Regulating Toxin-Antitoxin Expression: Controlled Detonation of Intracellular Molecular Timebombs

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: A Brief Overview of Toxin-Antitoxin Complexes
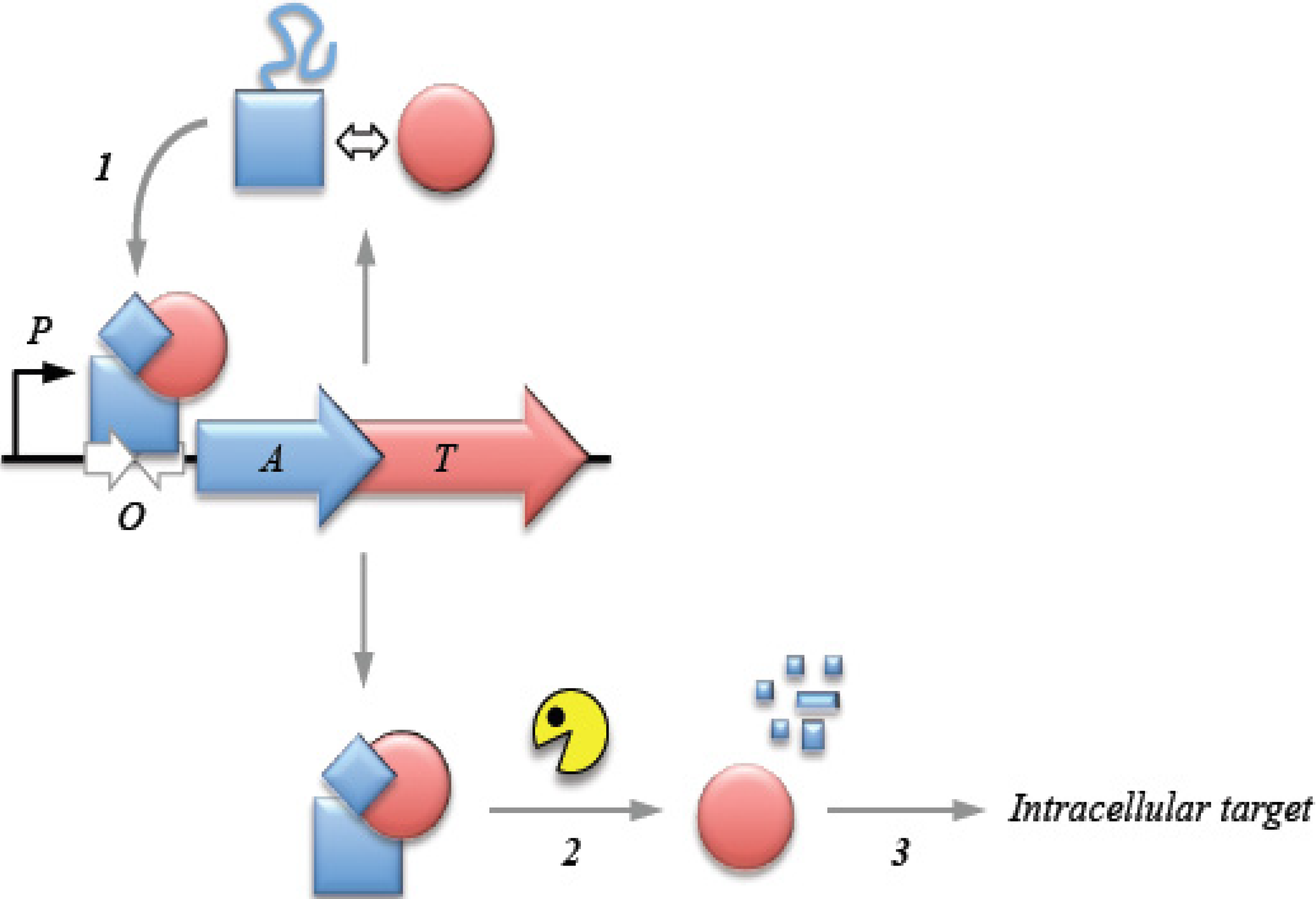
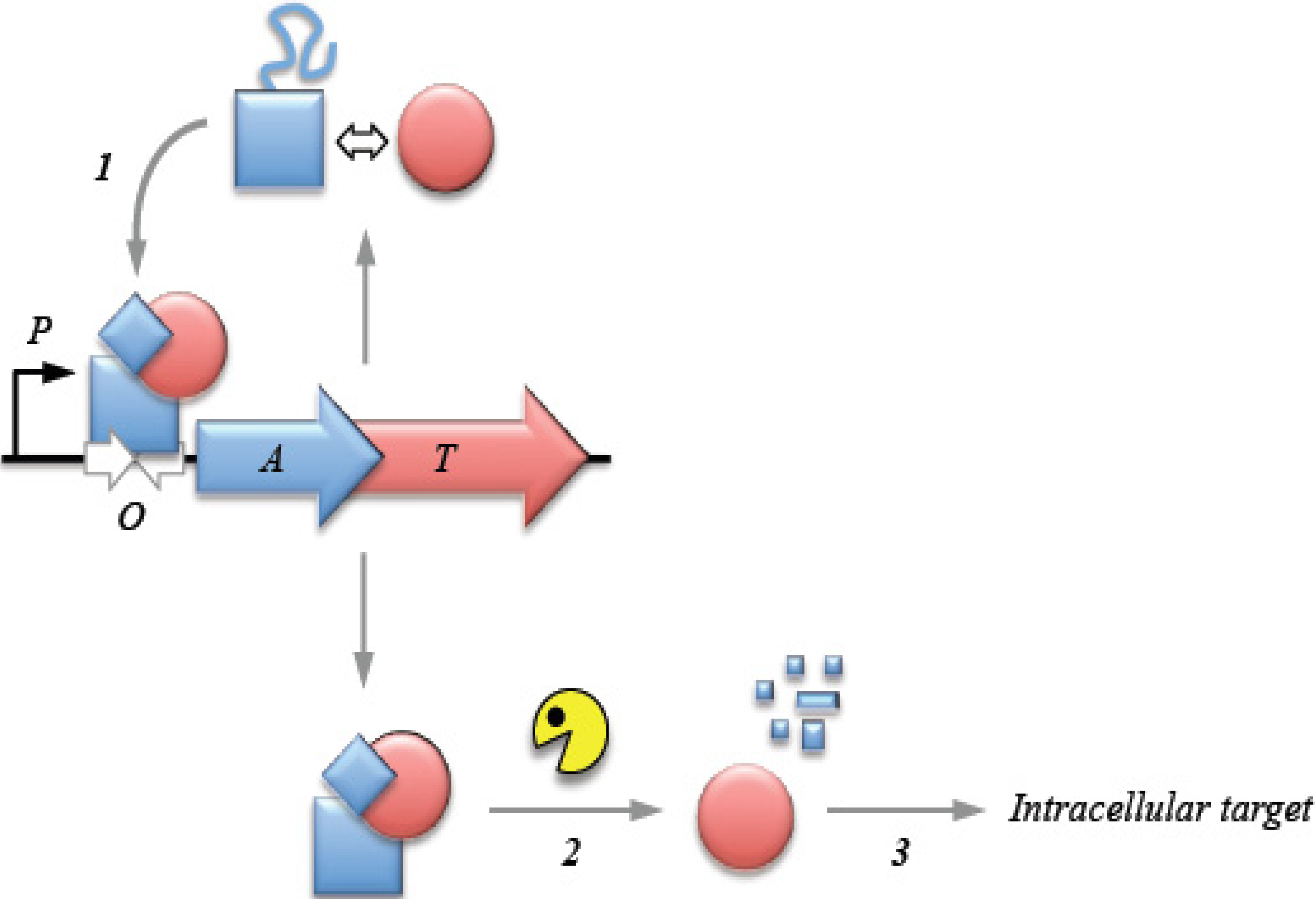
2. Transcriptional Autoregulation Is a Characteristic Feature of Type II TA Cassettes
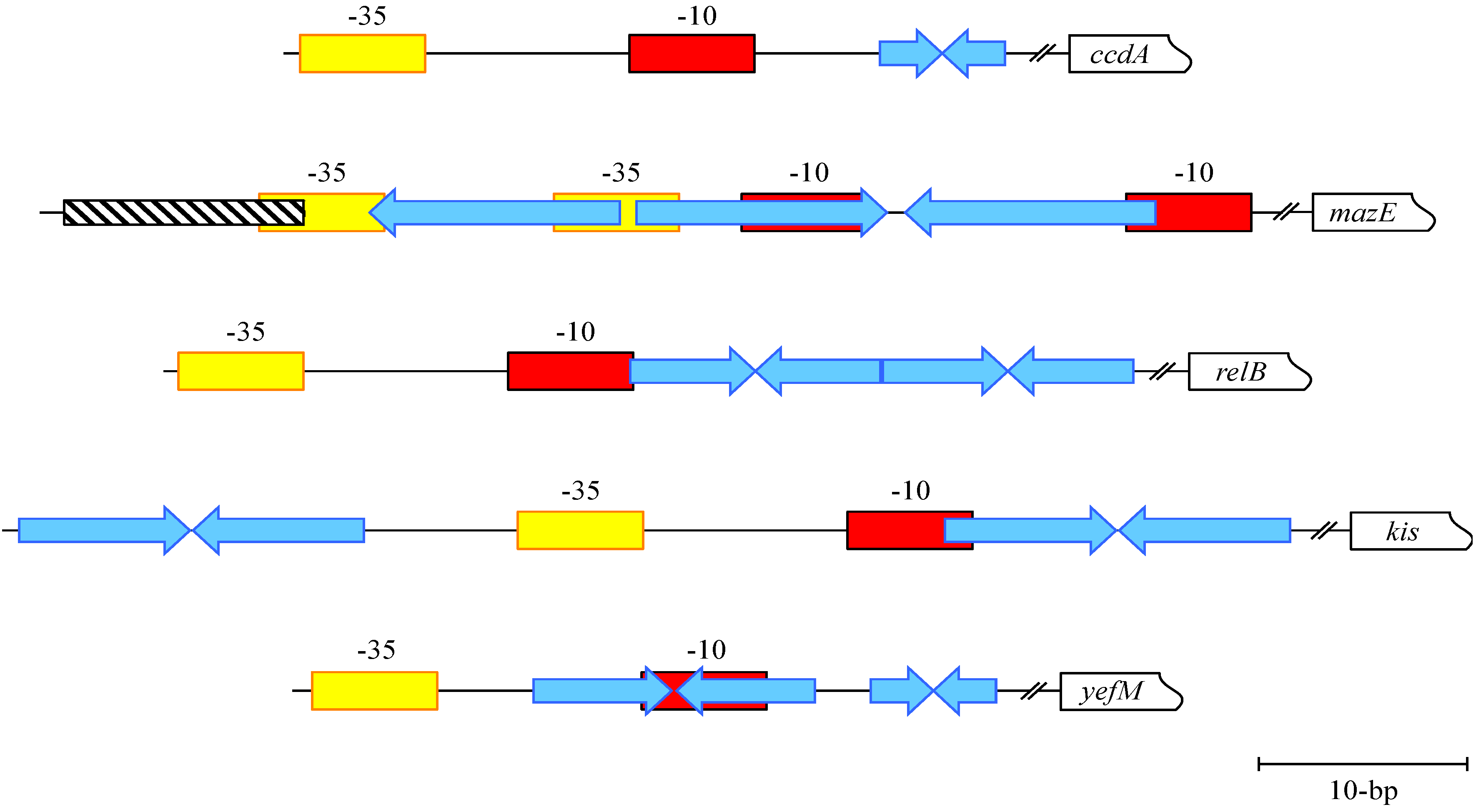
3. CcdA-CcdB: Transcriptional Repression of a DNA Gyrase Poison
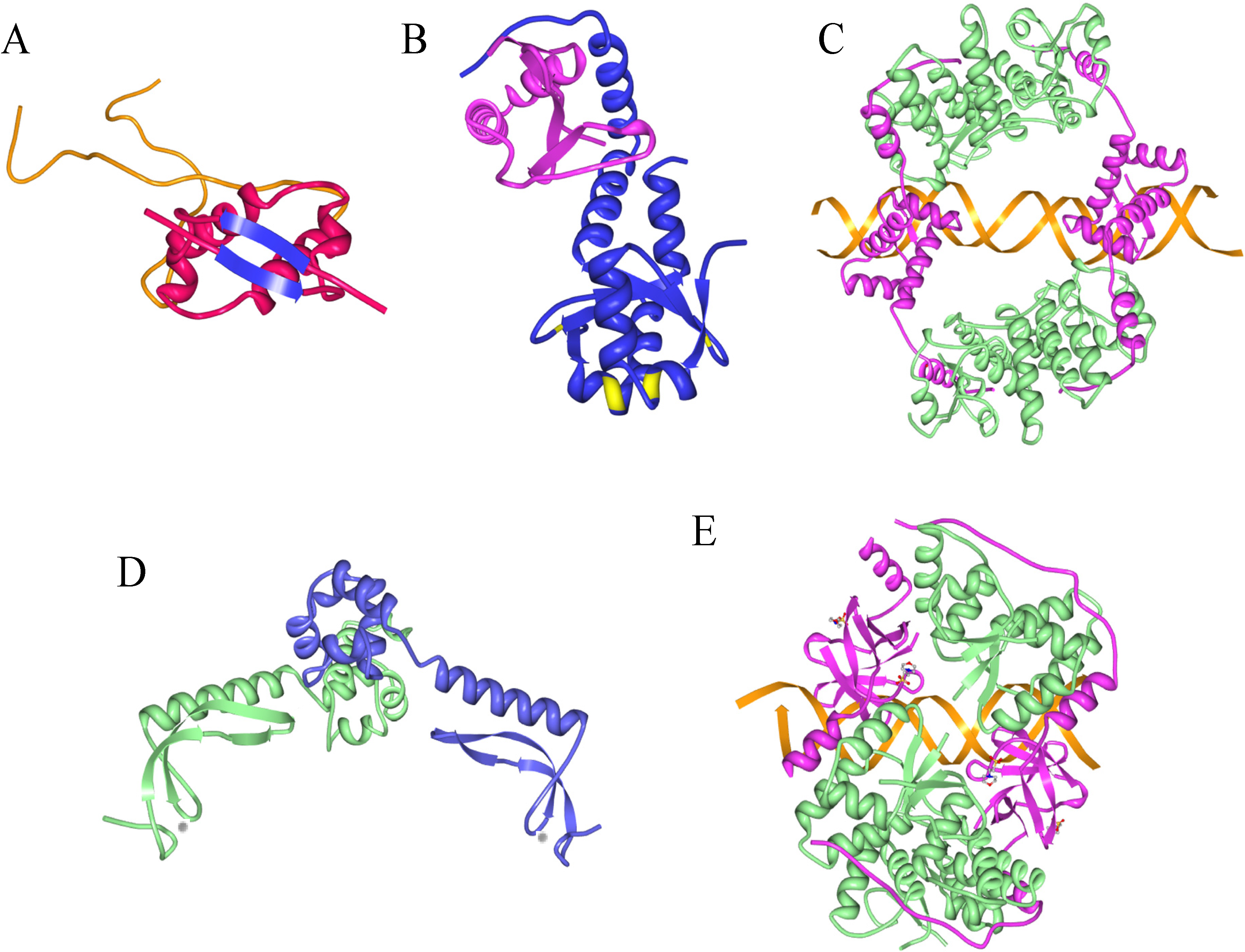
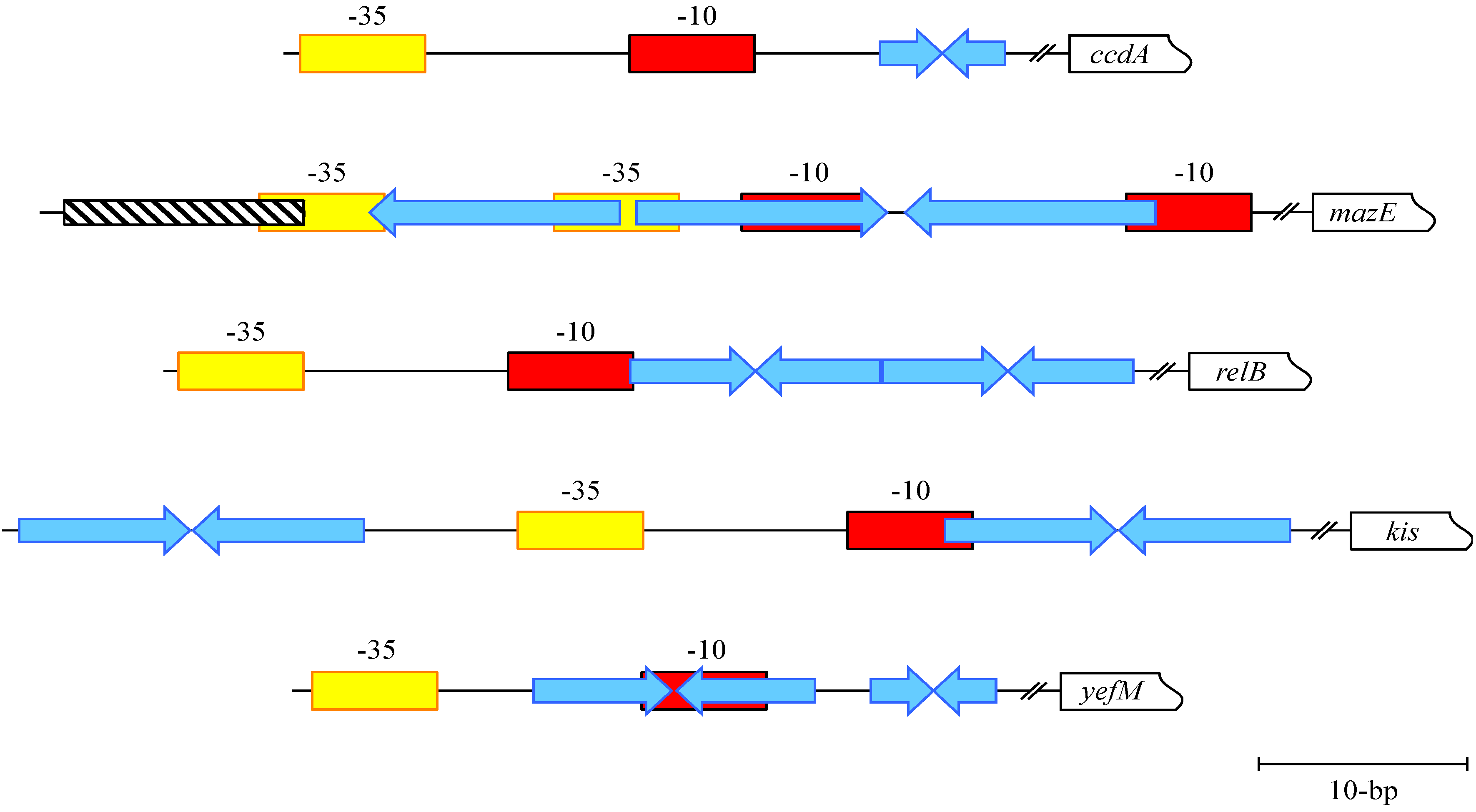
4. MazE-MazF: Autoregulation of a Chromosomally Encoded Type II Paradigm
5. Phd-Doc, RelB-RelE and Kis-Kid: Transcriptional Regulation and Conditional Cooperativity
6. YefM-YoeB and Axe-Txe: Diverse Transcriptional Control of Homologous TA Complexes
7. Diverging from the Canonical Pattern of Type II TA Transcriptional Control: Tripartite Protein Complexes and a Toxin that Directly Binds DNA
8. Type II TAs that Regulate Other Genes or Do Not Display Autoregulation
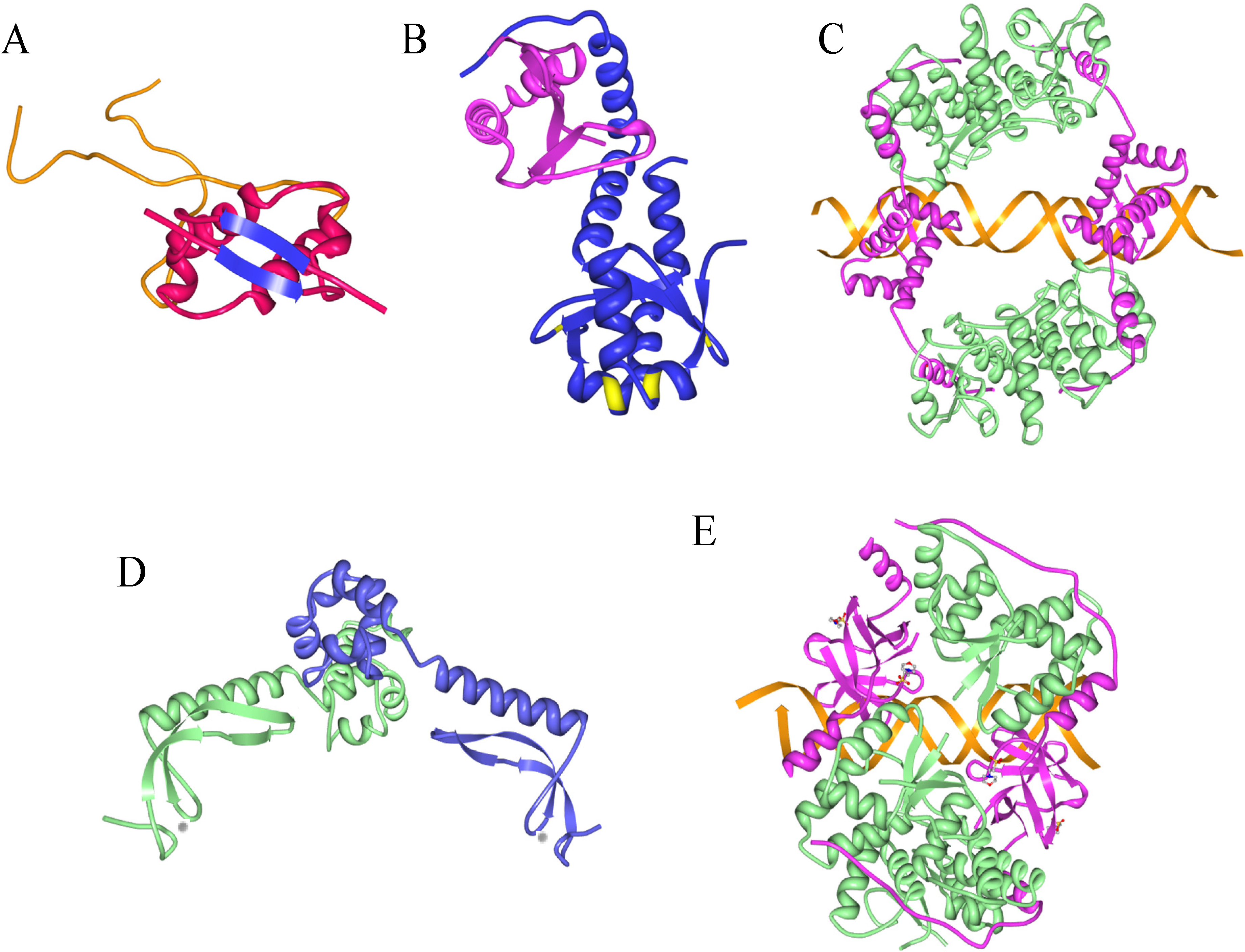
9. Information from Toxin-Antitoxin-DNA Costructures
10. New Antibiotics that Interfere with TA Transcriptional Autoregulation to Detonate Toxins Artificially
Acknowledgments
Conflicts of Interest
References
- Blower, T.R.; Salmond, G.P.; Luisi, B.F. Balancing at survival’s edge: The structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr. Opin. Struct. Biol. 2011, 21, 109–118. [Google Scholar] [CrossRef]
- Fozo, E.M.; Makarova, K.S.; Shabalina, S.A.; Yutin, N.; Koonin, E.V.; Storz, G. Abundance of type I toxin-antitoxin systems in bacteria: Searches for new candidates and discovery of novel families. Nucleic Acids Res. 2010, 38, 3743–3759. [Google Scholar] [CrossRef]
- Hayes, F.; van Melderen, L. Toxins-antitoxins: Diversity, evolution and function. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 386–408. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 2009, 4, 19. [Google Scholar] [CrossRef]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in freeliving but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Inouye, M. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol. 2011, 9, 779–790. [Google Scholar] [CrossRef]
- Hayes, F. Toxins-antitoxins: Plasmid maintenance, programmed cell death, and cell cycle arrest. Science 2003, 301, 1496–1499. [Google Scholar] [CrossRef]
- Saavedra De Bast, M.; Mine, N.; van Melderen, L. Chromosomal toxin-antitoxin systems may act as anti-addiction modules. J. Bacteriol. 2008, 190, 4603–4609. [Google Scholar] [CrossRef]
- Van Melderen, L. Toxin-antitoxin systems: Why so many, what for? Curr. Opin. Microbiol. 2010, 13, 781–785. [Google Scholar] [CrossRef]
- Van Melderen, L.; Saavedra De Bast, M. Bacterial toxin-antitoxin systems: More than selfish entities? PLoS Genet. 2009, 5, e000437. [Google Scholar]
- Gerdes, K.; Maisonneuve, E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012, 66, 103–123. [Google Scholar] [CrossRef]
- Magnuson, R.D. Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 2007, 189, 6089–6092. [Google Scholar] [CrossRef]
- Wang, X.; Wood, T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef]
- Schuster, C.F.; Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 2013, 340, 73–85. [Google Scholar] [CrossRef]
- Mruk, I.; Kobayashi, I. To be or not to be: Regulation of restriction-modification systems and other toxin-antitoxin systems. Nucleic Acids Res. 2014, 42, 70–86. [Google Scholar] [CrossRef]
- Fozo, E.M.; Hemm, M.R.; Storz, G. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 2008, 72, 579–589. [Google Scholar] [CrossRef]
