The E. coli CNF1 as a Pioneering Therapy for the Central Nervous System Diseases




{kind=link}
Abstract
:1. Introduction
2. About CNF1
2.1. The Molecular Point of View
2.2. CNF1 and Eukaryotic Cells
3. CNF1 on Neuronal Cells and Its Promising Therapeutic Effects
3.1. Improvement of the Analgesic Activity
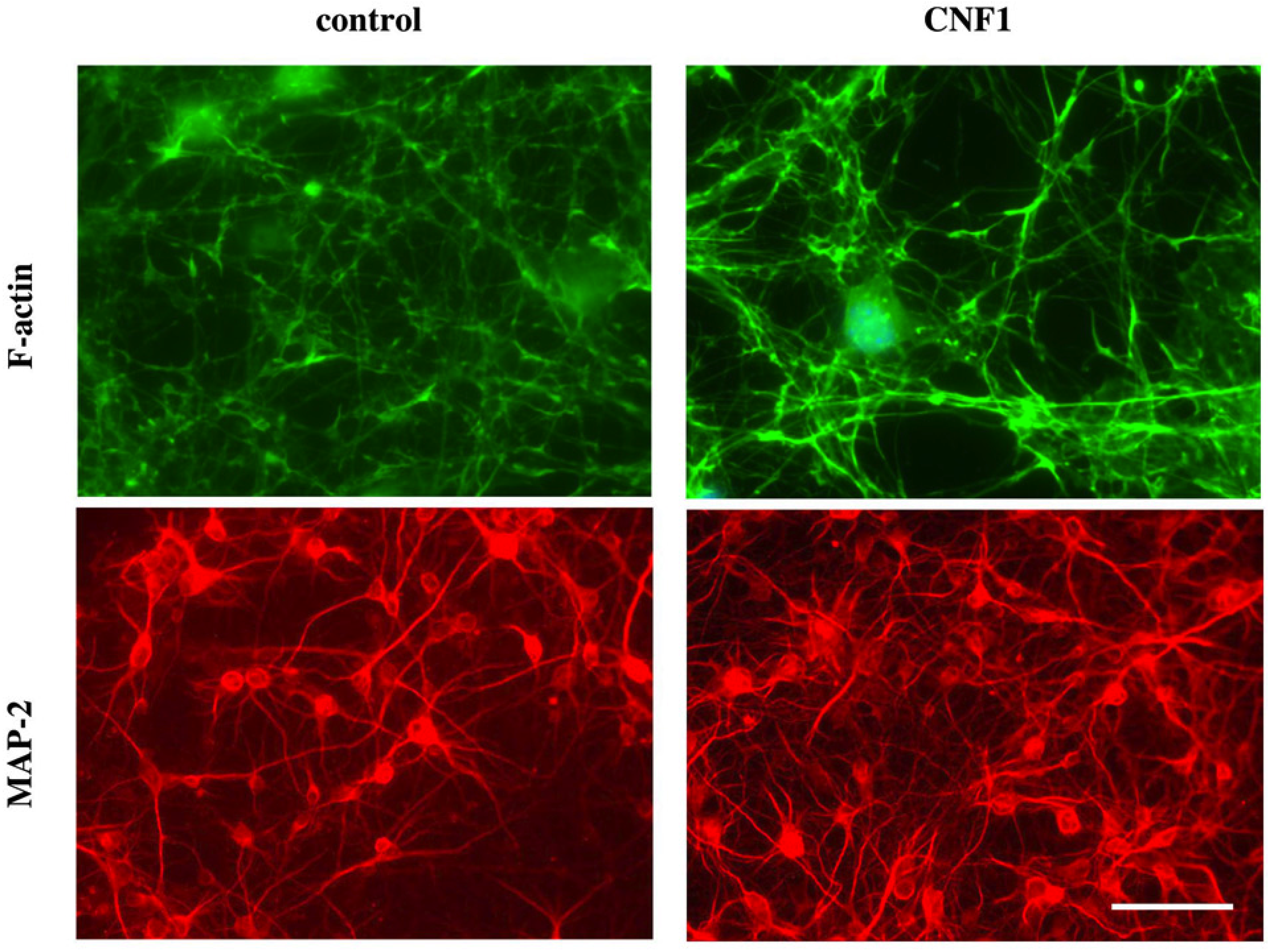
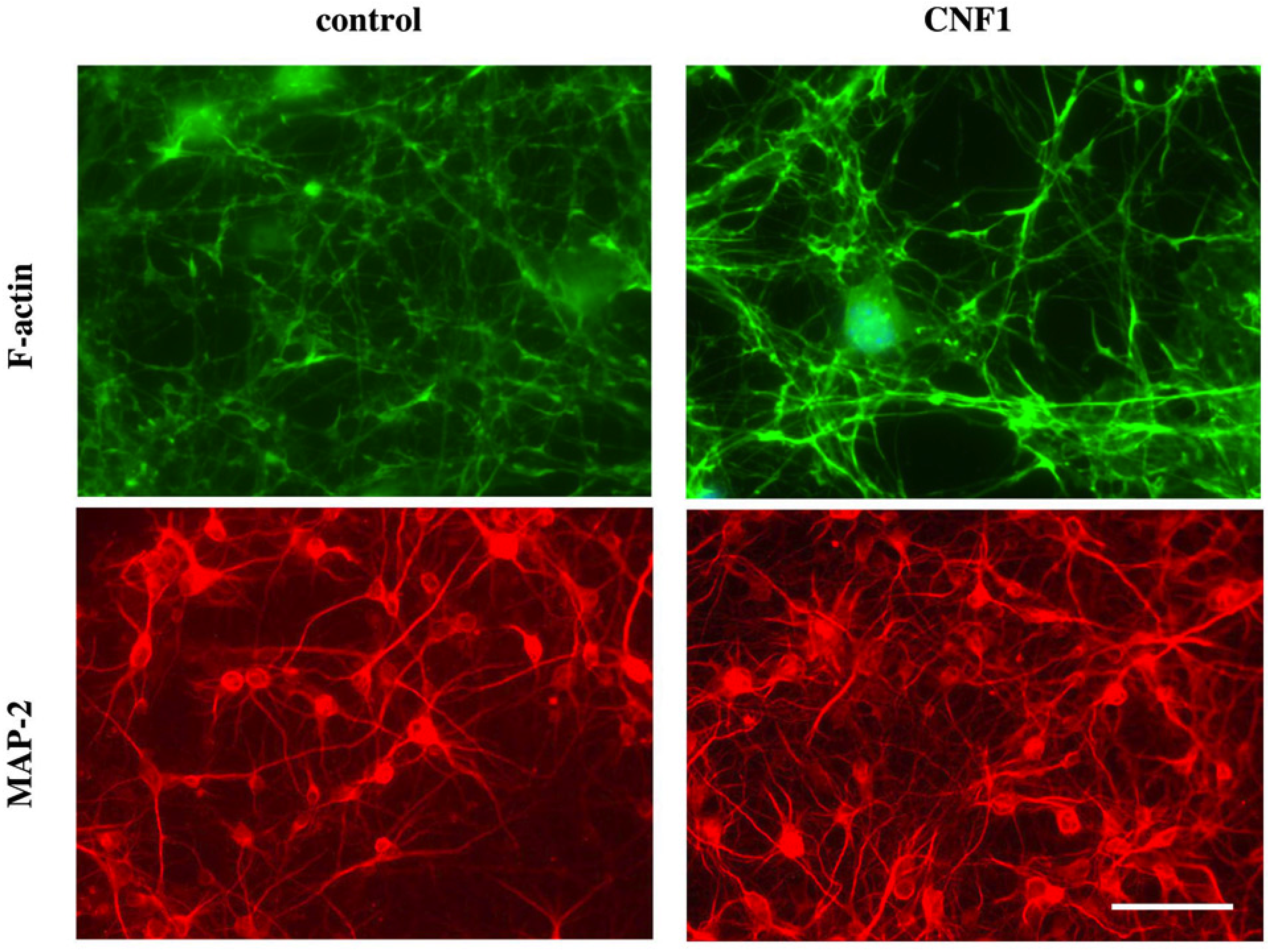
3.2. CNF1 and Functional Plasticity in the Adult Rat Visual Cortex
3.3. CNF1 and Rett Syndrome
3.4. CNF1 and Alzheimer’s Disease
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Houdouin, V.; Bonacorsi, S.; Brahimi, N.; Clermont, O.; Nassif, X.; Bingen, E. A uropathogenicity island contributes to the pathogenicity of Escherichia coli strains that cause neonatal meningitis. Infect. Immun. 2002, 70, 5865–5869. [Google Scholar] [CrossRef]
- Blanco, J.; Alonso, M.P.; González, E.A.; Blanco, M.; Garabal, J.I. Virulence factors of bacteraemic Escherichia coli with particular reference to production of cytotoxic necrotising factor (CNF) by P-fimbriate strains. J. Med. Microbiol. 1990, 31, 175–183. [Google Scholar] [CrossRef]
- Hacker, J.; Blum-Oehler, G.; Mühldorfer, I.; Tschäpe, H. Pathogenicity islands of virulent bacteria: Structure, function and impact on microbial evolution. Mol. Microbiol. 1997, 23, 1089–1097. [Google Scholar] [CrossRef]
- De Rycke, J.; Milon, A.; Oswald, E. Necrotoxic Escherichia coli (NTEC): Two emerging categories of human and animal pathogens. Vet. Res. 1999, 30, 221–233. [Google Scholar]
- Landraud, L.; Gauthier, M.; Fosse, T.; Boquet, P. Frequency of Escherichia coli strains producing the cytotoxic necrotizing factor (CNF1) in nosocomial urinary tract infections. Lett. Appl. Microbiol. 2000, 30, 213–216. [Google Scholar] [CrossRef]
- Boquet, P. The cytotoxic necrotizing factor 1 (CNF1) from Escherichia coli. Toxicon 2001, 39, 1673–1680. [Google Scholar] [CrossRef]
- Falzano, L.; Fiorentini, C.; Donelli, G.; Michel, E.; Kocks, C.; Cossart, P.; Cabanié, L.; Oswald, E.; Boquet, P. Induction of phagocytic behaviour in human epithelial cells by Escherichia coli cytotoxic necrotizing factor type 1. Mol. Microbiol. 1993, 9, 1247–1254. [Google Scholar] [CrossRef]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Donelli, G.; Boquet, P.; Malorni, W. Rho-dependent cell spreading activated by E.coli cytotoxic necrotizing factor 1 hinders apoptosis in epithelial cells. Cell. Death Differ. 