Reduction of Streptolysin O (SLO) Pore-Forming Activity Enhances Inflammasome Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
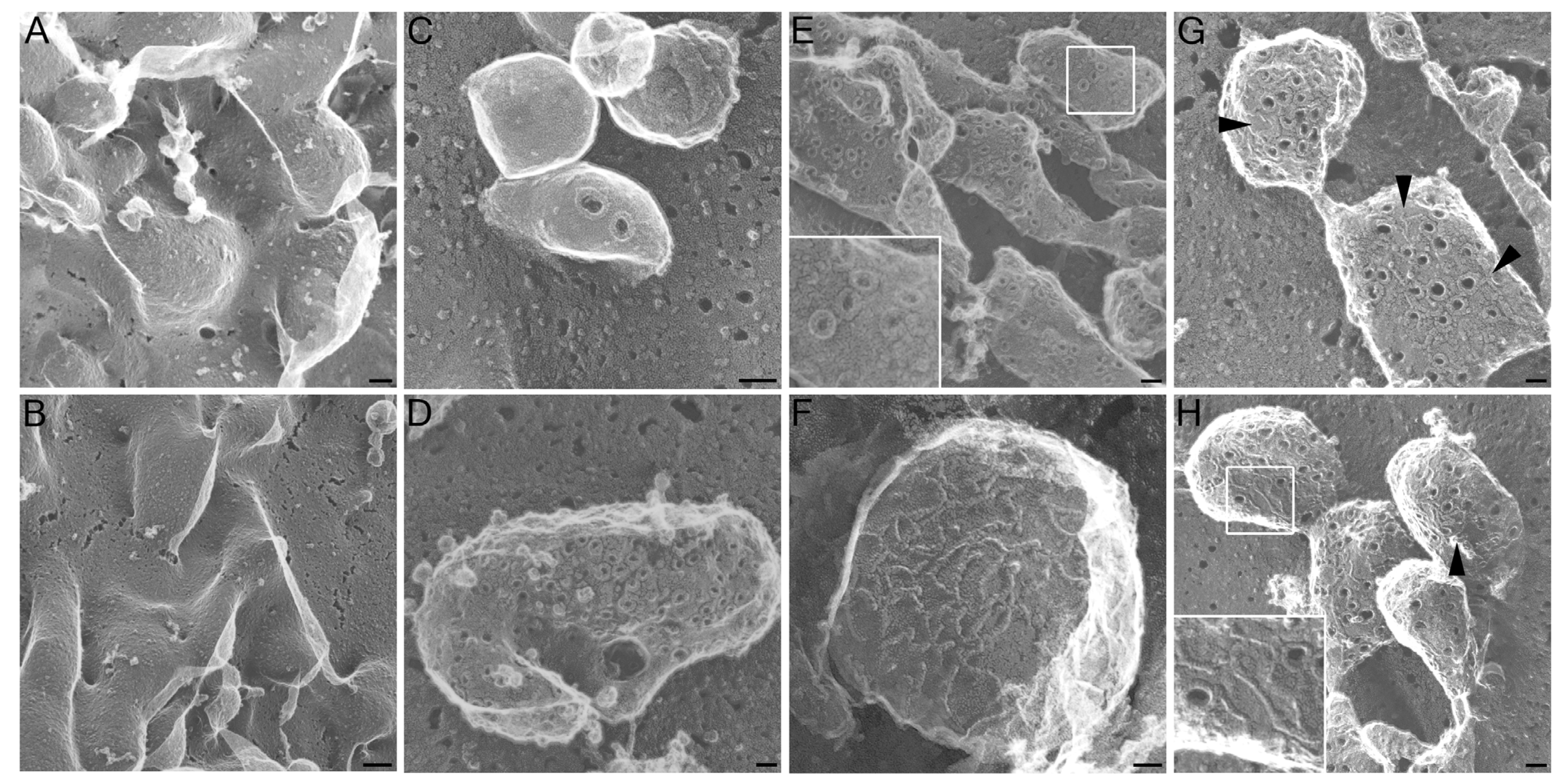
2.1. Altered Pore Formation in SLO N402C Mutant
