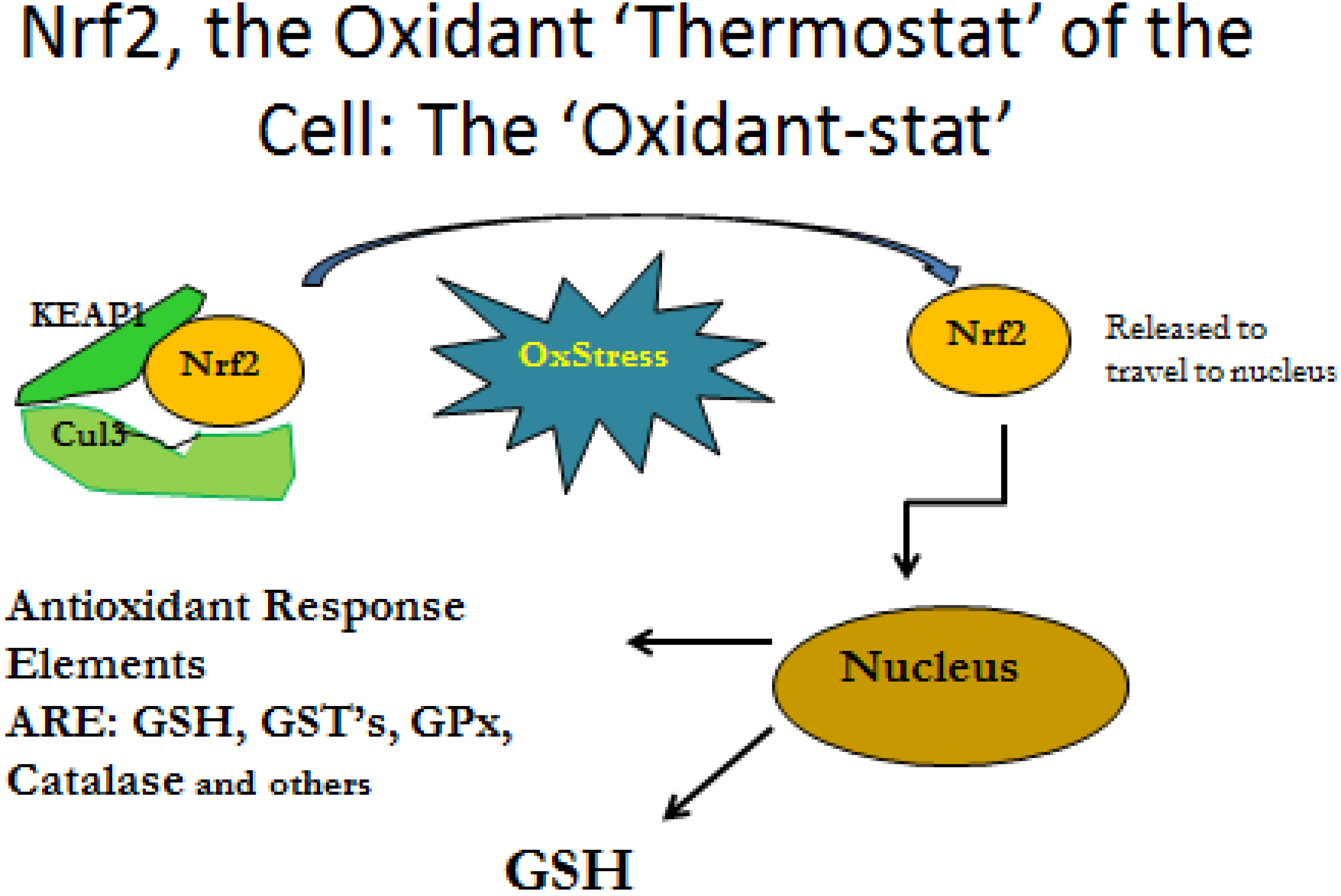
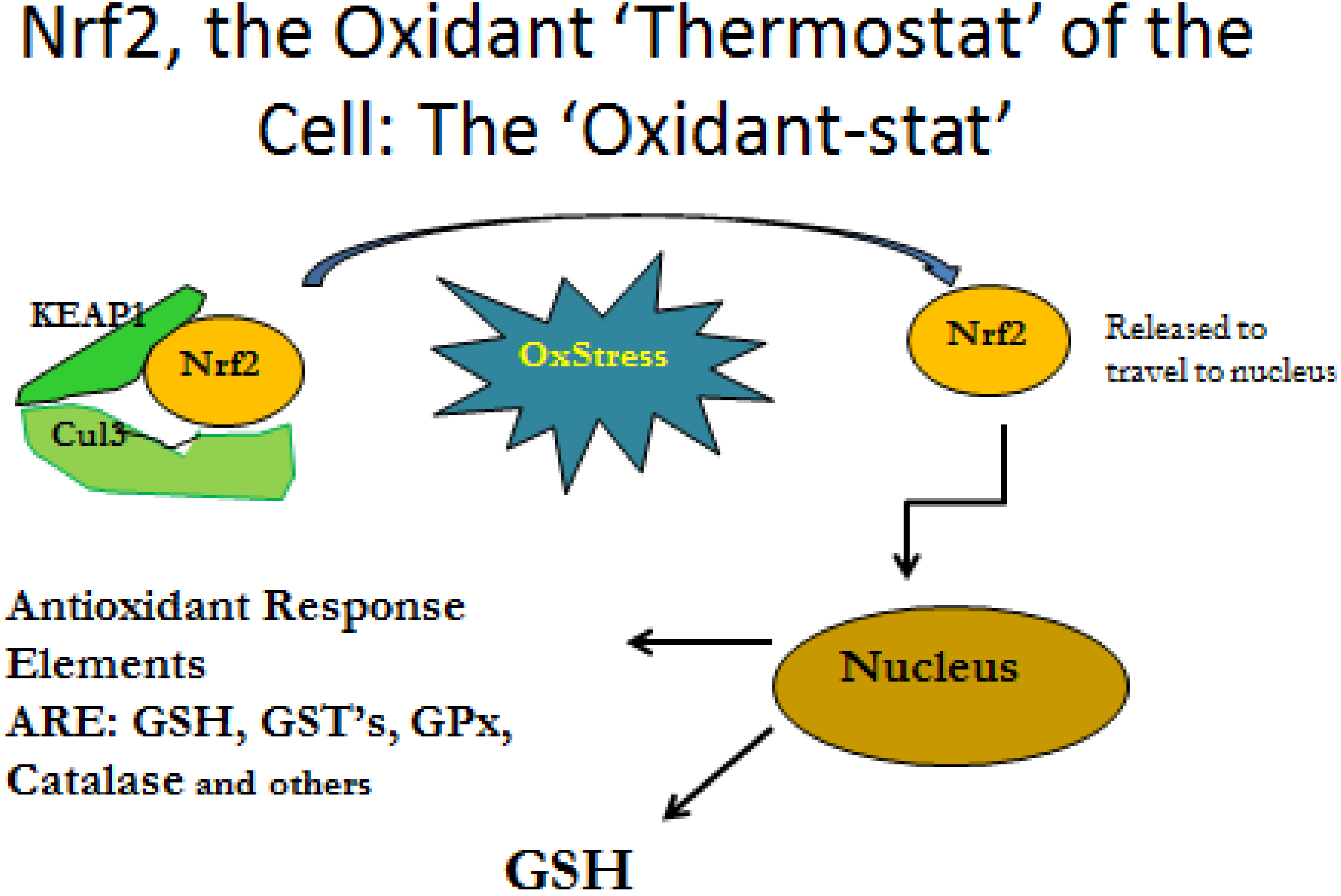
3.2. Cytokine Influence on GSH Production
3.3. Glutathione S-Transferase
3.4. GST Genetic Polymorphisms
4. Identification of Mycotoxin-Related Oxidant Stress
5. Mycotoxin Effect on Immunity is Oxidative Stress Related
6. Conclusions
Conflicts of Interest
References
- Council for Agricultural Science and Technology. Mycotoxins: Risks in Plant, Animal, and Human Systems; Council for Agricultural Science and Technology: Ames, IA, USA, 2002. [Google Scholar]
- Hussein, H.S.; Brasel, J.M. Toxicity, metabolism, and impact of mycotoxins on humans and animals. Toxicology 2001, 167, 101–134. [Google Scholar] [CrossRef]
- Yazar, S.; Omurtag, G.Z. Fumonisins, trichothecenes and zearalenone in cereals. Int. J. Mol. Sci. 2008, 9, 2062–2090. [Google Scholar] [CrossRef]
- Sondergaard, T.E.; Klitgaard, L.G.; Purup, S.; Kobayashi, H.; Giese, H.; Sorensen, J.L. Estrogenic effects of fusarielins in human breast cancer cell lines. Toxicol. Lett. 2012, 214, 259–262. [Google Scholar] [CrossRef]
- Carey, S.A.; Plopper, C.G.; Hyde, D.M.; Islam, Z.; Pestka, J.J.; Harkema, J.R. Satratoxin-G from the black mold Stachybotrys chartarum induces rhinitis and apoptosis of olfactory sensory neurons in the nasal airways of rhesus monkeys. Toxicol. Pathol. 2012, 40, 887–898. [Google Scholar] [CrossRef]
- Van der Merwe, K.J.; Steyn, P.S.; Fourie, L.; Scott, D.B.; Theron, J.J. Ochratoxin A, a toxic metabolite produced by Aspergillus ochraceus Wilh. Nature 1965, 205, 1112–1113. [Google Scholar] [CrossRef]
- Larsen, T.O.; Svendsen, A.; Smedsgaard, J. Biochemical characterization of ochratoxin A—Producing strains of the genus penicillium. Appl. Environ. Microbiol. 2001, 67, 3630–3635. [Google Scholar] [CrossRef]
- Abbas, H.K. Aflatoxin and Food Safety; Taylor & Francis: Boca Raton, Fl, USA, 2005. [Google Scholar]
- Wang, J.S.; Groopman, J.D. DNA damage by mycotoxins. Mutat. Res. 1999, 424, 167–181. [Google Scholar] [CrossRef]
- Ellis, E.M. Protection against aflatoxin B1 in rat—A new look at the link between toxicity, carcinogenicity, and metabolism. Toxicol. Sci. 2009, 109, 1–3. [Google Scholar] [CrossRef]
- Roebuck, B.D.; Johnson, D.N.; Sutter, C.H.; Egner, P.A.; Scholl, P.F.; Friesen, M.D.; Baumgartner, K.J.; Ware, N.M.; Bodreddigari, S.; Groopman, J.D.; et al. Transgenic expression of aflatoxin aldehyde reductase (akr7a1) modulates aflatoxin B1 metabolism but not hepatic carcinogenesis in the rat. Toxicol. Sci. 2009, 109, 41–49. [Google Scholar] [CrossRef]
- Eaton, D.L.; Gallagher, E.P. Mechanisms of aflatoxin carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 135–172. [Google Scholar] [CrossRef]
