Cytotoxic Necrotizing Factor 1 Contributes to Escherichia coli Meningitis

1
Division of Pediatric Urology, Johns Hopkins University School of Medicine, 1800 Orleans Street, Bloomberg, 7308, Baltimore, MD 21287, USA
2
Division of Pediatric Infectious Diseases, Johns Hopkins University School of Medicine, 200 North Wolfe St, Room 3157, Baltimore, MD 21287, USA
*
Author to whom correspondence should be addressed.
Toxins 2013, 5(11), 2270-2280; https://doi.org/10.3390/toxins5112270
Submission received: 1 October 2013
/
Revised: 13 November 2013
/
Accepted: 18 November 2013
/
Published: 22 November 2013
(This article belongs to the Special Issue Cytotoxic Necrotizing Factors)
Abstract
:E. coli is the most common Gram-negative bacteria causing neonatal meningitis, and E. coli meningitis continues to be an important cause of mortality and morbidity throughout the world. Recent reports of E. coli meningitis caused by antimicrobial resistant strains are a particular concern. These findings indicate that a novel strategy is needed to identify new targets for prevention and therapy of E. coli meningitis. Cytotoxic necrotizing factor 1 (CNF1) is a bacterial virulence factor associated principally with E. coli strains causing urinary tract infection and meningitis. We have shown that CNF1 contributes to E. coli invasion of the blood-brain barrier and penetration into the brain, the essential step in the development of E. coli meningitis, and identified the host receptor for CNF1, 37-kDa laminin receptor precursor (37LRP). CNF1, however, is a cytoplasmic protein and its contribution to E. coli invasion of the blood-brain barrier requires its secretion from the bacterial cytoplasm. No signal peptide is found in the CNF1 sequence. CNF1 secretion is, therefore, a strategy utilized by meningitis-causing E. coli to invade the blood-brain barrier. Elucidation of the mechanisms involved in CNF1 secretion, as shown in this report with the involvement of Fdx and YgfZ provides the novel information on potential targets for prevention and therapy of E. coli meningitis by virtue of targeting the secretion of CNF1.
1. Introduction
Neonatal Gram-negative bacillary meningitis continues to be an important cause of mortality and morbidity throughout the world. Case fatality rates have ranged between 15% and 40%, and approximately 50% of survivors sustain neurological sequelae [1,2,3,4,5]. A major contributing factor to such mortality and morbidity is our incomplete understanding of the pathogenesis of this disease [6,7,8,9,10,11].
E. coli is the most common Gram-negative bacillary organism that causes meningitis, particularly during the neonatal period. Both clinical and experimental data indicate limited efficacy with antimicrobial therapy alone for the treatment of Gram-negative bacillary meningitis [11,12,13]. Recent reports of neonatal meningitis caused by E. coli strains producing CTX-M-type or TEM-type extended-spectrum β-lactamases are a particular concern [14,15]. Drug-resistant clonal group ST131 is prevalent among E. coli strains causing extraintestinal infection and some carry CTX-M-type extended-spectrum β-lactamases [14]. Cerebrospinal fluid (CSF) is devoid of sufficient opsonic and phagocytic activity against meningitis-causing bacteria and eradication of the infecting pathogens from the CSF is entirely dependent on antibiotics [10,11]. An emergence of antibiotic resistance is, therefore, an important issue in the treatment of E. coli meningitis. Taken together, these findings indicate that a novel strategy is needed to enhance our knowledge on the pathogenesis and also to identify new targets for prevention and therapy of E. coli meningitis.
2. E. coli Penetration of the Blood-Brain Barrier
Several lines of evidence from human cases of E. coli meningitis and animal models of experimental hematogenous E. coli meningitis indicate that E. coli invasion into the brain follows a high level of bacteremia and cerebral capillaries are the portal of entry into the brain [16,17,18], but how meningitis-causing E. coli strains invade the blood-brain barrier and penetrate into the brain remains incompletely understood.
The development of both in vitro and in vivo models of the blood-brain barrier has facilitated our investigations on the mechanisms of microbial traversal of the blood-brain barrier, a key step required for the development of E. coli meningitis [6,7,8,9,10,11,19,20]. The blood-brain barrier protects the brain from microbes circulating in the blood, but meningitis-causing pathogens have been shown to penetrate the blood-brain barrier transcellularly, paracellularly and/or by means of infected phagocytic cells (Trojan-horse mechanism) [9]. Recent studies have demonstrated that meningitis-causing E. coli strains exhibit the ability to penetrate the blood-brain barrier by transcellular mechanism. This concept was shown by the demonstration that meningitis-causing E. coli strains invade human brain mircovascular endothelial cells (HBMEC) without affecting the HBMEC integrity, and penetrate into the brain without affecting the blood-brain barrier permeability and accompanying phagocytes [6,7,8,9,10,11,18,21].