1998, 5, 921–929. [Google Scholar]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Fabbri, A.; Donelli, G.; Cossarizza, A.; Boquet, P.; Malorni, W. Toxin-induced activation of Rho GTP-binding protein increases Bcl-2 expression and influences mitochondrial homeostasis. Exp. Cell. Res. 1998, 242, 341–350. [Google Scholar] [CrossRef]
- Fiorentini, C.; Falzano, L.; Fabbri, A.; Stringaro, A.; Logozzi, M.; Travaglione, S.; Contamin, S.; Arancia, G.; Malorni, W.; Fais, S. Activation of rho GTPases by cytotoxic necrotizing factor 1 induces macropinocytosis and scavenging activity in epithelial cells. Mol. Biol. Cell. 2001, 12, 2061–2073. [Google Scholar] [CrossRef]
- Boyer, L.; Travaglione, S.; Falzano, L.; Gauthier, N.C.; Popoff, M.R.; Lemichez, E.; Fiorentini, C.; Fabbri, A. Rac GTPase instructs nuclear factor-kappaB activation by conveying the SCF complex and IkBalpha to the ruffling membranes. Mol. Biol. Cell. 2004, 15, 1124–1133. [Google Scholar]
- Travaglione, S.; Messina, G.; Fabbri, A.; Falzano, L.; Giammarioli, A.M.; Grossi, M.; Rufini, S.; Fiorentini, C. Cytotoxic necrotizing factor 1 hinders skeletal muscle differentiation in vitro by perturbing the activation/deactivation balance of Rho GTPases. Cell. Death Differ. 2005, 12, 78–86. [Google Scholar] [CrossRef]
- Falzano, L.; Filippini, P.; Travaglione, S.; Miraglia, A.G.; Fabbri, A.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 blocks cell cycle G2/M transition in uroepithelial cells. Infect. Immun. 2006, 74, 3765–3772. [Google Scholar] [CrossRef]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. J. Biol. Chem. 1997, 272, 19532–19537. [Google Scholar]
- Schmidt, G.; Sher, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef]
- Lerm, M.; Selzer, J.; Hoffmeyer, A.; Rapp, U.R.; Aktories, K.; Schmidt, G. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: Activation of the C-Jun N-terminal kinase in HeLa cells. Infect. Immun. 1999, 67, 496–503. [Google Scholar]
- Hall, A. Rho family GTPases. Biochem. Soc. Trans. 2012, 40, 1378–1382. [Google Scholar] [CrossRef]
- Luo, L. Rho GTPases in neuronal morphogenesis. Nat. Rev. Neurosci. 2000, 1, 173–180. [Google Scholar] [CrossRef]
- Tashiro, A.; Yuste, R. Role of Rho GTPases in the morphogenesis and motility of dendritic spines. Methods Enzymol. 2008, 439, 285–302. [Google Scholar]