- Croy, R.G.; Essigmann, J.M.; Reinhold, V.N.; Wogan, G.N. Identification of the principal aflatoxin B1-DNA adduct formed in vivo in rat liver. Proc. Natl. Acad. Sci. USA 1978, 75, 1745–1749. [Google Scholar] [CrossRef]
- Essigmann, J.M.; Croy, R.G.; Nadzan, A.M.; Busby, W.F.; Reinhold, V.N.; Büchi, G.; Wogan, G.N. Structural identification of the major DNA adduct formed by aflatoxin B1 in vitro. Proc. Natl. Acad. Sci. USA 1977, 74, 1870–1874. [Google Scholar] [CrossRef]
- Hayes, J.D.; Pulford, D.J.; Ellis, E.M.; McLeod, R.; James, R.F.; Seidegard, J.; Mosialou, E.; Jernstrom, B.; Neal, G.E. Regulation of rat glutathione S-transferase A5 by cancer chemopreventive agents: Mechanisms of inducible resistance to aflatoxin B1. Chem. Biol. Interact. 1998, 111–112, 51–67. [Google Scholar] [CrossRef]
- Ilic, Z.; Crawford, D.; Vakharia, D.; Egner, P.A.; Sell, S. Glutathione-S-transferase A3 knockout mice are sensitive to acute cytotoxic and genotoxic effects of aflatoxin B1. Toxicol. Appl. Pharmacol. 2010, 242, 241–246. [Google Scholar] [CrossRef]
- Dvorackova, I. Aflatoxins & Human Health; Taylor & Francis: Boca Raton, Fl, USA, 1989. [Google Scholar]
- Wild, C.P.; Gong, Y.Y. Mycotoxins and human disease: A largely ignored global health issue. Carcinogenesis 2010, 31, 71–82. [Google Scholar] [CrossRef]
- Mally, A.; Zepnik, H.; Wanek, P.; Eder, E.; Dingley, K.; Ihmels, H.; Volkel, W.; Dekant, W. Ochratoxin A: Lack of formation of covalent DNA adducts. Chem. Res. Toxicol. 2004, 17, 234–242. [Google Scholar] [CrossRef]
- Turesky, R.J. Perspective: Ochratoxin A is not a genotoxic carcinogen. Chem. Res. Toxicol. 2005, 18, 1082–1090. [Google Scholar] [CrossRef]
- Hope, J.H.; Hope, B.E. A review of the diagnosis and treatment of ochratoxin a inhalational exposure associated with human illness and kidney disease including focal segmental glomerulosclerosis. J. Environ. Public Health 2012, 2012, 835059. [Google Scholar]
- Brewer, J.H.; Thrasher, J.D.; Straus, D.C.; Madison, R.A.; Hooper, D. Detection of mycotoxins in patients with chronic fatigue syndrome. Toxins 2013, 5, 605–617. [Google Scholar]
- Gautier, J.C.; Holzhaeuser, D.; Markovic, J.; Gremaud, E.; Schilter, B.; Turesky, R.J. Oxidative damage and stress response from ochratoxin a exposure in rats. Free Radic. Biol. Med. 2001, 30, 1089–1098. [Google Scholar] [CrossRef]
- Petrik, J.; Zanic-Grubisic, T.; Barisic, K.; Pepeljnjak, S.; Radic, B.; Ferencic, Z.; Cepelak, I. Apoptosis and oxidative stress induced by ochratoxin a in rat kidney. Arch. Toxicol. 2003, 77, 685–693. [Google Scholar] [CrossRef]
- Schilter, B.; Marin-Kuan, M.; Delatour, T.; Nestler, S.; Mantle, P.; Cavin, C. Ochratoxin A: Potential epigenetic mechanisms of toxicity and carcinogenicity. Food Addit. Contam. 2005, 22, 88–93. [Google Scholar] [CrossRef]