3. Cytotoxic Necrotizing Factor 1 (CNF1) Contributes to E. coli Invasion of HBMEC and Penetration into the Brain
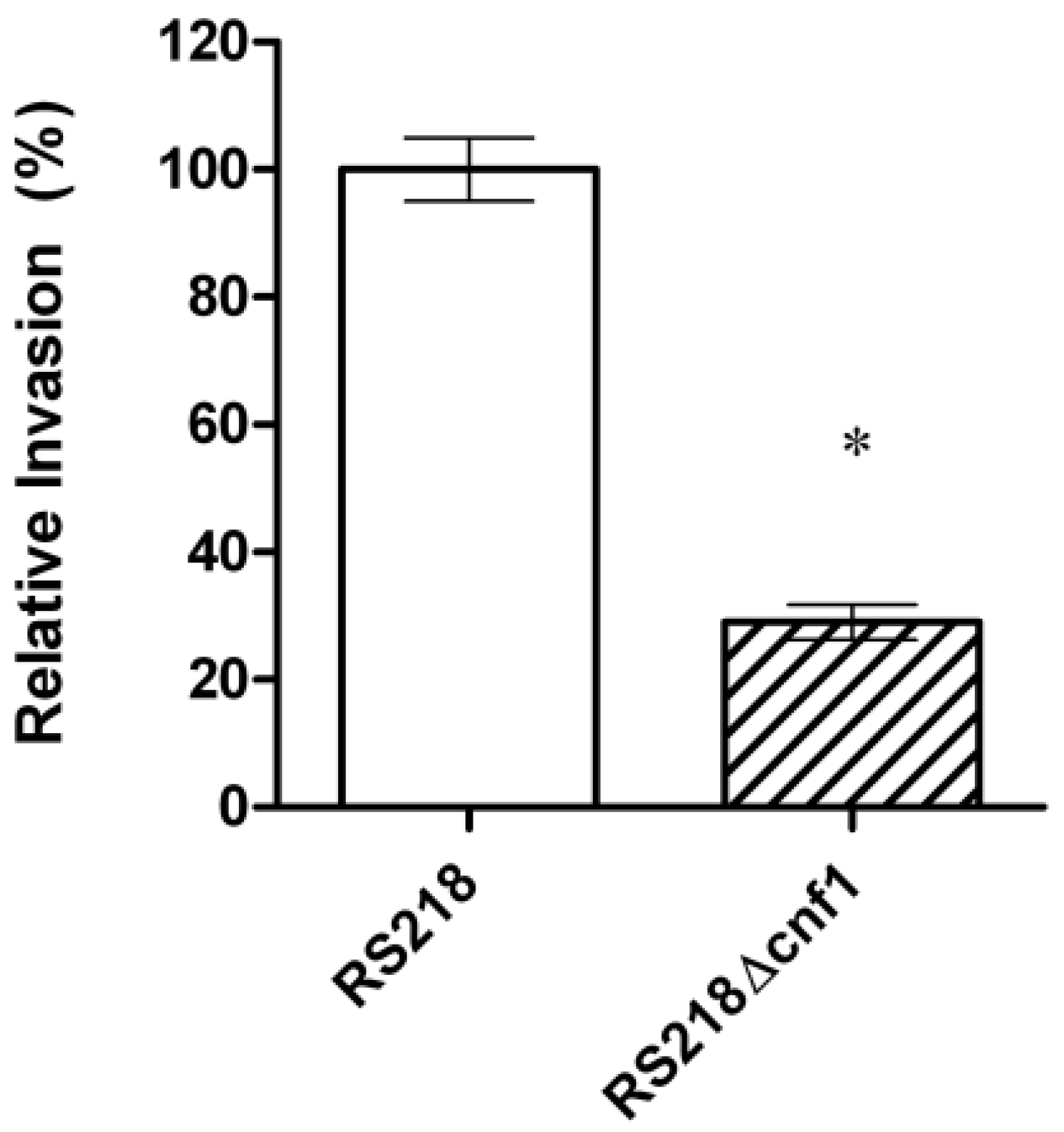
CNF1 is a bacterial virulence factor associated principally with extraintestinal pathogenic E. coli strains causing urinary tract infection and meningitis [22]. Our previous studies using signature-tagged mutagenesis identified CNF1 that contributes to E. coli invasion of HBMEC monolayer and penetration into the brain in the infant rat model of experimental hematogenous E. coli meningitis, which mimicks the pathogenesis of E. coli meningitis in humans, e.g., hematogenous invasion of the meninges [23,24,25]. An isogenic mutant deleted of cnf1 was significantly less invasive in HBMEC (Figure 1) and less able to penetrate into the brain in vivo, and its invasive defect was restored to the level of the parent strain by complementation with respective wild-type gene [24].
Figure 1.
Relative invasion frequency (mean ± SD of three separate experiments in triplicate) of meningitis-causing E. coli strain RS218 and its CNF1 deletion mutant in human brain mircovascular endothelial cells (HBMEC) monolayer. The ΔCNF1 mutant was significantly defective (*p < 0.02 compared to the parent strain RS218, which was set at 100%) in HBMEC invasion.
Figure 1.
Relative invasion frequency (mean ± SD of three separate experiments in triplicate) of meningitis-causing E. coli strain RS218 and its CNF1 deletion mutant in human brain mircovascular endothelial cells (HBMEC) monolayer. The ΔCNF1 mutant was significantly defective (*p < 0.02 compared to the parent strain RS218, which was set at 100%) in HBMEC invasion.
CNF1 has been shown to activate RhoGTPases such as RhoA, Rac1 and Cdc42 and induce uptake of latex beads, bacteria and apoptotic bodies into non-professional phagocytes such as epithelial and endothelial cells [26,27,28,29]. The above-mentioned in vitro and in vivo effects of CNF1 in E. coli penetration of the blood-brain barrier, however, are dependent upon CNF activation of RhoA [24]. These conclusions were based on our demonstration that (a) the CNF1 deletion mutant exhibited significantly decreased invasion and RhoA activation compared to the parent strain in HBMEC and (b) the CNF1 mutant’s invasion frequency was restored to the level of the parent strain in HBMEC expressing constitutively active RhoA. Rac1 activation has been shown to contribute E. coli invasion of HBMEC, but Rac1 activation occurred in response to non-CNF1 E. coli factors (IbeA and OmpA) and Rac1-mediated invasion of HBMEC was shown to be under the control of STAT3 [30].