- Gerdes, K.; Wagner, E. RNA antitoxins. Curr. Opin. Microbiol. 2007, 10, 117–124. [Google Scholar] [CrossRef]
- Darfeuille, F.; Unoson, C.; Vogel, J.; Wagner, E.G. An antisense RNA inhibits translation by competing with standby ribosomes. Mol. Cell 2007, 26, 381–392. [Google Scholar] [CrossRef]
- Blower, T.R.; Short, F.L.; Rao, F.; Mizuguchi, K.; Pei, X.Y.; Fineran, P.C.; Luisi, B.F.; Salmond, G.P. Identification and classification of bacterial type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012, 40, 6158–6173. [Google Scholar] [CrossRef]
- Masuda, H.; Tan, Q.; Awano, N.; Wu, K.P.; Inouye, M. YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli. Mol. Microbiol. 2012, 84, 979–989. [Google Scholar] [CrossRef]
- Tan, Q.; Awano, N.; Inouye, M. YeeV is an Escherichia coli toxin that inhibits cell division by targeting the cytoskeleton proteins, FtsZ and MreB. Mol. Microbiol. 2011, 79, 109–118. [Google Scholar] [CrossRef]
- Wang, X.; Lord, D.M.; Cheng, H.Y.; Osbourne, D.O.; Hong, S.H.; Sanchez-Torres, V.; Quiroga, C.; Zheng, K.; Herrmann, T.; Peti, W.; et al. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 2012, 8, 855–861. [Google Scholar] [CrossRef]
- Cook, G.M.; Robson, J.R.; Frampton, R.A.; McKenzie, J.; Przybilski, R.; Fineran, P.C.; Arcus, V.L. Ribonucleases in bacterial toxin-antitoxin systems. Biochim. Biophys. Acta 2013, 1829, 523–531. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Prog. Mol. Biol. Transl. Sci. 2009, 85, 467–500. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Y.; Inouye, M.; Woychik, N.A. Bacterial addiction module toxin Doc inhibits translation elongation through its association with the 30S ribosomal subunit. Proc. Natl. Acad. Sci. USA 2008, 105, 5885–5890. [Google Scholar]
- Schumacher, M.A.; Piro, K.M.; Xu, W.; Hansen, S.; Lewis, K.; Brennan, R.G. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 2009, 323, 396–401. [Google Scholar] [CrossRef]
- Winther, K.S.; Gerdes, K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. USA 2011, 108, 7403–7407. [Google Scholar] [CrossRef]
- Bernard, P.; Couturier, M. Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J. Mol. Biol. 1992, 226, 735–745. [Google Scholar] [CrossRef]
- Jiang, Y.; Pogliano, J.; Helinski, D.R.; Konieczny, I. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 2002, 44, 971–979. [Google Scholar] [CrossRef]
- Yuan, J.; Sterckx, Y.; Mitchenall, L.A.; Maxwell, A.; Loris, R.; Waldor, M.K. Vibrio cholerae ParE2 poisons DNA gyrase via a mechanism distinct from other gyrase inhibitors. J. Biol. Chem. 2010, 285, 40397–40408. [Google Scholar]
- Aakre, C.D.; Phung, T.N.; Huang, D.; Laub, M.T. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the β sliding clamp. Mol. Cell 2013, 52, 617–628. [Google Scholar] [CrossRef]
- Mutschler, H.; Gebhardt, M.; Shoeman, R.L.; Meinhart, A. A novel mechanism of programmed cell death in bacteria by toxin-antitoxin systems corrupts peptidoglycan synthesis. PLoS Biol. 2011, 9, e1001033. [Google Scholar] [CrossRef]
- Ogura, T.; Hiraga, S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. USA 1983, 80, 4784–4788. [Google Scholar] [CrossRef]
- Guérout, A.M.; Iqbal, N.; Mine, N.; Ducos-Galand, M.; van Melderen, L.; Mazel, D. Characterization of the phd-doc and ccd toxin-antitoxin cassettes from Vibrio superintegrons. J. Bacteriol. 2013, 195, 2270–2283. [Google Scholar] [CrossRef]
- Smith, A.B.; Maxwell, A. A strand-passage conformation of DNA gyrase is required to allow the bacterial toxin, CcdB, to access its binding site. Nucleic Acids Res. 2006, 34, 4667–4676. [Google Scholar] [CrossRef]