- Kamp, H.G.; Eisenbrand, G.; Janzowski, C.; Kiossev, J.; Latendresse, J.R.; Schlatter, J.; Turesky, R.J. Ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Mol. Nutr. Food Res. 2005, 49, 1160–1167. [Google Scholar] [CrossRef]
- Omar, R.F.; Hasinoff, B.B.; Mejilla, F.; Rahimtula, A.D. Mechanism of ochratoxin A stimulated lipid peroxidation. Biochem. Pharmacol. 1990, 40, 1183–1191. [Google Scholar] [CrossRef]
- Sauvant, C.; Holzinger, H.; Gekle, M. The nephrotoxin ochratoxin A induces key parameters of chronic interstitial nephropathy in renal proximal tubular cells. Cell. Physiol. Biochem. 2005, 15, 125–134. [Google Scholar] [CrossRef]
- Schaaf, G.J.; Nijmeijer, S.M.; Maas, R.F.; Roestenberg, P.; de Groene, E.M.; Fink-Gremmels, J. The role of oxidative stress in the ochratoxin A—Mediated toxicity in proximal tubular cells. Biochim. Biophys. Acta 2002, 1588, 149–158. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as a molecular mechanism of ochratoxin a carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar]
- Marin-Kuan, M.; Ehrlich, V.; Delatour, T.; Cavin, C.; Schilter, B. Evidence for a role of oxidative stress in the carcinogenicity of ochratoxin A. J. Toxicol. 2011, 2011, 645361. [Google Scholar]
- Alpsoy, L.; Yildirim, A.; Agar, G. The antioxidant effects of vitamin A, C, and E on aflatoxin B1-induced oxidative stress in human lymphocytes. Toxicol. Ind. Health 2009, 25, 121–127. [Google Scholar]
- Zhang, L.; Ye, Y.; An, Y.; Tian, Y.; Wang, Y.; Tang, H. Systems responses of rats to aflatoxin B1 exposure revealed with metabonomic changes in multiple biological matrices. J. Proteome Res. 2010, 10, 614–623. [Google Scholar]
- Akcam, M.; Artan, R.; Yilmaz, A.; Ozdem, S.; Gelen, T.; Naziroglu, M. Caffeic acid phenethyl ester modulates aflatoxin B1-induced hepatotoxicity in rats. Cell Biochem. Funct. 2013, 31, 692–697. [Google Scholar]
- Gerschman, R.; Gilbert, D.; Nye, S.W.; Dwyer, P.; Fenn, W.O. Oxygen poisoning and x-irradiation: A mechanism in common. Science 1954, 119, 623–626. [Google Scholar]
- Commoner, B.; Townsend, J.; Pake, G.E. Free radicals in biological materials. Nature 1954, 174, 689–691. [Google Scholar]
- Lavoisier, A.L. Traité élémentaire de chimie: Présenté dans un ordre nouveau et d’après les découvertes modernes; Cuchet: Paris, France, 1789. [Google Scholar]
- Weissmann, G. Free radicals can kill you: Lavoisier’s oxygen revolution. FASEB J. 2010, 24, 649–652. [Google Scholar]
- Fitzpatrick, A.M.; Jones, D.P.; Brown, L.A. Glutathione redox control of asthma: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2012, 17, 375–408. [Google Scholar]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar]