CNF1-producing E. coli infections have also been hypothesized to contribute to cancer development by counteracting apoptosis, and inducing pro-inflammatory cytokines’ release, COX2 expression, NF-kB activation [31]. Of interest, the host receptor for CNF1, 37-kDa laminin receptor precursor (37LRP)/67-kDa laminin receptor (67LR) is also shown to be associated with the progression of a wide variety of cancers [32,33]. It remains speculative to suggest whether CNF1-producing E. coli infection and/or its interaction with 37LRP/67LR may be a risk factor for cancer development such as colon cancer.
4. Identification of the Host Receptor for CNF1
CNF1 is an AB-type toxin, composed of the N-terminal cell binding domain and the C-terminal catalytic domain possessing a deaminase activity through the site-specific deamination of a Gln residue to Glu [34,35].
CNF1 has been suggested to be internalized via receptor-mediated endocytosis upon binding to a cell surface receptor [22], but the identity of its receptor was unknown. We have identified the HBMEC receptor for CNF1 by yeast two-hybrid screening of the HBMEC cDNA library using the N-terminal cell-binding domain of CNF1 as bait [33]. This receptor, 37-kDa laminin receptor precursor (37LRP) interacted with the N-terminal CNF1 and full-length CNF1 but not with the C-terminal CNF1. CNF1-mediated RhoA activation and bacterial uptake were inhibited by 37LRP antisense oligodeoxynucleotides, whereas they were increased in 37LRP-overexpressing cells, demonstrating a direct correlation between effects of CNF1 and levels of 37LRP expression in HBMEC [33]. These findings indicate that CNF1 interaction with its receptor, 37LRP, is the initial step required for CNF1-mediated RhoA activation and bacterial uptake in eukaryotic cells. The 37LRP is a ribosome-associated cytoplasmic protein and shown to be a precursor of 67-kDa laminin receptor (67LR). It is unclear how 67LR is matured and synthesized from the 37LRP, but mature 67LR is shown to be present on the cell surface and functions as a membrane receptor for the adhesive basement membrane protein laminin [36].
We have shown that incubation of HBMEC with CNF1-expressing E. coli up-regulates 67LR expression and recruits 67LR to the site of invading E. coli in a CNF1-dependent manner [37], supporting that 67LR participates in CNF1-expressing E. coli invasion of HBMEC. Of interest, 37LRP is also shown to be a cellular target for other CNS-infecting microorganisms, including S. pneumoniae, N. meningitidis, H. influenzae type b, dengue virus, adeno-associated virus, Venezuelan equine encephalitis virus, and prion protein [10]. The mechanisms by which the same receptor is involved in CNS penetration by different organisms, however, remain to be established.
5. Secretion of CNF1 across the Bacterial Inner and Outer Membrane
As indicated above, CNF1 is a key microbial factor contributing to E. coli invasion of HBMEC monolayer and penetration into the brain via the interaction with its receptor (37LRP) on HBMEC [24,33]. CNF1, however, is a cytoplasmic protein and execution of its contribution to E. coli invasion of the blood-brain barrier requires its secretion from the bacterial cytoplasm across the bacterial inner and outer membranes. No signal peptide is present in the CNF1 sequence. CNF1 secretion is, therefore, a strategy utilized by meningitis-causing E. coli to invade the blood-brain barrier. CNF1 is closely associated with α-hemolysin, but type 1 secretion system responsible for secretion of α-hemolysin did not affect CNF1 secretion. CNF1 secretion across the bacterial inner membrane did not involve the Sec and Tat secretion systems, and none of currently known secretion systems are involved in CNF1 secretion across the bacterial outer membrane. Recent studies have shown that CNF1 is transported to the culture supernatant in a complex with outer membrane vesicles in uropathogenic E. coli strains [38,39], but the underlying mechanisms remain unclear.
6. Identification of Tn5 Mutants Defective in CNF1 Secretion
We determined the genes involved in CNF1 secretion in meningitis-causing E. coli. For this study, we constructed NBC (CNF1-Bla-CAT) strain, where β-lactamase (Bla) was used as the reporter gene and translationally fused to the C-terminal of cnf1 gene in the chromosome of meningitis-causing E. coli strain RS218, as described previously [40,41]. In strain NBC, Bla secretion is entirely dependent upon CNF1 secretion and the secretion of CNF1 can be monitored by measuring β-lactamase activity. Our screen of Tn5 transposon mutants of NBC strain identified two mutants with defects in Bla activity but without affecting CNF1 expression (NBC-14H2 and NBC-1E6) (Figure 2).
Figure 2.
Bla activity in the culture supernatants of strain RS218 (negative control), strain NBC (positive control) and two Tn5 mutants (NBC-14H2 and NBC-1E6). The Bla activity (mean ± SD) represents the absorbance measurements of three experiments in triplicate. (modified from [40,41]).

NBC-14H2 and NBC-1E6 were found to have Tn5 insertions in fdx and ygfZ, respectively. We have subsequently shown that ferredoxin (Fdx) is involved in CNF1 secretion across the bacterial inner membrane and YgfZ is involved in CNF1 secretion into outer membrane vesicles [40,41].