- Saneyoshi, T.; Fortin, D.A.; Soderling, T.R. Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr. Opin. Neurobiol. 2010, 20, 108–115. [Google Scholar] [CrossRef]
- O’Kane, E.M.; Stone, T.W.; Morris, B.J. Activation of Rho GTPases by synaptic transmission in the hippocampus. J. Neurochem. 2003, 87, 1309–1312. [Google Scholar] [CrossRef]
- Wang, H.G.; Lu, F.M.; Jin, I.; Udo, H.; Kandel, E.R.; de Vente, J.; Walter, U.; Lohmann, S.M.; Hawkins, R.D.; Antonova, I. Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron 2005, 45, 389–403. [Google Scholar] [CrossRef]
- Kasri, N.N.; van Aelst, L. Rho-linked genes and neurological disorders. Pflugers Arch. 2008, 455, 787–797. [Google Scholar] [CrossRef]
- Rex, C.S.; Chen, L.Y.; Sharma, A.; Liu, J.; Babayan, A.H.; Gall, C.M.; Lynch, G. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J. Cell. Biol. 2009, 186, 85–97. [Google Scholar] [CrossRef]
- Asrar, S.; Meng, Y.; Zhou, Z.; Todorovski, Z.; Huang, W.W.; Jia, Z. Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 2009, 56, 73–80. [Google Scholar]
- Fortin, D.A.; Davare, M.A.; Srivastava, T.; Brady, J.D.; Nygaard, S.; Derkach, V.A.; Soderling, T.R. Long term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J. Neurosci. 2010, 30, 11565–11575. [Google Scholar]
- Ramakers, G.J. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002, 25, 191–199. [Google Scholar] [CrossRef]
- Malchiodi-Albedi, F.; Paradisi, S.; Di Nottia, M.; Simone, D.; Travaglione, S.; Falzano, L.; Guidotti, M.; Frank, C.; Cutarelli, A.; Fabbri, A.; et al. CNF1 improves astrocytic ability to support neuronal growth and differentiation in vitro. PLoS ONE 2012, 7, e34115. [Google Scholar]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of rett syndrome. Neuropsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef]
- Loizzo, S.; Rimondini, R.; Travaglione, S.; Fabbri, A.; Guidotti, M.; Ferri, A.; Campana, G.; Fiorentini, C. CNF1 Increases brain energy level, counteracts neuroinflammatory markers and rescues cognitive deficits in a murine model of Alzheimer’s disease. PLoS ONE 2013, 8, e65898. [Google Scholar]
- Hackett, R.; Kam, P.C. Botulinum toxin: Pharmacology and clinical developments: A literature review. Med. Chem. 2007, 3, 333–345. [Google Scholar] [CrossRef]