- Zhang, H.; Forman, H.J.; Choi, J. Gamma-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005, 401, 468–483. [Google Scholar] [CrossRef]
- Hanigan, M.H. Gamma-glutamyl transpeptidase, a glutathionase: Its expression and function in carcinogenesis. Chem. Biol. Interact. 1998, 111–112, 333–342. [Google Scholar]
- Heisterkamp, N.; Groffen, J.; Warburton, D.; Sneddon, T.P. The human gamma-glutamyltransferase gene family. Hum. Genet. 2008, 123, 321–332. [Google Scholar]
- Fitzpatrick, A.M.; Stephenson, S.T.; Hadley, G.R.; Burwell, L.; Penugonda, M.; Simon, D.M.; Hansen, J.; Jones, D.P.; Brown, L.A. Thiol redox disturbances in children with severe asthma are associated with posttranslational modification of the transcription factor nuclear factor (erythroid-derived 2)-like 2. J. Allergy Clin. Immunol. 2011, 127, 1604–1611. [Google Scholar] [CrossRef]
- Morris, D.; Guerra, C.; Khurasany, M.; Guilford, F.; Saviola, B.; Huang, Y.; Venketaraman, V. Glutathione supplementation improves macrophage functions in HIV. J. Interferon Cytokine Res. 2013, 33, 270–279. [Google Scholar] [CrossRef]
- Hayes, J.D.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef]
- Reddy, N.M.; Kleeberger, S.R.; Yamamoto, M.; Kensler, T.W.; Scollick, C.; Biswal, S.; Reddy, S.P. Genetic dissection of the Nrf2-dependent redox signaling-regulated transcriptional programs of cell proliferation and cytoprotection. Physiol. Genomics 2007, 32, 74–81. [Google Scholar] [CrossRef]
- Comhair, S.A.; Erzurum, S.C. Redox control of asthma: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 93–124. [Google Scholar]
- Fitzpatrick, A.M.; Holguin, F.; Teague, W.G.; Brown, L.A. Alveolar macrophage phagocytosis is impaired in children with poorly controlled asthma. J. Allergy Clin. Immunol. 2008, 121, 1372–1378. [Google Scholar]
- Bishayee, A.; Bhatia, D.; Thoppil, R.J.; Darvesh, A.S.; Nevo, E.; Lansky, E.P. Pomegranate-mediated chemoprevention of experimental hepatocarcinogenesis involves Nrf2-regulated antioxidant mechanisms. Carcinogenesis 2011, 32, 888–896. [Google Scholar]
- Morris, D.; Guerra, C.; Donohue, C.; Oh, H.; Khurasany, M.; Venketaraman, V. Unveiling the mechanisms for decreased glutathione in individuals with HIV infection. Clin. Dev. Immunol. 2012, 2012, 734125. [Google Scholar]
- Bakin, A.V.; Stourman, N.V.; Sekhar, K.R.; Rinehart, C.; Yan, X.; Meredith, M.J.; Arteaga, C.L.; Freeman, M.L. Smad3-ATF3 signaling mediates TGF-beta suppression of genes encoding Phase II detoxifying proteins. Free Radic. Biol. Med. 2005, 38, 375–387. [Google Scholar] [CrossRef]