- De Jonge, N.; Garcia-Pino, A.; Buts, L.; Haesaerts, S.; Charlier, D.; Zangger, K.; Wyns, L.; de Greve, H.; Loris, R. Rejuvenation of CcdB-poisoned gyrase by an intrinsically disordered protein domain. Mol. Cell 2009, 35, 154–163. [Google Scholar] [CrossRef]
- Afif, H.; Allali, N.; Couturier, M.; van Melderen, L. The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison-antidote system. Mol. Microbiol. 2001, 41, 73–82. [Google Scholar] [CrossRef]
- De Feyter, R.; Wallace, C.; Lane, D. Autoregulation of the ccd operon in the F plasmid. Mol. Gen. Genet. 1989, 218, 481–486. [Google Scholar] [CrossRef]
- Tam, J.E.; Kline, B.C. Control of the ccd operon in plasmid F. J. Bacteriol. 1989, 171, 2353–2360. [Google Scholar]
- Tam, J.E.; Kline, B.C. The F plasmid ccd autorepressor is a complex of CcdA and CcdB proteins. Mol. Gen. Genet. 1989, 219, 26–32. [Google Scholar]
- Dao-Thi, M.H.; Charlier, D.; Loris, R.; Maes, D.; Messens, J.; Wyns, L.; Backmann, J. Intricate interactions within the ccd plasmid addiction system. J. Biol. Chem. 2002, 277, 3733–3742. [Google Scholar]
- Salmon, M.A.; van Melderen, L.; Bernard, P.; Couturier, M. The antidote and autoregulatory functions of the F plasmid CcdA protein: A genetic and biochemical survey. Mol. Gen. Genet. 1994, 244, 530–538. [Google Scholar] [CrossRef]
- Schreiter, E.R.; Drennan, C.L. Ribbon-helix-helix transcription factors: Variations on a theme. Nat. Rev. Microbiol. 2007, 5, 710–720. [Google Scholar] [CrossRef]
- Madl, T.; van Melderen, L.; Mine, N.; Respondek, M.; Oberer, M.; Keller, W.; Khatai, L.; Zangger, K. Structural basis for nucleic acid and toxin recognition of the bacterial antitoxin CcdA. J. Mol. Biol. 2006, 364, 170–185. [Google Scholar] [CrossRef]
- Kamada, K.; Hanaoka, F. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol. Cell 2005, 19, 497–509. [Google Scholar] [CrossRef]
- Bailey, S.E.S.; Hayes, F. Influence of operator site geometry on transcriptional control by the YefM-YoeB toxin-antitoxin complex. J. Bacteriol. 2009, 191, 762–772. [Google Scholar] [CrossRef]
- Mattison, K.; Wilbur, J.S.; So, M.; Brennan, R.G. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J. Biol. Chem. 2006, 281, 37942–37951. [Google Scholar] [CrossRef]
- Brown, B.L.; Grigoriu, S.; Kim, Y.; Arruda, J.M.; Davenport, A.; Wood, T.K.; Peti, W.; Page, R. Three dimensional structure of the MqsR:MqsA complex: A novel TA pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog. 2009, 5, e1000706. [Google Scholar] [CrossRef]
- Maté, M.J.; Vincentelli, R.; Foos, N.; Raoult, D.; Cambillau, C.; Ortiz-Lombardía, M. Crystal structure of the DNA-bound VapBC2 antitoxin/toxin pair from Rickettsia felis. Nucleic Acids Res. 2012, 40, 3245–3258. [Google Scholar] [CrossRef]
- Moreland, J.L.; Gramada, A.; Buzko, O.V.; Zhang, Q.; Bourne, P.E. The Molecular Biology Toolkit (MBT): A modular platform for developing molecular visualization applications. BMC Bioinformatics 2005, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Aizenman, E.; Engelberg-Kulka, H.; Glaser, G. An Escherichia coli chromosomal “addiction module” regulated by guanosine 3',5'-bispyrophosphate: A model for programmed bacterial cell death. Proc. Natl. Acad. Sci. USA 1996, 93, 6059–6063. [Google Scholar]
- Kamada, K.; Hanaoka, F.; Burley, S.K. Crystal structure of the MazE/MazF complex: Molecular bases of antidote-toxin recognition. Mol. Cell 2003, 11, 875–884. [Google Scholar] [CrossRef]
- Erental, A.; Sharon, I.; Engelberg-Kulka, H. Two programmed cell death systems in Escherichia coli: An apoptotic-like death is inhibited by the mazEF-mediated death pathway. PLoS Biol. 2012, 10, e1001281. [Google Scholar] [CrossRef]