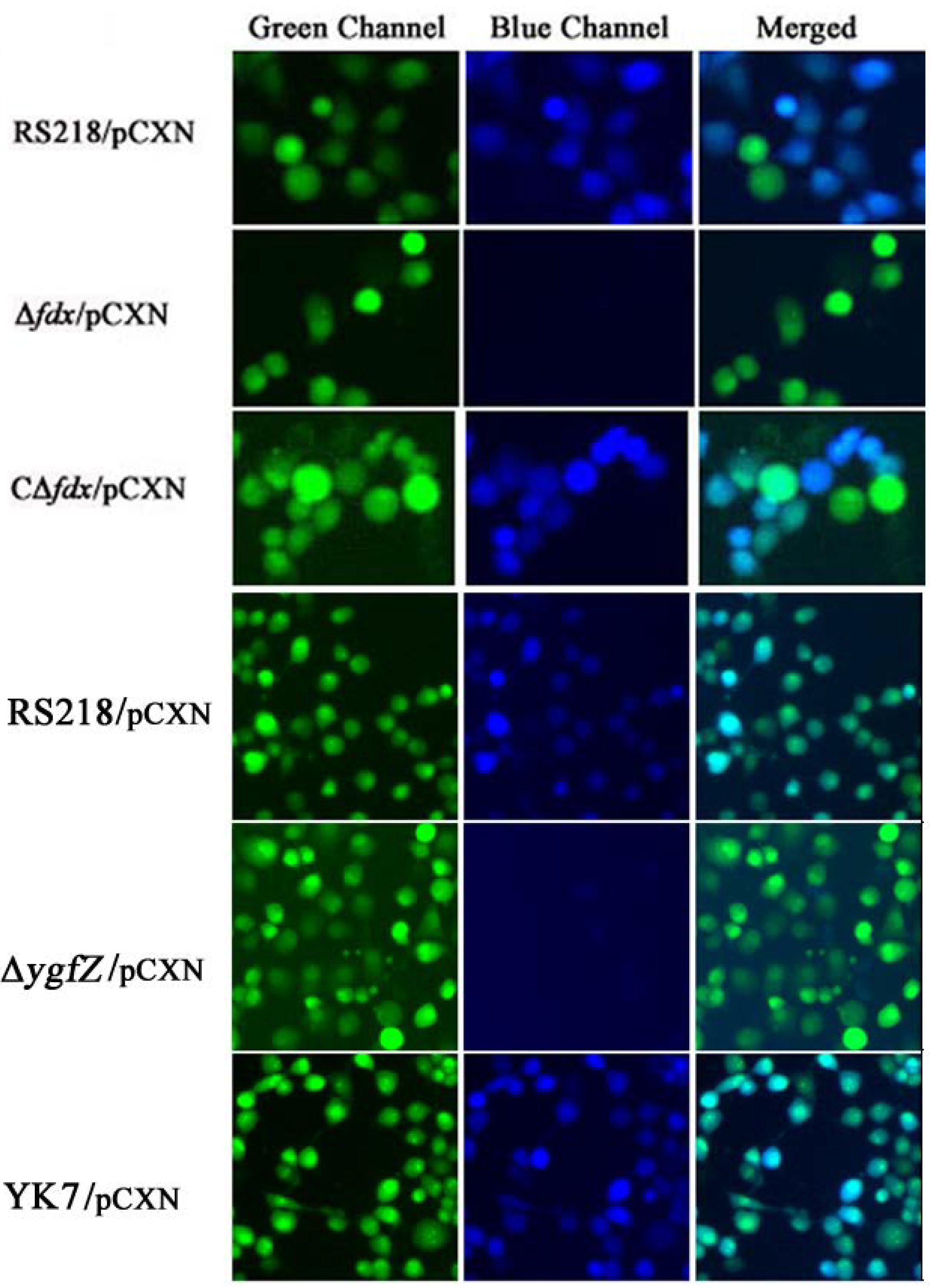
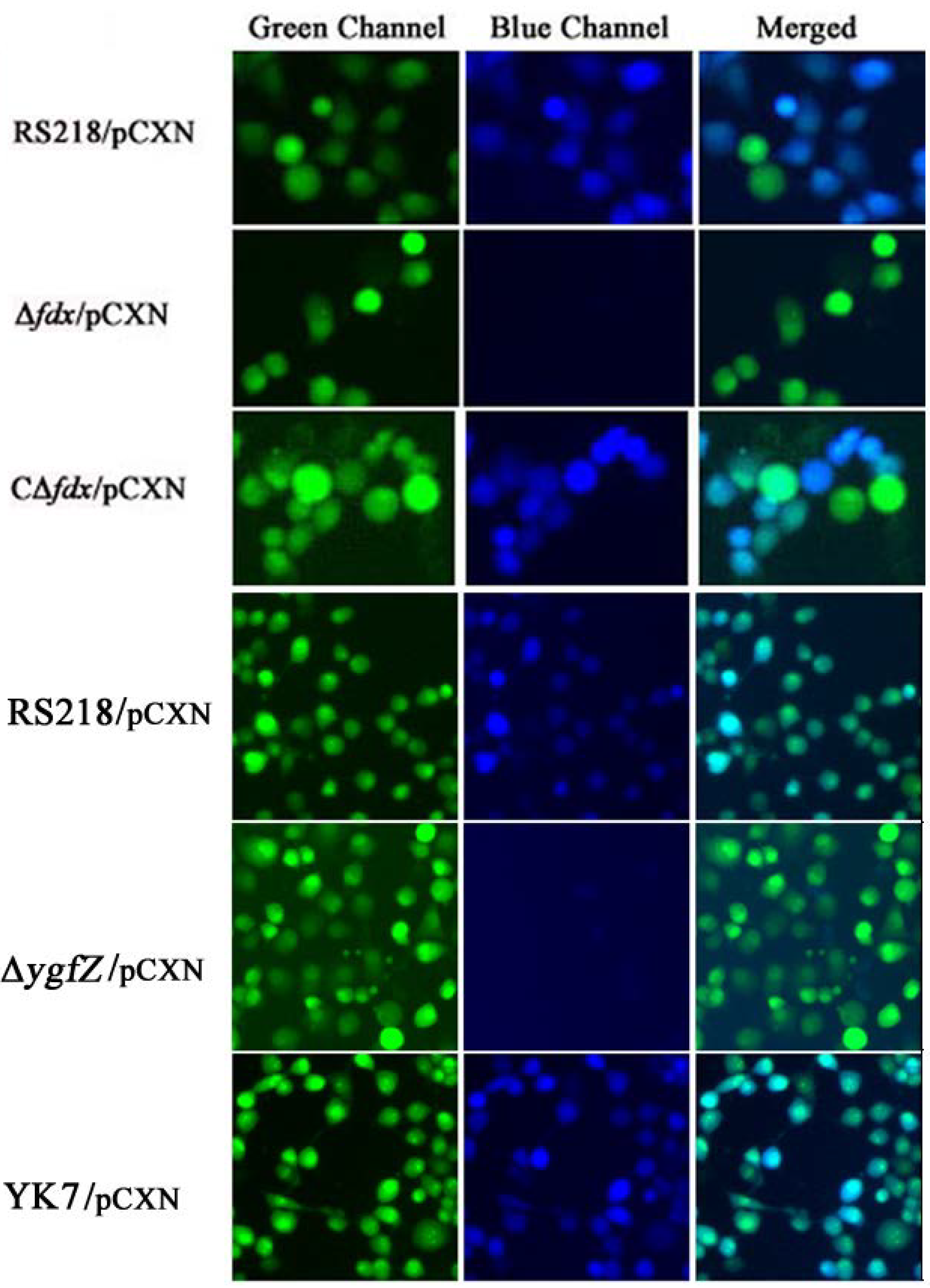
We also showed that CNF1 translocation into HBMEC monolayer did not occur in the fdx and ygfZ deletion mutants, and these defects were restored by complementation with fdx and ygfZ, respectively (Figure 3). Briefly, CNF1 translocation into HBMEC monolayer was examined using E. coli strains harboring the plasmid pCXN, as previously described [40,41]. In pCXN, CNF1 is translationally fused with Bla, and the expression of the CNF1-Bla fusion from pCXN was induced with 1 mM IPTG. On the day of infection, HBMEC were pre-loaded with CCF4/AM dye and incubated with IPTG-treated bacteria. Non-fluorescent esterified CCF4/AM, upon entry into HBMEC monolayer is converted to fluorescent green CCF4 by cellular esterases. Translocation of CNF1-Bla into HBMEC monolayer induces catalytic cleavage of the CCF4 β-lactam ring, which produces an easily detectable color change in CCF4 fluorescence from green to blue emission. After 45 min of infection, the translocation of CNF1-Bla into HBMEC monolayer was examined under fluorescent microscope (Figure 3).
Figure 3.