- Franklin, C.C.; Rosenfeld-Franklin, M.E.; White, C.; Kavanagh, T.J.; Fausto, N. TGFβ1-induced suppression of glutathione antioxidant defenses in hepatocytes: Caspase-dependent post-translational and caspase-independent transcriptional regulatory mechanisms. FASEB J. 2003, 17, 1535–1537. [Google Scholar]
- Liu, R.M.; Gaston Pravia, K.A. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef]
- Armstrong, R.N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997, 10, 2–18. [Google Scholar] [CrossRef]
- Eaton, D.L.; Bammler, T.K. Concise review of the glutathione S-transferases and their significance to toxicology. Toxicol. Sci. 1999, 49, 156–164. [Google Scholar]
- Yin, Z.; Ivanov, V.N.; Habelhah, H.; Tew, K.; Ronai, Z. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res. 2000, 60, 4053–4057. [Google Scholar]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar]
- Romieu, I.; Sienra-Monge, J.J.; Ramirez-Aguilar, M.; Moreno-Macias, H.; Reyes-Ruiz, N.I.; Estela del Rio-Navarro, B.; Hernandez-Avila, M.; London, S.J. Genetic polymorphism of GSTM1 and antioxidant supplementation influence lung function in relation to ozone exposure in asthmatic children in Mexico City. Thorax 2004, 59, 8–10. [Google Scholar]
- Gross-Steinmeyer, K.; Stapleton, P.L.; Tracy, J.H.; Bammler, T.K.; Strom, S.C.; Eaton, D.L. Sulforaphane- and phenethyl isothiocyanate-induced inhibition of aflatoxin B1-mediated genotoxicity in human hepatocytes: Role of GSTM1 genotype and CYP3A4 gene expression. Toxicol. Sci. 2010, 116, 422–432. [Google Scholar] [CrossRef]
- Wang, J.; Deng, Y.; Cheng, J.; Ding, J.; Tokudome, S. GST genetic polymorphisms and lung adenocarcinoma susceptibility in a Chinese population. Cancer Lett. 2003, 201, 185–193. [Google Scholar]
- Deng, Z.L.; Wei, Y.P.; Ma, Y. Polymorphism of glutathione S-transferase mu 1 and theta 1 genes and hepatocellular carcinoma in southern Guangxi, China. World J. Gastroenterol. 2005, 11, 272–274. [Google Scholar]
- Chen, S.Y.; Chen, C.J.; Tsai, W.Y.; Ahsan, H.; Liu, T.Y.; Lin, J.T.; Santella, R.M. Associations of plasma aflatoxin B1-albumin adduct level with plasma selenium level and genetic polymorphisms of glutathione S-transferase M1 and T1. Nutr. Cancer 2000, 38, 179–185. [Google Scholar] [CrossRef]
- Sun, C.A.; Wang, L.Y.; Chen, C.J.; Lu, S.N.; You, S.L.; Wang, L.W.; Wang, Q.; Wu, D.M.; Santella, R.M. Genetic polymorphisms of glutathione S-transferases M1 and T1 associated with susceptibility to aflatoxin-related hepatocarcinogenesis among chronic hepatitis B carriers: A nested case-control study in Taiwan. Carcinogenesis 2001, 22, 1289–1294. [Google Scholar] [CrossRef]
- Piacentini, S.; Polimanti, R.; Squitti, R.; Ventriglia, M.; Cassetta, E.; Vernieri, F.; Rossini, P.M.; Manfellotto, D.; Fuciarelli, M. GSTM1 null genotype as risk factor for late-onset Alzheimer’s disease in italian patients. J. Neurol. Sci. 2012, 317, 137–140. [Google Scholar] [CrossRef]