- Wheeler, A.; Smith, H.S. Botulinum toxins: Mechanisms of action, antinociception and clinical applications. Toxicology 2013, 306, 124–146. [Google Scholar] [CrossRef]
- Phillips, D.D.; Fattah, R.J.; Crown, D.; Zhang, Y.; Liu, S.; Moayeri, M.; Fischer, E.R.; Hansen, B.T.; Ghirlando, R.; Nestorovich, E.M.; et al. Engineering anthrax toxin variants that exclusively form octamers and their application to targeting tumors. J. Biol. Chem. 2013, 288, 9058–9065. [Google Scholar] [CrossRef]
- McCluskey, A.J.; Olive, A.J.; Starnbach, M.N.; Collier, R.J. Targeting HER2-positive cancer cells with receptor-redirected anthrax protective antigen. Mol. Oncol. 2013, 7, 440–451. [Google Scholar] [CrossRef]
- Alfano, M.; Rizzi, C.; Corti, D.; Adduce, L.; Poli, G. Bacterial toxins: Potential weapons against HIV infection. Curr. Pharm. Des. 2005, 11, 2909–2926. [Google Scholar] [CrossRef]
- Alfano, M.; Pushkarsky, T.; Poli, G.; Bukrinsky, M. The B-oligomer of pertussis toxin inhibits human immunodeficiency virus type 1 replication at multiple stages. J. Virol. 2000, 74, 8767–8770. [Google Scholar] [CrossRef]
- Lemichez, E.; Flatau, G.; Bruzzone, M.; Boquet, P.; Gauthier, M. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol. Microbiol. 1997, 24, 1061–1070. [Google Scholar]
- Buetow, L.; Flatau, G.; Chiu, K.; Boquet, P.; Ghosh, P. Structure of the Rho-activating domain of Escherichia coli cytotoxic necrotizing factor 1. Nat. Struct. Biol. 2001, 8, 584–588. [Google Scholar] [CrossRef]
- Contamin, S.; Galmiche, A.; Doye, A.; Flatau, G.; Benmerah, A.; Boquet, P. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol. Biol. Cell. 2000, 11, 1775–1787. [Google Scholar] [CrossRef]
- Chung, J.W.; Hong, S.J.; Kim, K.J.; Goti, D.; Stins, M.F.; Shin, S.; Dawson, V.L.; Dawson, T.M.; Kim, K.S. 37-kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J. Biol. Chem. 2003, 278, 16857–16862. [Google Scholar] [CrossRef]
- Kim, K.J.; Chung, J.W.; Kim, K.S. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 2005, 280, 1360–1368. [Google Scholar]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [CrossRef]
- Knust, Z.; Blumenthal, B.; Aktories, K.; Schmidt, G. Cleavage of Escherichia coli cytotoxic necrotizing factor 1 is required for full biologic activity. Infect. Immun. 2009, 77, 1835–1841. [Google Scholar] [CrossRef]