- Tsilibaris, V.; Maenhaut-Michel, G.; Mine, N.; van Melderen, L. What is the benefit to Escherichia coli of having multiple toxin-antitoxin systems in its genome? J. Bacteriol. 2007, 189, 6101–6108. [Google Scholar] [CrossRef]
- Gross, M.; Marianovsky, I.; Glaser, G. MazG-a regulator of programmed cell death in Escherichia coli. Mol. Microbiol. 2006, 59, 590–601. [Google Scholar] [CrossRef]
- Marianovsky, I.; Aizenman, E.; Engelberg-Kulka, H.; Glaser, G. The regulation of the Escherichia coli mazEF promoter involves an unusual alternating palindrome. J. Biol. Chem. 2001, 276, 5975–5984. [Google Scholar]
- Schneider, R.; Lurz, R.; Lüder, G.; Tolksdorf, C.; Travers, A.; Muskhelishvili, G. An architectural role of the Escherichia coli chromatin protein FIS in organising DNA. Nucleic Acids Res. 2001, 29, 5107–5114. [Google Scholar] [CrossRef]
- Skoko, D.; Yoo, D.; Bai, H.; Schnurr, B.; Yan, J.; McLeod, S.M.; Marko, J.F.; Johnson, R.C. Mechanism of chromosome compaction and looping by the Escherichia coli nucleoid protein Fis. J. Mol. Biol. 2006, 364, 777–798. [Google Scholar] [CrossRef]
- Dorman, C.J. Nucleoid-associated proteins and bacterial physiology. Adv. Appl. Microbiol. 2009, 67, 47–64. [Google Scholar] [CrossRef]
- Belitsky, M.; Avshalom, H.; Erental, A.; Yelin, I.; Kumar, S.; London, N.; Sperber, M.; Schueler-Furman, O.; Engelberg-Kulka, H. The Escherichia coli extracellular death factor EDF induces the endoribonucleolytic activities of the toxins MazF and ChpBK. Mol. Cell 2011, 41, 625–635. [Google Scholar]
- Dorsey-Oresto, A.; Lu, T.; Mosel, M.; Wang, X.; Salz, T.; Drlica, K.; Zhao, X. YihE kinase is a central regulator of programmed cell death in bacteria. Cell Rep. 2013, 3, 528–337. [Google Scholar] [CrossRef]
- Cline, S.D.; Saleem, S.; Daines, D.A. Regulation of the vapBC-1 toxin-antitoxin locus in nontypeable Haemophilus influenzae. PLoS One 2012, 7, e32199. [Google Scholar] [CrossRef]
- Magnuson, R.; Lehnherr, H.; Mukhopadhyay, G.; Yarmolinsky, M.B. Autoregulation of the plasmid addiction operon of bacteriophage P1. J. Biol. Chem. 1996, 271, 18705–18710. [Google Scholar] [CrossRef]
- Garcia-Pino, A.; Balasubramanian, S.; Wyns, L.; Gazit, E.; de Greve, H.; Magnuson, R.D.; Charlier, D.; van Nuland, N.A.; Loris, R. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 2010, 142, 101–111. [Google Scholar] [CrossRef]
- Bøggild, A.; Sofos, N.; Andersen, K.R.; Feddersen, A.; Easter, A.D.; Passmore, L.A.; Brodersen, D.E. The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure 2012, 20, 1641–1648. [Google Scholar] [CrossRef]
- Overgaard, M.; Borch, J.; Jørgensen, M.G.; Gerdes, K. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 2008, 69, 841–857. [Google Scholar] [CrossRef]
- Johnson, E.P.; Strom, A.R.; Helinski, D.R. Plasmid RK2 toxin protein ParE: Purification and interaction with the ParD antitoxin protein. J. Bacteriol. 1996, 178, 1420–1429. [Google Scholar]
- Magnuson, R.; Yarmolinsky, M.B. Corepression of the P1 addiction operon by Phd and Doc. J. Bacteriol. 1998, 180, 6342–6351. [Google Scholar]
- Monti, M.C.; Hernandez-Arriaga, A.M.; Kamphuis, M.B.; Lopez-Villarejo, J.; Heck, A.J.R.; Boelens, R.; Díaz-Orejas, R.; van den Heuvel, R.H. Interactions of Kid-Kis toxin-antitoxin complexes with the parD operator-promoter region of plasmid R1 are piloted by the Kis antitoxin and tuned by the stoichiometry of Kid-Kis oligomers. Nucleic Acids Res. 2007, 35, 1737–1749. [Google Scholar] [CrossRef]