- Doi, K.; Uetsuka, K. Mechanisms of mycotoxin-induced neurotoxicity through oxidative stress-associated pathways. Int .J. Mol. Sci. 2011, 12, 5213–5237. [Google Scholar] [CrossRef]
- Boorman, G.A. National Toxicology Program. Toxicology and carcinogenesis studies of ochratoxin A (CAS No. 303-47-9) in F344/N rats (gavage studies). Natl. Toxicol. Program Tech. Rep. Ser. 1989, 358, 1–142. [Google Scholar]
- Dietrich, D.R.; Heussner, A.H.; O’Brien, E. Ochratoxin A: Comparative pharmacokinetics and toxicological implications (experimental and domestic animals and humans). Food Addit. Contam. 2005, 22, 45–52. [Google Scholar] [CrossRef]
- Dirheimer, G.; Creppy, E.E. Mechanism of action of ochratoxin A. IARC Sci. Publ. 1991, 171–186. [Google Scholar]
- Gekle, M.; Sauvant, C.; Schwerdt, G. Ochratoxin A at nanomolar concentrations: A signal modulator in renal cells. Mol. Nutr. Food Res. 2005, 49, 118–130. [Google Scholar] [CrossRef]
- Marin-Kuan, M.; Nestler, S.; Verguet, C.; Bezencon, C.; Piguet, D.; Mansourian, R.; Holzwarth, J.; Grigorov, M.; Delatour, T.; Mantle, P.; et al. A toxicogenomics approach to identify new plausible epigenetic mechanisms of ochratoxin A carcinogenicity in rat. Toxicol. Sci. 2006, 89, 120–134. [Google Scholar]
- Huang, Q.; Dunn, R.T., 2nd; Jayadev, S.; DiSorbo, O.; Pack, F.D.; Farr, S.B.; Stoll, R.E.; Blanchard, K.T. Assessment of cisplatin-induced nephrotoxicity by microarray technology. Toxicol. Sci. 2001, 63, 196–207. [Google Scholar] [CrossRef]
- Misawa, H.; Yamaguchi, M. Involvement of nuclear factor-1 (NF1) binding motif in the regucalcin gene expression of rat kidney cortex: The expression is suppressed by cisplatin administration. Mol. Cell. Biochem. 2001, 219, 29–37. [Google Scholar] [CrossRef]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2—Are pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef]
- Schwerdt, G.; Bauer, K.; Gekle, M.; Silbernagl, S. Accumulation of ochratoxin A in rat kidney in vivo and in cultivated renal epithelial cells in vitro. Toxicology 1996, 114, 177–185. [Google Scholar] [CrossRef]
- Zepnik, H.; Volkel, W.; Dekant, W. Toxicokinetics of the mycotoxin ochratoxin A in F 344 rats after oral administration. Toxicol. Appl. Pharmacol. 2003, 192, 36–44. [Google Scholar] [CrossRef]
- Cavin, C.; Delatour, T.; Marin-Kuan, M.; Holzhauser, D.; Higgins, L.; Bezencon, C.; Guignard, G.; Junod, S.; Richoz-Payot, J.; Gremaud, E.; et al. Reduction in antioxidant defenses may contribute to ochratoxin a toxicity and carcinogenicity. Toxicol. Sci. 2007, 96, 30–39. [Google Scholar]