- Pei, S.; Doye, A.; Boquet, P. Mutation of specific acidic residues of the CNF1 T domain into lysine alters cell membrane translocation of the toxin. Mol. Microbiol. 2001, 41, 1237–1247. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Rittinger, K.; Walker, P.A.; Eccleston, J.F.; Nurmahomed, K.; Owen, D.; Laue, E.; Gamblin, S.J.; Smerdon, S.J. Crystal structure of a small G protein in complex with the GTPase-activating protein rhoGAP. Nature 1997, 388, 693–697. [Google Scholar] [CrossRef]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clément, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar] [CrossRef]
- Weissman, A.M. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell. Biol. 2001, 2, 169–178. [Google Scholar] [CrossRef]
- Marín, I. Animal HECT ubiquitin ligases: Evolution and functional implications. BMC Evol. Biol. 2010, 10, 56. [Google Scholar] [CrossRef]
- Boyer, L.; Turchi, L.; Desnues, B.; Doye, A.; Ponzio, G.; Mege, J.L.; Yamashita, M.; Zhang, Y.E.; Bertoglio, J.; Flatau, G.; et al. CNF1-induced ubiquitylation and proteasome destruction of activated RhoA is impaired in Smurf1−/− cells. Mol. Biol. Cell. 2006, 17, 2489–2497. [Google Scholar] [CrossRef]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell. Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Fiorentini, C.; Arancia, G.; Caprioli, A.; Falbo, V.; Ruggeri, F.M.; Donelli, G. Cytoskeletal changes induced in HEp-2 cells by the cytotoxic necrotizing factor of Escherichia coli. Toxicon 1988, 26, 1047–1056. [Google Scholar] [CrossRef]
- Fiorentini, C.; Fabbri, A.; Flatau, G.; Donelli, G.; Matarrese, P.; Lemichez, E.; Falzano, L.; Boquet, P. Escherichia coli cytotoxic necrotizing factor 1 (CNF1), a toxin that activates the Rho GTPase. J. Biol. Chem. 1997, 272, 19532–19537. [Google Scholar] [CrossRef]
- Lacerda, H.M.; Pullinger, G.D.; Lax, A.J.; Rozengurt, E. Cytotoxic necrotizing factor 1 from Escherichia coli and dermonecrotic toxin from Bordetella bronchiseptica induce p21(rho)-dependent tyrosine phosphorylation of focal adhesion kinase and paxillin in Swiss 3T3 cells. J. Biol. Chem. 1997, 272, 9587–9596. [Google Scholar] [CrossRef]
- Falzano, L.; Quaranta, M.G.; Travaglione, S.; Filippini, P.; Fabbri, A.; Viora, M.; Donelli, G.; Fiorentini, C. Cytotoxic necrotizing factor 1 enhances reactive oxygen species-dependent transcription and secretion of proinflammatory cytokines in human uroepithelial cells. Infect. Immun. 2003, 71, 4178–4181. [Google Scholar] [CrossRef]