- Cataudella, I.; Trusina, A.; Sneppen, K.; Gerdes, K.; Mitarai, N. Conditional cooperativity in toxin-antitoxin regulation prevents random toxin activation and promotes fast translational recovery. Nucleic Acids Res. 2012, 40, 6424–6434. [Google Scholar] [CrossRef]
- Anantharaman, V.; Aravind, L. New connections in the prokaryotic toxin-antitoxin network: Relationship with the eukaryotic nonsense-mediated RNA decay system. Genome Biol. 2003, 4, R81. [Google Scholar] [CrossRef]
- Leplae, R.; Geeraerts, D.; Hallez, R.; Guglielmini, J.; Dreze, P.; van Melderen, L. Diversity of bacterial type II toxin-antitoxin systems: A comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011, 39, 5513–5525. [Google Scholar] [CrossRef]
- Goeders, N.; Drèze, P.L.; van Melderen, L. Relaxed cleavage specificity within the RelE toxin family. J. Bacteriol. 2013, 195, 2541–2549. [Google Scholar] [CrossRef]
- Neubauer, C.; Gao, Y.G.; Andersen, K.R.; Dunham, C.M.; Kelley, A.C.; Hentschel, J.; Gerdes, K.; Ramakrishnan, V.; Brodersen, D.E. The structural basis for mRNA recognition and cleavage by the ribosome-dependent endonuclease RelE. Cell 2009, 139, 1084–1095. [Google Scholar] [CrossRef]
- Pedersen, K.; Zavialov, A.V.; Pavlov, M.Y.; Elf, J.; Gerdes, K.; Ehrenberg, M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 2003, 112, 131–140. [Google Scholar] [CrossRef]
- Li, G.Y.; Zhang, Y.; Inouye, M.; Ikura, M. Structural mechanism of transcriptional autorepression of the Escherichia coli RelB/RelE antitoxin/toxin module. J. Mol. Biol. 2008, 380, 107–119. [Google Scholar] [CrossRef]
- Overgaard, M.; Borch, J.; Gerdes, K. RelB and RelE of Escherichia coli form a tight complex that represses transcription via the ribbon-helix-helix motif in RelB. J. Mol. Biol. 2009, 394, 183–196. [Google Scholar] [CrossRef]
- Li, G.Y.; Zhang, Y.; Inouye, M.; Ikura, M. Inhibitory mechanism of Escherichia coli RelE-RelB toxin-antitoxin module involves a helix displacement near an mRNA interferase active site. J. Biol. Chem. 2009, 284, 14628–14636. [Google Scholar] [CrossRef]
- Gotfredsen, M.; Gerdes, K. The Escherichia coli relBE genes belong to a new toxin-antitoxin gene family. Mol. Microbiol. 1998, 29, 1065–1076. [Google Scholar] [CrossRef]
- Diago-Navarro, E.; Hernandez-Arriaga, A.M.; López-Villarejo, J.; Muñoz-Gómez, A.J.; Kamphuis, M.B.; Boelens, R.; Lemonnier, M.; Díaz-Orejas, R. parD toxin-antitoxin system of plasmid R1-basic contributions, biotechnological applications and relationships with closely-related toxin-antitoxin systems. FEBS J. 2010, 277, 3097–3117. [Google Scholar] [CrossRef]
- Kamphuis, M.B.; Monti, M.C.; van den Heuvel, R.H.; Santos-Sierra, S.; Folkers, G.E.; Lemonnier, M.; Díaz-Orejas, R.; Heck, A.J.; Boelens, R. Interactions between the toxin Kid of the bacterial parD system and the antitoxins Kis and MazE. Proteins 2007, 67, 219–231. [Google Scholar] [CrossRef]
- Ruiz-Echevarria, M.J.; Berzal-Herranz, A.; Gerdes, K.; Diaz-Orejas, R. The kis and kid genes of the parD maintenance system of plasmid R1 form an operon that is autoregulated at the level of transcription by the co-ordinated action of the Kis and Kid proteins. Mol. Microbiol. 1991, 5, 2685–2693. [Google Scholar] [CrossRef]
- Ruiz-Echevarría, M.J.; de la Cueva, G.; Díaz-Orejas, R. Translational coupling and limited degradation of a polycistronic messenger modulate differential gene expression in the parD stability system of plasmid R1. Mol. Gen. Genet. 1995, 248, 599–609. [Google Scholar] [CrossRef]
- López-Villarejo, J.; Diago-Navarro, E.; Hernández-Arriaga, A.M.; Díaz-Orejas, R. Kis antitoxin couples plasmid R1 replication and parD (kis,kid) maintenance modules. Plasmid 2012, 67, 118–127. [Google Scholar] [CrossRef]