Analysis of CNF1 secretion into HBMEC monolayer. Translocation of CNF1-Bla into HBMEC induces catalytic cleavage of the CCF4 β-lactam ring, which produces an easily detectable color change in CCF4 fluorescence from green to blue emission. In the parent strain RS218/pCXN, CNF1-Bla was, as expected, successfully translocated into HBMEC, as shown by the emission of blue fluorescence. In contrast, Δfdx and ΔygfZ mutants harboring pCXN showed no blue emission, indicating no translocation of CNF1 into HBMEC. Of note, CΔfdx and YK7 represent the complemented strains of Δfdx and ΔygfZ mutants with wild type fdx and ygfZ, respectively, which showed predominant blue emission, indicating the translocation of CNF1 into HBMEC (modified from [40,41]).
Figure 3.
Analysis of CNF1 secretion into HBMEC monolayer. Translocation of CNF1-Bla into HBMEC induces catalytic cleavage of the CCF4 β-lactam ring, which produces an easily detectable color change in CCF4 fluorescence from green to blue emission. In the parent strain RS218/pCXN, CNF1-Bla was, as expected, successfully translocated into HBMEC, as shown by the emission of blue fluorescence. In contrast, Δfdx and ΔygfZ mutants harboring pCXN showed no blue emission, indicating no translocation of CNF1 into HBMEC. Of note, CΔfdx and YK7 represent the complemented strains of Δfdx and ΔygfZ mutants with wild type fdx and ygfZ, respectively, which showed predominant blue emission, indicating the translocation of CNF1 into HBMEC (modified from [40,41]).