- Miraglia, A.G.; Travaglione, S.; Meschini, S.; Falzano, L.; Matarrese, P.; Quaranta, M.G.; Viora, M.; Fiorentini, C.; Fabbri, A. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IkappaB kinase pathway: Role of nuclear factor-kappaB and Bcl-2. Mol. Biol. Cell. 2007, 18, 2735–2744. [Google Scholar] [CrossRef]
- Hotulainen, P.; Hoogenraad, C.C. Actin in dendritic spines: Connecting dynamics to function. J. Cell. Biol. 2010, 189, 619–629. [Google Scholar] [CrossRef]
- Gilmore, J.H.; Jarskog, L.F.; Vadlamudi, S.; Lauder, J.M. Prenatal infection and risk for schizophrenia: IL-1beta, IL-6, and TNFalpha inhibit cortical neuron dendrite development. Neuropsychopharmacology 2004, 29, 1221–1229. [Google Scholar] [CrossRef]
- Menet, V.; Gimenez y Ribotta, M.; Chauvet, N.; Drian, M.J.; Lannoy, J.; Colucci-Guyon, E.; Privat, A. Inactivation of the glial fibrillary acidic protein gene, but not that of vimentin, improves neuronal survival and neurite growth by modifying adhesion molecule expression. J. Neurosci. 2001, 21, 6147–6158. [Google Scholar]
- Pavone, F.; Luvisetto, S.; Marinelli, S.; Straface, E.; Fabbri, A.; Falzano, L.; Fiorentini, C.; Malorni, W. The Rac GTPase-activating bacterial protein toxin CNF1 induces analgesia up-regulating mu-opioid receptors. Pain 2009, 145, 219–229. [Google Scholar] [CrossRef]
- Bhave, G.; Gereau, R.W. Growing pains: The cytoskeleton as a critical regulator of pain plasticity. Neuron 2003, 39, 577–579. [Google Scholar] [CrossRef]
- Dina, O.A.; McCarter, G.C.; de Coupade, C.; Levine, J.D. Role of the sensory neuron cytoskeleton in second messenger signaling for inflammatory pain. Neuron 2003, 39, 613–624. [Google Scholar] [CrossRef]
- Goswami, C.; Dreger, M.; Otto, H.; Schwappach, B.; Hucho, F. Rapid disassembly of dynamic microtubules upon activation of the capsaicin receptor TRPV1. J. Neurochem. 2006, 96, 254–266. [Google Scholar] [CrossRef]
- Przewlocki, R.; Przewlocka, B. Opioids in chronic pain. Eur. J. Pharmacol. 2001, 429, 79–91. [Google Scholar] [CrossRef]
- Van Aelst, L.; Cline, H.T. Rho GTPases and activity-dependent dendrite development. Curr. Opin. Neurobiol. 2004, 14, 297–304. [Google Scholar] [CrossRef]
- Cerri, C.; Fabbri, A.; Vannini, E.; Spolidoro, M.; Costa, M.; Maffei, L.; Fiorentini, C.; Caleo, M. Activation of Rho GTPases triggers structural remodeling and functional plasticity in the adult rat visual cortex. J. Neurosci. 2011, 31, 15163–15172. [Google Scholar] [CrossRef]