- Bartsch, H.; Nair, J. Oxidative stress and lipid peroxidation-derived DNA-lesions in inflammation driven carcinogenesis. Cancer Detect. Prev. 2004, 28, 385–391. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Kong, A.N. Anti-oxidative stress regulator NF-E2-related factor 2 mediates the adaptive induction of antioxidant and detoxifying enzymes by lipid peroxidation metabolite 4-hydroxynonenal. Cell Biosci. 2012, 2, 40. [Google Scholar] [CrossRef]
- Boesch-Saadatmandi, C.; Wagner, A.E.; Graeser, A.C.; Hundhausen, C.; Wolffram, S.; Rimbach, G. Ochratoxin A impairs Nrf2-dependent gene expression in porcine kidney tubulus cells. J. Anim. Physiol. Anim. Nutr. 2009, 93, 547–554. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef]
- Chaudhary, M.; Rao, P.V. Brain oxidative stress after dermal and subcutaneous exposure of T-2 toxin in mice. Food Chem. Toxicol. 2010, 48, 3436–3442. [Google Scholar] [CrossRef]
- Meissonnier, G.M.; Pinton, P.; Laffitte, J.; Cossalter, A.M.; Gong, Y.Y.; Wild, C.P.; Bertin, G.; Galtier, P.; Oswald, I.P. Immunotoxicity of aflatoxin B1: Impairment of the cell-mediated response to vaccine antigen and modulation of cytokine expression. Toxicol. Appl. Pharmacol. 2008, 231, 142–149. [Google Scholar] [CrossRef]
- Seeboth, J.; Solinhac, R.; Oswald, I.P.; Guzylack-Piriou, L. The fungal T-2 toxin alters the activation of primary macrophages induced by TLR-agonists resulting in a decrease of the inflammatory response in the pig. Vet. Res. 2012, 43, 35. [Google Scholar] [CrossRef]
- Mary, V.S.; Theumer, M.G.; Arias, S.L.; Rubinstein, H.R. Reactive oxygen species sources and biomolecular oxidative damage induced by aflatoxin B1 and fumonisin B1 in rat spleen mononuclear cells. Toxicology 2012, 302, 299–307. [Google Scholar] [CrossRef]
- Theumer, M.G.; Canepa, M.C.; Lopez, A.G.; Mary, V.S.; Dambolena, J.S.; Rubinstein, H.R. Subchronic mycotoxicoses in Wistar rats: Assessment of the in vivo and in vitro genotoxicity induced by fumonisins and aflatoxin b(1), and oxidative stress biomarkers status. Toxicology 2010, 268, 104–110. [Google Scholar] [CrossRef]
- Grenier, B.; Loureiro-Bracarense, A.P.; Lucioli, J.; Pacheco, G.D.; Cossalter, A.M.; Moll, W.D.; Schatzmayr, G.; Oswald, I.P. Individual and combined effects of subclinical doses of deoxynivalenol and fumonisins in piglets. Mol. Nutr. Food Res. 2011, 55, 761–771. [Google Scholar]
- Schutze, N.; Lehmann, I.; Bonisch, U.; Simon, J.C.; Polte, T. Exposure to mycotoxins increases the allergic immune response in a murine asthma model. Am. J. Respir. Crit. Care Med. 2010, 181, 1188–1199. [Google Scholar] [CrossRef]
- Wichmann, G.; Herbarth, O.; Lehmann, I. The mycotoxins citrinin, gliotoxin, and patulin affect interferon-gamma rather than interleukin-4 production in human blood cells. Environ. Toxicol. 2002, 17, 211–218. [Google Scholar] [CrossRef]