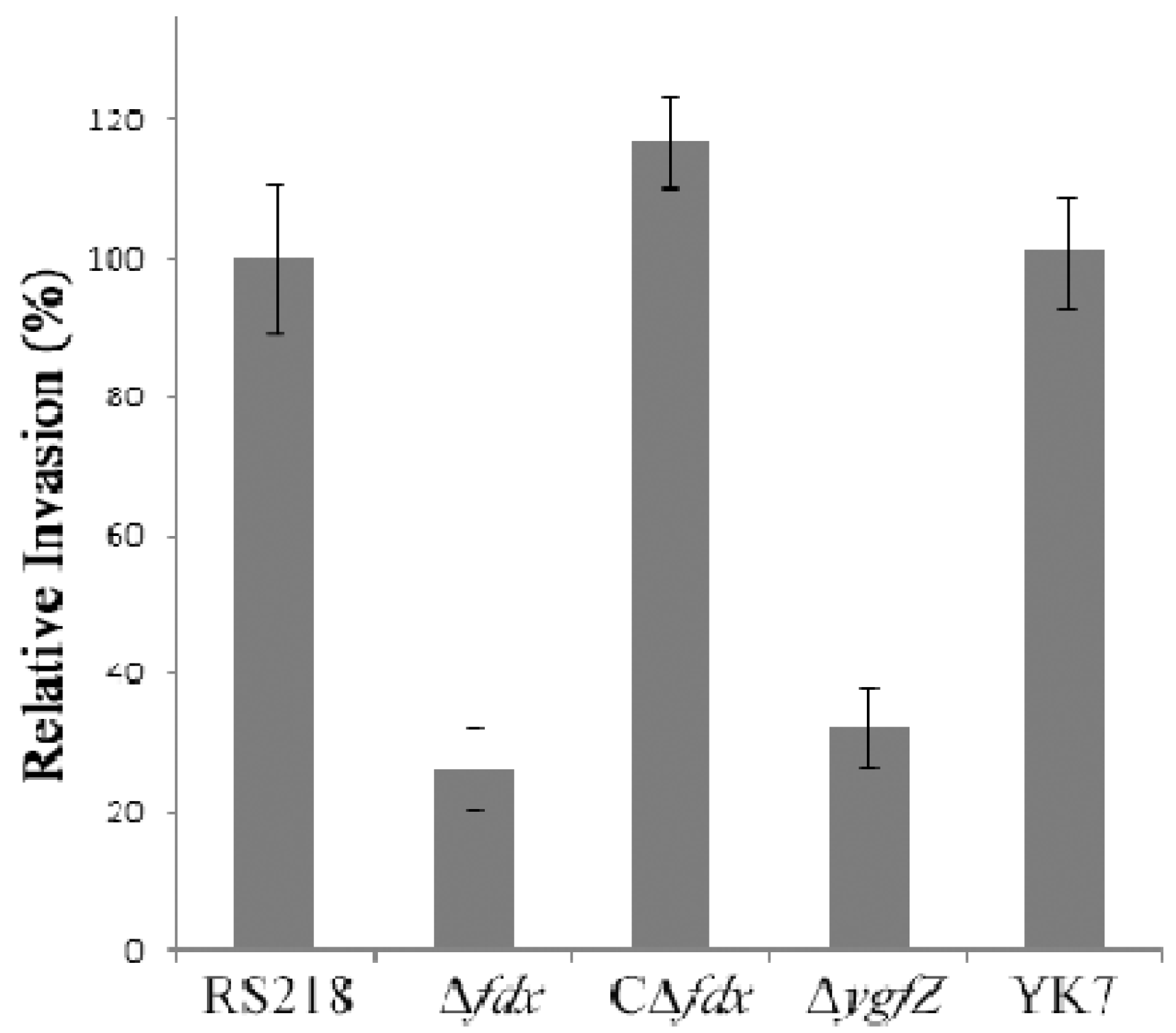
We next showed that the fdx and ygfZ mutants were significantly defective in invasion of HBMEC compared to the parent strain by virtue of their failure to secrete CNF1, and their invasion defects were restored to the levels of the parent strain by complementation with respective wild type genes (Figure 4).
Elucidation of the mechanisms involved in CNF1 secretion will, therefore, help in enhancing our knowledge on the pathogenesis of E. coli meningitis and also in developing a novel strategy targeting CNF1 secretion in prevention of E. coli meningitis. Taken together, these findings suggest that modulation of bacterial secretion systems (CNF1 secretion) is likely to represent a novel approach for investigating the pathogenesis and prevention of E. coli meningitis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
7. Modulation of Host Receptor and Signaling Molecules for Prevention of E. coli Invasion of the Blood-Brain Barrier
As indicated above, meningitis-causing E. coli penetration into the brain requires E. coli invasion of HBMEC, involving specific microbial-host interactions (so-called ligand-receptor interactions) and specific host cell signaling molecules [9,10,11]. We have shown that CNF1-mediated E. coli uptake and RhoA activation in HBMEC were inhibited by 37LRP antisense oligodeoxynucleotides, whereas they were increased in 37LRP-overexpressing cells [33]. These findings support the concept that the expression level of host cell receptor(s) dictates the fate of E. coli invasion of HBMEC in a bacterial ligand-dependent manner. In addition, pharmacological inhibition of the host cell receptor (e.g., 37LRP) and host cell signaling molecules (e.g., RhoA) involved in E. coli invasion of HBMEC was efficient in preventing E. coli penetration of the blood-brain barrier [42]. These findings demonstrate that pharmacological inhibition of the HBMEC receptors that interact with E. coli factors and host cell signaling molecules contributing to E. coli invasion of HBMEC might be a novel strategy for prevention of E. coli meningitis.
8. Conclusion
E. coli meningitis continues to be an important cause of mortality and morbidity, and recent reports of E. coli meningitis caused by antimicrobial resistant strains are a particular concern. These findings indicate that a novel strategy is needed to identify new targets for prevention and therapy of E. coli meningitis. CNF1 is a key microbial factor contributing to E. coli penetration of the blood-brain barrier, the essential step in the development of E. coli meningitis, but CNF1 is a cytoplasmic protein, and CNF1 secretion is needed to interact with the host receptor on the blood-brain barrier. CNF secretion and CNF1 interaction with the host receptor are likely to represent a novel target for prevention and therapy of E. coli meningitis.