- Brown, B.L.; Lord, D.M.; Grigoriu, S.; Peti, W.; Page, R. The Escherichia coli toxin MqsR destabilizes the transcriptional repression complex formed between the antitoxin MqsA and the mqsRA operon promoter. J. Biol. Chem. 2013, 288, 1286–1294. [Google Scholar]
- Pomerantsev, A.P.; Golovliov, I.R.; Ohara, Y.; Mokrievich, A.N.; Obuchi, M.; Norqvist, A.; Kuoppa, K.; Pavlov, V.M. Genetic organization of the Francisella plasmid pFNL10. Plasmid 2001, 46, 210–222. [Google Scholar] [CrossRef]
- Grady, R.; Hayes, F. Axe-Txe, a broad spectrum proteic toxin-antitoxin system specified by a multidrug-resistant, clinical isolate of Enterococcus faecium. Mol. Microbiol. 2003, 47, 1419–1432. [Google Scholar] [CrossRef]
- Połom, D.; Boss, L.; Węgrzyn, G.; Hayes, F.; Kędzierska, B. Amino acids crucial for specificity of toxin-antitoxin interactions in the homologous Axe-Txe and YefM-YoeB complexes. FEBS J. 2013, 280, 5906–5918. [Google Scholar] [CrossRef]
- Zhang, Y.; Inouye, M. The inhibitory mechanism of protein synthesis by YoeB, an Escherichia coli toxin. J. Biol. Chem. 2009, 284, 6627–6638. [Google Scholar] [CrossRef]
- Kędzierska, B.; Lian, L.Y.; Hayes, F. Toxin-antitoxin regulation: Bimodal interaction of YefM-YoeB with paired DNA palindromes exerts transcriptional autorepression. Nucleic Acids Res. 2007, 35, 325–339. [Google Scholar] [CrossRef]
- Boss, L.; Labudda, Ł.; Węgrzyn, G.; Hayes, F.; Kędzierska, B. The Axe-Txe complex of Enterococcus faecium presents a multilayered mode of toxin-antitoxin gene expression regulation. PLoS One 2013, 8, e73569. [Google Scholar]
- Chan, W.T.; Nieto, C.; Harikrishna, J.A.; Khoo, S.K.; Othman, R.Y.; Espinosa, M.; Yeo, C.C. Genetic regulation of the yefM-yoeB toxin-antitoxin locus of Streptococcus pneumoniae. J. Bacteriol. 2011, 193, 4612–4625. [Google Scholar] [CrossRef]
- Fiebig, A.; Castro Rojas, C.M.; Siegal-Gaskins, D.; Crosson, S. Interaction specificity, toxicity and regulation of a paralogous set of ParE/RelE-family toxin-antitoxin systems. Mol. Microbiol. 2010, 77, 236–251. [Google Scholar] [CrossRef]
- Smith, A.S.G.; Rawlings, D.E. The poison antidote stability system of the broad-host range Thiobacillus ferrooxidans plasmid pTF-FC2. Mol. Microbiol. 1997, 26, 261–270. [Google Scholar] [CrossRef]
- Smith, A.S.G.; Rawlings, D.E. Autoregulation of the pTF-FC2 proteic poison-antidote plasmid addiction system (pas) is essential for plasmid stabilization. J. Bacteriol. 1998, 180, 5463–5465. [Google Scholar]
- Hallez, R.; Geeraerts, D.; Sterckx, Y.; Mine, N.; Loris, R.; van Melderen, L. New toxins homologous to ParE belonging to three-component toxin-antitoxin systems in Escherichia coli O157:H7. Mol. Microbiol. 2010, 76, 719–732. [Google Scholar]
- De la Hoz, A.B.; Ayora, S.; Sitkiewicz, I.; Fernandez, S.; Pankiewicz, R.; Alonso, J.C.; Cegłowski, P. Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc. Natl. Acad. Sci. USA 2000, 97, 728–733. [Google Scholar] [CrossRef]
- De la Hoz, A.B.; Pratto, F.; Misselwitz, R.; Speck, C.; Weihofen, W.; Welfle, K.; Saenger, W.; Welfle, H.; Alonso, J.C. Recognition of DNA by ω protein from the broad-host range Streptococcus pyogenes plasmid pSM19035: Analysis of binding to operator DNA with one to four heptad repeats. Nucleic Acids Res. 2004, 32, 3136–3147. [Google Scholar] [CrossRef]
- Camacho, A.G.; Misselwitz, R.; Behlke, J.; Ayora, S.; Welfle, K.; Meinhart, A.; Lara, B.; Saenger, W.; Welfle, H.; Alonso, J.C. In vitro and in vivo stability of the ε2ζ2 protein complex of the broad host-range Streptococcus pyogenes pSM19035 addiction system. Biol. Chem. 2002, 383, 1701–1713. [Google Scholar]