- Luft, P.; Oostingh, G.J.; Gruijthuijsen, Y.; Horejs-Hoeck, J.; Lehmann, I.; Duschl, A. Patulin influences the expression of Th1/Th2 cytokines by activated peripheral blood mononuclear cells and T cells through depletion of intracellular glutathione. Environ. Toxicol. 2008, 23, 84–95. [Google Scholar] [CrossRef]
- Johannessen, L.N.; Nilsen, A.M.; Lovik, M. Mycotoxin-induced depletion of intracellular glutathione and altered cytokine production in the human alveolar epithelial cell line A549. Toxicol. Lett. 2007, 168, 103–112. [Google Scholar]
- Peterson, J.D.; Herzenberg, L.A.; Vasquez, K.; Waltenbaugh, C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 3071–3076. [Google Scholar] [CrossRef]
- Murata, Y.; Ohteki, T.; Koyasu, S.; Hamuro, J. IFN-γand pro-inflammatory cytokine production by antigen-presenting cells is dictated by intracellular thiol redox status regulated by oxygen tension. Eur. J. Immunol. 2002, 32, 2866–2873. [Google Scholar] [CrossRef]
- Fitzpatrick, A.M.; Teague, W.G.; Burwell, L.; Brown, M.S.; Brown, L.A. Glutathione oxidation is associated with airway macrophage functional impairment in children with severe asthma. Pediatr. Res. 2011, 69, 154–159. [Google Scholar] [CrossRef]
- Morris, D.; Guerra, C.; Khurasany, M.; Guilford, F.; Saviola, B.; Huang, Y.; Venketaraman, V. Glutathione supplementation improves macrophage functions in hiv. J. Interferon Cytokine Res. 2013, 33, 270–279. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar]
- Novi, A.M.; Florke, R.; Stukenkemper, M. Glutathione and aflatoxin-B1-induced liver tumors: Requirement for an intact glutathione molecule for regression of malignancy in neoplastic tissue. Ann. N. Y. Acad. Sci. 1982, 397, 62–71. [Google Scholar] [CrossRef]
- Hope, J. A review of the mechanism of injury and treatment approaches for illness resulting from exposure to water-damaged buildings, mold, and mycotoxins. Sci. World J. 2013. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guilford, F.T.; Hope, J. Deficient Glutathione in the Pathophysiology of Mycotoxin-Related Illness. Toxins 2014, 6, 608-623. https://doi.org/10.3390/toxins6020608
Guilford FT, Hope J. Deficient Glutathione in the Pathophysiology of Mycotoxin-Related Illness. Toxins. 2014; 6(2):608-623. https://doi.org/10.3390/toxins6020608
Chicago/Turabian StyleGuilford, Frederick T., and Janette Hope. 2014. "Deficient Glutathione in the Pathophysiology of Mycotoxin-Related Illness" Toxins 6, no. 2: 608-623. https://doi.org/10.3390/toxins6020608
APA StyleGuilford, F. T., & Hope, J. (2014). Deficient Glutathione in the Pathophysiology of Mycotoxin-Related Illness. Toxins, 6(2), 608-623. https://doi.org/10.3390/toxins6020608