Acknowledgements
This work was supported in part by NIH grants, NS26310 and AI84984
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gladstone, I.M.; Ehrenkranz, R.A.; Edberg, S.C.; Baltimore, R.S. A ten-year review of neonatal sepsis and comparison with the previous fifty-year experience. Pediatr. Infect. Dis. J. 1990, 9, 819–825. [Google Scholar] [CrossRef]
- Unhanand, M.; Musatafa, M.M.; McCracken, G.H.; Nelson, J.D. Gram-negative enteric bacillary meningitis: A twenty-one year experience. J. Pediatr. 1993, 122, 15–21. [Google Scholar] [CrossRef]
- Dawson, K.G.; Emerson, J.C.; Burns, J.L. Fifteen years of experience with bacterial meningitis. Pediatr. Infect. Dis. J. 1999, 18, 816–822. [Google Scholar] [CrossRef]
- Klinger, G.; Chin, C.-N.; Beyene, J.; Perlman, M. Predicting the outcome of neonatal bacterial meningitis. Pediatrics 2000, 106, 477–482. [Google Scholar] [CrossRef]
- Stevens, J.P.; Eames, M.; Kent, A.; Halket, S.; Holt, D.; Harvey, D. Long term outcome of neonatal meningitis. Arch. Dis. Child. Fetal Neonatal Ed. 2003, 88, F179–F184. [Google Scholar] [CrossRef]
- Kim, K.S. E. coli translocation at the blood-brain barrier. Infect. Immun. 2001, 69, 5217–5222. [Google Scholar] [CrossRef]
- Kim, K.S. Strategy of E. coli for crossing the blood-brain barrier. J. Infect. Dis. 2002, 186, S220–S224. [Google Scholar] [CrossRef]
- Kim, K.S. Neurological diseases: Pathogenesis of bacterial meningitis: From bacteremia to neuronal injury. Nat. Rev. Neurosci. 2003, 4, 376–385. [Google Scholar]
- Kim, K.S. Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 2008, 6, 625–634. [Google Scholar] [CrossRef]
- Kim, K.S. Acute bacterial meningitis in infants and children. Lancet Infect. Dis. 2010, 10, 32–42. [Google Scholar] [CrossRef]
- Kim, K.S. Current concepts on the pathogenesis of E. coli meningitis: Implications for prevention and therapy. Curr. Opin. Infect. Dis. 2012, 25, 273–278. [Google Scholar] [CrossRef]
- McCracken, G.H., Jr.; Threlkeld, N.; Mize, S.; Baker, C.J.; Kapal, S.L.; Fraingezicht, I.; Feldman, W.F.; Schad, U.; The Neonatal Meningitis Cooperative Study Group. Moxalactam therapy for neonatal meningitis due to gram-negative sepsis enteric bacilli. JAMA 1984, 252, 1427–1432. [Google Scholar] [CrossRef]
- Kim, K.S. Comparison of cefotaxime, imipenem-cilastatin, ampicillin-gentamicin and ampicillin-chloramphenicol in the treatment of experimental E. coli bacteremia and meningitis. Antimicrob. Agents Chemother. 1985, 28, 433–436. [Google Scholar] [CrossRef]
- Blanco, J.; Mora, A.; Mamani, R.; Lopez, C.; Blanco, M.; Dabhi, G.; Herrea, A.; Blanco, J.E.; Alonso, M.P.; Garcia-Garrote, F.; et al. National survey of Escherichia coli causing extraintestinal infections reveal the spread of drug-resistant clonal groups O25b:H4-B2-ST131, O15:H5-D-ST393 and CGA-D-ST69 with high virulence gene content in Spain. J. Antimicrob. Chemother. 2011, 66, 2011–2012. [Google Scholar] [CrossRef]
- Moissenet, D.; Slauze, B.; Clermont, O.; Bingen, E.; Arlet, G.; Denamur, E.; Merens, A.; Mitanchez, D.; Vu-Thien, H. Meningitis caused by Escherichia coli producing TEM-52 extended-spectrum β-lactamase within an extensive outbreak in a neonatal ward: epidemiological investigation and characterization of the strain. J. Clin. Microbiol. 2011, 48, 2459–2463. [Google Scholar]
- Dietzman, D.E.; Fischer, G.W.; Schoenknecht, F.D. Neonatal Escherichia coli septicemia—Bacterial counts in blood. J. Pediatr. 1974, 85, 128–130. [Google Scholar] [CrossRef]
- Berman, P.H.; Banker, B.Q. Neonatal meningitis: A clinical and pathologic study of 29 cases. Pediatrics 1966, 38, 6–24. [Google Scholar]
- Kim, K.S.; Itabashi, H.; Gemski, P.; Sadoff, J.; Warren, R.L.; Cross, A.S. The K1 capsule is the critical determinant in the development of Escherichia coli meningitis in the rat. J. Clin. Invest. 1992, 90, 897–905. [Google Scholar] [CrossRef]
- Stins, M.F.; Gilles, F.; Kim, K.S. Selective expression of adhesion molecules on human brain microvascular endothelial cells. J. Neuroimmunol. 1997, 76, 81–90. [Google Scholar] [CrossRef]
- Stins, M.F.; Badger, J.L.; Kim, K.S. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 2001, 30, 19–28. [Google Scholar] [CrossRef]
- Kim, K.S.; Wass, C.A.; Cross, A.S. Blood-brain barrier permeability during the development of experimental bacterial meningitis in the rat. Exp. Neurol. 1997, 145, 253–257. [Google Scholar] [CrossRef]
- Boquet, P. The cytotoxic necrotizing factor 1 (CNF1) from Escherichia coli. Toxicon 2001, 39, 1673–1680. [Google Scholar] [CrossRef]
- Badger, J.; Wass, C.; Weissman, S.; Kim, K.S. Application of signature-tagged mutagenesis for the identification of E. coli K1 genes that contribute to invasion of the blood-brain barrier. Infect. Immun. 2000, 68, 5056–5061. [Google Scholar] [CrossRef]