- Meinhart, A.; Alonso, J.C.; Strater, N.; Saenger, W. Crystal structure of the plasmid maintenance system ε/ζ: Functional mechanism of toxin ζ and inactivation by ε2ζ2 complex formation. Proc. Natl. Acad. Sci. USA 2003, 100, 1661–1666. [Google Scholar]
- Murayama, K.; Orth, P.; de la Hoz, A.B.; Alonso, J.C.; Saenger, W. Crystal structure of ω transcriptional repressor encoded by Streptococcus pyogenes plasmid pSM19035 at 1.5 Å resolution. J. Mol. Biol. 2001, 314, 789–796. [Google Scholar] [CrossRef]
- Arcus, V.L.; McKenzie, J.L.; Robson, J.; Cook, G.M. The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng. Des. Sel. 2011, 24, 33–40. [Google Scholar] [CrossRef]
- Robson, J.; McKenzie, J.L.; Cursons, R.; Cook, G.M.; Arcus, V.L. The vapBC operon from Mycobacterium smegmatis is an autoregulated toxin-antitoxin module that controls growth via inhibition of translation. J. Mol. Biol. 2009, 390, 353–367. [Google Scholar] [CrossRef]
- Winther, K.S.; Gerdes, K. Regulation of enteric vapBC transcription: Induction by VapC toxin dimer-breaking. Nucleic Acids Res. 2012, 40, 4347–4357. [Google Scholar] [CrossRef]
- Tian, Q.B.; Hayashi, T.; Murata, T.; Terawaki, Y. Gene product identification and promoter analysis of hig locus of plasmid Rts1. Biochem. Biophys. Res. Commun. 1996, 225, 679–684. [Google Scholar] [CrossRef]
- Jorgensen, M.G.; Pandey, D.P.; Jaskolska, M.; Gerdes, K. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 2009, 191, 1191–1199. [Google Scholar] [CrossRef]
- Brown, B.L.; Wood, T.K.; Peti, W.; Page, R. Structure of the Escherichia coli antitoxin MqsA (YgiT/b3021) bound to its gene promoter reveals extensive domain rearrangements and the specificity of transcriptional regulation. J. Biol. Chem. 2011, 286, 2285–2296. [Google Scholar] [CrossRef]
- Kim, Y.; Wang, X.; Zhang, X.S.; Grigoriu, S.; Page, R.; Peti, W.; Wood, T.K. Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin Csp. Environ. Microbiol. 2010, 12, 1105–1121. [Google Scholar] [CrossRef]
- Wang, X.; Kim, Y.; Hong, S.H.; Ma, Q.; Brown, B.L.; Pu, M.; Tarone, A.M.; Benedik, M.J.; Peti, W.; Page, R.; et al. Antitoxin MqsA helps mediate the bacterial general stress response. Nat. Chem. Biol. 2011, 7, 359–366. [Google Scholar] [CrossRef]
- Soo, V.W.; Wood, T.K. Antitoxin MqsA represses curli formation through the master biofilm regulator CsgD. Sci. Rep. 2013, 3, 3186. [Google Scholar]
- González Barrios, A.F.; Zuo, R.; Hashimoto, Y.; Yang, L.; Bentley, W.E.; Wood, T.K. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J. Bacteriol. 2006, 188, 305–316. [Google Scholar] [CrossRef]
- Christensen-Dalsgaard, M.; Jørgensen, M.G.; Gerdes, K. Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol. Microbiol. 2010, 75, 333–348. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Park, J.H.; Inouye, M. MqsR, a crucial regulator for quorum sensing and biofilm formation, is a GCU-specific mRNA interferase in Escherichia coli. J. Biol. Chem. 2009, 284, 28746–28753. [Google Scholar] [CrossRef]
- Donegan, N.P.; Cheung, A.L. Regulation of the mazEF toxin-antitoxin module in Staphylococcus aureus and its impact on sigB expression. J. Bacteriol. 2009, 191, 2795–2805. [Google Scholar] [CrossRef]
- Cheung, A.L.; Nishina, K.A.; Trotonda, M.P.; Tamber, S. The SarA protein family of Staphylococcus aureus. Int. J. Biochem. Cell Biol. 2008, 40, 355–361. [Google Scholar] [CrossRef]
- Black, D.S.; Irwin, B.; Moyed, H.S. Autoregulation of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J. Bacteriol. 1994, 176, 4081–4091. [Google Scholar]