- Frenkel, M.Y.; Bear, M.F. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron 2004, 44, 917–923. [Google Scholar] [CrossRef]
- Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005, 33, 891–895. [Google Scholar] [CrossRef]
- Van Galen, E.J.; Ramakers, G.J. Rho proteins, mental retardation and the neurobiological basis of intelligence. Prog. Brain Res. 2005, 147, 295–317. [Google Scholar] [CrossRef]
- Chen, Q.; Zhu, Y.C.; Yu, J.; Miao, S.; Zheng, J.; Xu, J.; Zhou, Y.; Li, D.; Zhang, C.; Tao, J.; et al. CDKL5, a protein associated with Rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J. Neurosci. 2011, 30, 12777–12786. [Google Scholar]
- Diana, G.; Valentini, G.; Travaglione, S.; Falzano, L.; Pieri, M.; Zona, C.; Meschini, S.; Fabbri, A.; Fiorentini, C. Enhancement of learning and memory after activation of cerebral Rho GTPases. Proc. Natl. Acad. Sci. USA 2007, 104, 636–641. [Google Scholar] [CrossRef]
- De Viti, S.; Martino, A.; Musilli, M.; Fiorentini, C.; Diana, G. The Rho GTPase activating CNF1 improves associative working memory for object-in-place. Behav. Brain Res. 2010, 212, 78–83. [Google Scholar] [CrossRef]
- Shahbazian, M.; Young, J.; Yuva-Paylor, L.; Spencer, C.; Antalffy, B.; Noebels, J.; Armstrong, D.; Paylor, R.; Zoghbi, H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 2002, 35, 243–254. [Google Scholar] [CrossRef]
- Moretti, P.; Bouwknecht, J.A.; Teague, R.; Paylor, R.; Zoghbi, H.Y. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 2005, 14, 205–220. [Google Scholar]
- Moretti, P.; Levenson, J.M.; Battaglia, F.; Atkinson, R.; Teague, R.; Antalffy, B.; Armstrong, D.; Arancio, O.; Sweatt, J.D.; Zoghbi, H.Y. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 2006, 26, 319–327. [Google Scholar] [CrossRef]
- De Filippis, B.; Ricceri, L.; Laviola, G. Early postnatal behavioral changes in the Mecp2-308 truncation mouse model of Rett syndrome. Genes. Brain Behav. 2010, 9, 213–223. [Google Scholar] [CrossRef]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro 2010, 2, e00045. [Google Scholar]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar]
- Roses, A.D.; Saunders, A.M. APOE is a major susceptibility gene for alzheimer’s disease. Curr. Opin. Biotechnol. 1994, 5, 663–667. [Google Scholar] [CrossRef]