- Khan, N.A.; Wang, Y.; Kim, K.J.; Chung, J.W.; Wass, C.A.; Kim, K.S. Cytotoxic necrotizing factor 1 contributes to Escherichia coli K1 invasion of the central nervous system. J. Biol. Chem. 2002, 277, 15607–15612. [Google Scholar]
- Khan, N.A.; Shin, S.; Chung, J.W.; Kim, K.J.; Elliot, S.; Wang, Y.; Kim, K.S. Outer membrane protein A and cytotoxic necrotizing factor-1 use diverse signaling mechanisms for Escherichia coli K1 invasion of human brain microvascular endothelial cells. Microb. Pathog. 2003, 35, 35–42. [Google Scholar] [CrossRef]
- Fiorentini, C.; Fabbri, A.; Flatau, G.; Donelli, G.; Matarrese, P.; Lemichez, E.; Falzano, L.; Boquet, P. Escherichia coli cytotoxic necrotizing factor 1 (CNF1), a toxin that activates the Rho GTPase. J. Biol. Chem. 1997, 272, 19532–19537. [Google Scholar] [CrossRef]
- Fabbri, A.; Falzano, L.; Travaglione, S.; Stringaro, A.; Malorni, W.; Fais, S.; Fiorentini, C. Rho-activating Escherichia coli cytotoxic necrotizing factor 1: Macropinocytosis of apoptotic bodies in human epithelial cells. Int. J. Med. Microbiol. 2002, 291, 551–554. [Google Scholar]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clément, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar]
- Visvikis, O.; Boyer, L.; Torrino, S.; Doye, A.; Lemonnier, M.; Lorès, P.; Rolando, M.; Flatau, G.; Mettouchi, A.; Bouvard, D.; et al. Escherichia coli producing CNF1 toxin hijacks Tollip to trigger Rac1-dependent cell invasion. Traffic 2011, 12, 579–590. [Google Scholar] [CrossRef]
- Maruvada, R.; Kim, K.S. IbeA and OmpA of Escherichia coli K1 exploit Rac1 activation for invasion of human brain microvascular endothelial cells. Infect. Immun. 2012, 80, 2035–2041. [Google Scholar] [CrossRef]
- Fabbri, A.; Travaglione, S.; Ballan, G.; Loizzo, S.; Fiorentini, C. The cytotoxic necrotizing factor 1 from E. coli: A janus toxin playing with cancer regulators. Toxins 2013, 5, 1462–1474. [Google Scholar] [CrossRef]
- Stallmach, A.; Orzechowski, H.D.; Feldmann, P.; Riecken, E.O.; Zeitz, M.; Herbst, H. 32/67-kD laminin receptor expression in human colonic neoplasia: elevated transcript levels correlate with the degree of epithelial dysplasia. Am. J. Gastroenterol. 1999, 94, 3341–3347. [Google Scholar] [CrossRef]
- Chung, J.W.; Hong, S.J.; Kim, K.J.; Goti, D.; Stins, M.F.; Shin, S.; Dawson, V.L.; Dawson, T.M.; Kim, K.S. 37 kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J. Biol. Chem. 2003, 278, 16857–16862. [Google Scholar] [CrossRef]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 1997, 387, 729–733. [Google Scholar] [CrossRef]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-I. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef]
- Massia, S.P.; Rao, S.S.; Hubbell, J.A. Covalently immobilized laminin peptide Tyr-Ile-Gly-Ser-Arg (YIGSR) supports cell spreading and co-localization of the 67-kilodalton laminin receptor with alpha-actinin and vinculin. J. Biol. Chem. 1993, 268, 8053–8059. [Google Scholar]
- Kim, K.J.; Chung, J.W.; Kim, K.S. 67-kDa Laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 2005, 280, 1360–1368. [Google Scholar]
- Davis, J.M.; Carvalho, H.M.; Rasmussen, S.B.; O’Brien, A.D. Cytotoxic necrotizing factor type 1 delivered by outer membrane vesicles of uropathogenic Escherichia coli attenuates polymorphonuclear leukocyte antimicrobial activity and chemotaxis. Infect. Immun. 2006, 74, 4401–4408. [Google Scholar] [CrossRef]
- Kouokam, J.C.; Wai, S.N.; Fällman, M.; Dobrindt, U.; Hacker, J.; Uhlin, B.E. Active cytotoxic necrotizing factor 1 associated with outer membrane vesicles from uropathogenic Escherichia coli. Infect. Immun. 2006, 74, 2022–2030. [Google Scholar] [CrossRef]
- Yu, H.; Kim, K.S. Ferredoxin is involved in secretion of cytotoxic necrotizing factor 1 across the cytoplasmic membrane in Escherichia coli K1. Infect. Immun. 2010, 78, 838–844. [Google Scholar] [CrossRef]
- Yu, H.; Kim, K.S. YgfZ contributes to secretion of cytotoxic necrotizing factor 1 into outer membrane vesicles in Escherichia coli. Microbiology 2012, 158, 612–621. [Google Scholar] [CrossRef]
- Zhu, L.; Pearce, D.; Kim, K.S. Prevention of E. coli K1 penetration of the blood-brain barrier by counteracting host cell receptor and signaling molecule involved in E. coli invasion of human brain microvascular endothelial cells. Infect. Immun. 2010, 78, 3554–3559. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Wang, M.-H.; Kim, K.S. Cytotoxic Necrotizing Factor 1 Contributes to Escherichia coli Meningitis. Toxins 2013, 5, 2270-2280. https://doi.org/10.3390/toxins5112270
AMA Style
Wang M-H, Kim KS. Cytotoxic Necrotizing Factor 1 Contributes to Escherichia coli Meningitis. Toxins. 2013; 5(11):2270-2280. https://doi.org/10.3390/toxins5112270
Chicago/Turabian StyleWang, Ming-Hsien, and Kwang Sik Kim. 2013. "Cytotoxic Necrotizing Factor 1 Contributes to Escherichia coli Meningitis" Toxins 5, no. 11: 2270-2280. https://doi.org/10.3390/toxins5112270