- Alekshun, M.N.; Levy, S.B. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007, 128, 1037–1050. [Google Scholar] [CrossRef]
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef]
- Fauci, A.S.; Touchette, N.A.; Folkers, G.K. Emerging infectious diseases: A 10-year perspective from the National Institute of Allergy and Infectious Diseases. Emerg. Infect. Dis. 2005, 11, 519–525. [Google Scholar] [CrossRef]
- Goldberg, D.E.; Siliciano, R.F.; Jacobs, W.R., Jr. Outwitting evolution: Fighting drug-resistant TB, malaria, and HIV. Cell 2012, 148, 1271–1283. [Google Scholar] [CrossRef]
- Spellberg, B.; Guidos, R.; Gilbert, D.; Bradley, J.; Boucher, H.W.; Scheld, W.M.; Bartlett, J.G.; Edwards, J., Jr. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 46, 155–164. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Engelberg-Kulka, H.; Sat, B.; Reches, M.; Amitai, S.; Hazan, R. Bacterial programmed cell death systems as targets for antibiotics. Trends Microbiol. 2004, 12, 66–71. [Google Scholar] [CrossRef]
- Williams, J.J.; Hergenrother, P.J. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012, 20, 291–298. [Google Scholar] [CrossRef]
- Agarwal, S.; Mishra, N.K.; Bhatnagar, S.; Bhatnagar, R. PemK toxin of Bacillus anthracis is a ribonuclease: An insight into its active site, structure, and function. J. Biol. Chem. 2010, 285, 7254–7270. [Google Scholar]
- Chopra, N.; Agarwal, S.; Verma, S.; Bhatnagar, S.; Bhatnagar, R. Modeling of the structure and interactions of the B. anthracis antitoxin, MoxX: Deletion mutant studies highlight its modular structure and repressor function. J. Comput. Aided Mol. Des. 2011, 25, 275–291. [Google Scholar] [CrossRef]
- Lioy, V.S.; Rey, O.; Balsa, D.; Pellicer, T.; Alonso, J.C. A toxin-antitoxin module as a target for antimicrobial development. Plasmid 2010, 63, 31–39. [Google Scholar]
- Barbosa, L.C.; Garrido, S.S.; Garcia, A.; Delfino, D.B.; Santos Ldo, N.; Marchetto, R. Design and synthesis of peptides from bacterial ParE toxin as inhibitors of topoisomerases. Eur. J. Med. Chem. 2012, 54, 591–596. [Google Scholar] [CrossRef]
- Liskamp, R.M.; Rijkers, D.T.; Kruijtzer, J.A.; Kemmink, J. Peptides and proteins as a continuing exciting source of inspiration for peptidomimetics. Chembiochem 2011, 12, 1626–1653. [Google Scholar] [CrossRef]
- Gniazdowski, M.; Denny, W.A.; Nelson, S.M.; Czyz, M. Transcription factors as targets for DNA-interacting drugs. Curr. Med. Chem. 2003, 10, 909–924. [Google Scholar] [CrossRef]
- Gniazdowski, M.; Denny, W.A.; Nelson, S.M.; Czyz, M. Effects of anticancer drugs on transcription factor-DNA interactions. Expert Opin. Ther. Targets 2005, 9, 471–489. [Google Scholar] [CrossRef]
- Raskatov, J.A.; Meier, J.L.; Puckett, J.W.; Yang, F.; Ramakrishnan, P.; Dervan, P.B. Modulation of NF-kB-dependent gene transcription using programmable DNA minor groove binders. Proc. Natl. Acad. Sci. USA 2012, 109, 1023–1028. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hayes, F.; Kędzierska, B. Regulating Toxin-Antitoxin Expression: Controlled Detonation of Intracellular Molecular Timebombs. Toxins 2014, 6, 337-358. https://doi.org/10.3390/toxins6010337
Hayes F, Kędzierska B. Regulating Toxin-Antitoxin Expression: Controlled Detonation of Intracellular Molecular Timebombs. Toxins. 2014; 6(1):337-358. https://doi.org/10.3390/toxins6010337
Chicago/Turabian StyleHayes, Finbarr, and Barbara Kędzierska. 2014. "Regulating Toxin-Antitoxin Expression: Controlled Detonation of Intracellular Molecular Timebombs" Toxins 6, no. 1: 337-358. https://doi.org/10.3390/toxins6010337