- Johnson, L.A.; Arbones-Mainar, J.M.; Fox, R.G.; Pendse, A.A.; Altenburg, M.K.; Kim, H.S.; Maeda, N. Apolipoprotein E4 exaggerates diabetic dyslipidemia and atherosclerosis in mice lacking the LDL receptor. Diabetes 2011, 60, 2285–2294. [Google Scholar] [CrossRef]
- Shaftel, S.S.; Griffin, W.S.; O’Banion, M.K. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflamm. 2008, 5, 7. [Google Scholar] [CrossRef]
- Zhu, Y.; Nwabuisi-Heath, E.; Dumanis, S.B.; Tai, L.M.; Yu, C.; Rebeck, G.W.; Ladu, M.J. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 2012, 60, 559–569. [Google Scholar] [CrossRef]
- Chen, X.; Yan, S.D. Mitochondrial Abeta: A potential cause of metabolic dysfunction in Alzheimer’s disease. IUBMB Life 2006, 58, 686–694. [Google Scholar] [CrossRef]
- Chen, X.; Stern, D.; Yan, S.D. Mitochondrial dysfunction and Alzheimer’s disease. Curr. Alzheimer Res. 2006, 3, 515–520. [Google Scholar] [CrossRef]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef]
- Lu, Q.; Longo, F.M.; Zhou, H.; Massa, S.M.; Chen, Y.H. Signaling through Rho GTPase pathway as viable drug target. Curr. Med. Chem. 2009, 16, 1355–1365. [Google Scholar] [CrossRef]
- Raz, L.; Zhang, Q.G.; Zhou, C.F.; Han, D.; Gulati, P.; Yang, L.C.; Yang, F.; Wang, R.M.; Brann, D.W. Role of Rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PLoS ONE 2010, 5, e12606. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Travaglione, S.; Loizzo, S.; Ballan, G.; Fiorentini, C.; Fabbri, A. The E. coli CNF1 as a Pioneering Therapy for the Central Nervous System Diseases. Toxins 2014, 6, 270-282. https://doi.org/10.3390/toxins6010270
Travaglione S, Loizzo S, Ballan G, Fiorentini C, Fabbri A. The E. coli CNF1 as a Pioneering Therapy for the Central Nervous System Diseases. Toxins. 2014; 6(1):270-282. https://doi.org/10.3390/toxins6010270
Chicago/Turabian StyleTravaglione, Sara, Stefano Loizzo, Giulia Ballan, Carla Fiorentini, and Alessia Fabbri. 2014. "The E. coli CNF1 as a Pioneering Therapy for the Central Nervous System Diseases" Toxins 6, no. 1: 270-282. https://doi.org/10.3390/toxins6010270
APA StyleTravaglione, S., Loizzo, S., Ballan, G., Fiorentini, C., & Fabbri, A. (2014). The E. coli CNF1 as a Pioneering Therapy for the Central Nervous System Diseases. Toxins, 6(1), 270-282. https://doi.org/10.3390/toxins6010270