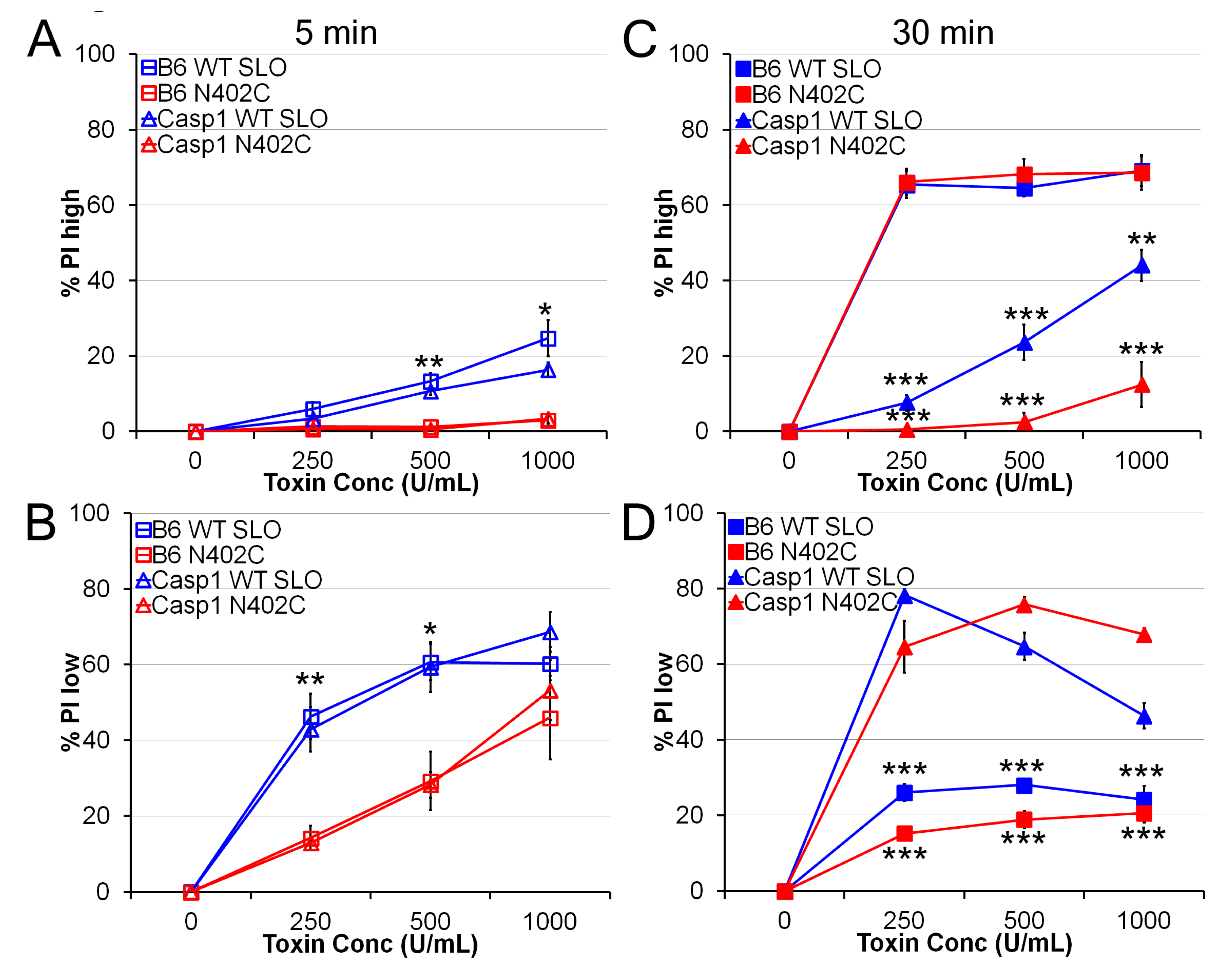
2.2. SLO N402C Has Reduced Toxicity in Mammalian Cells

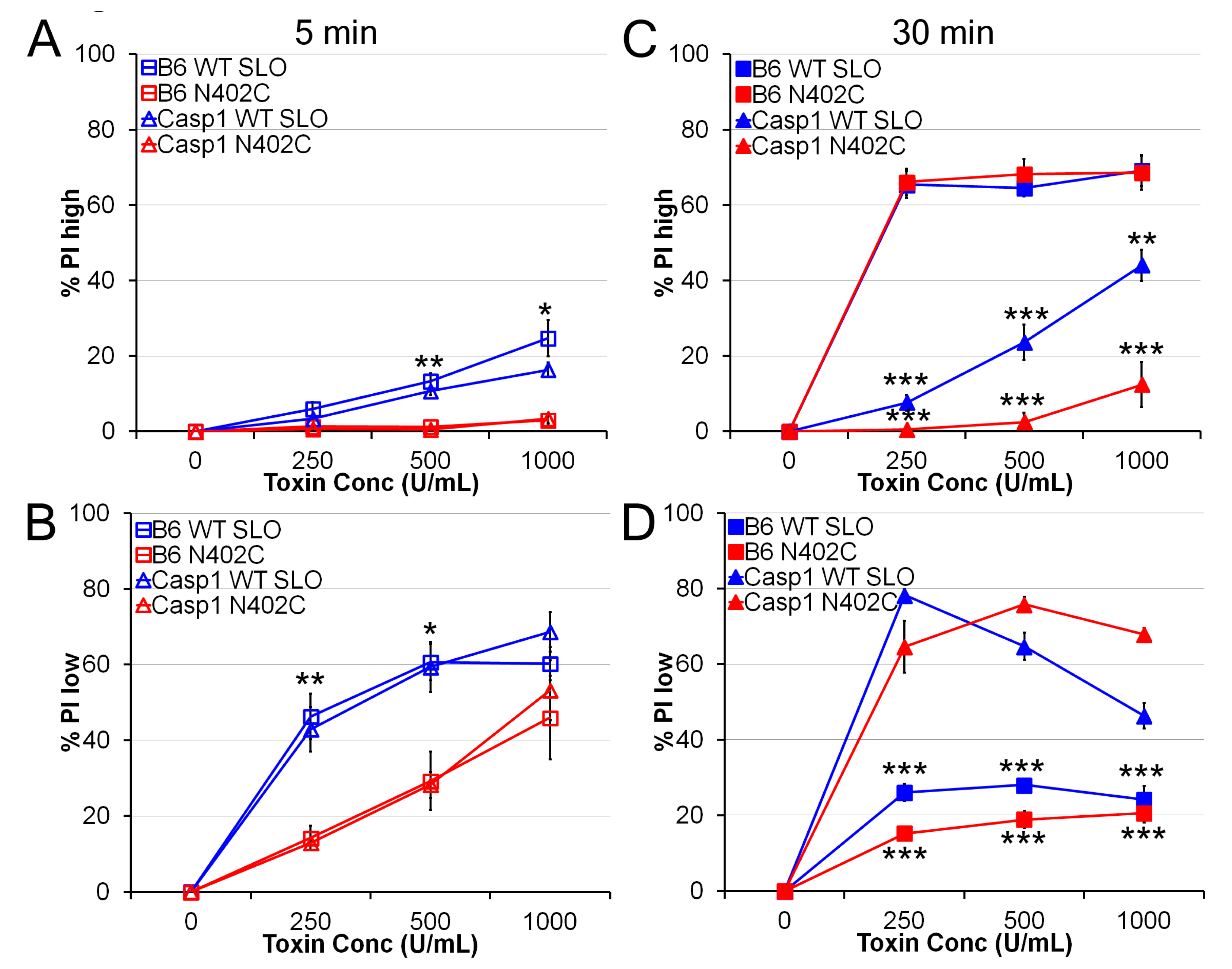
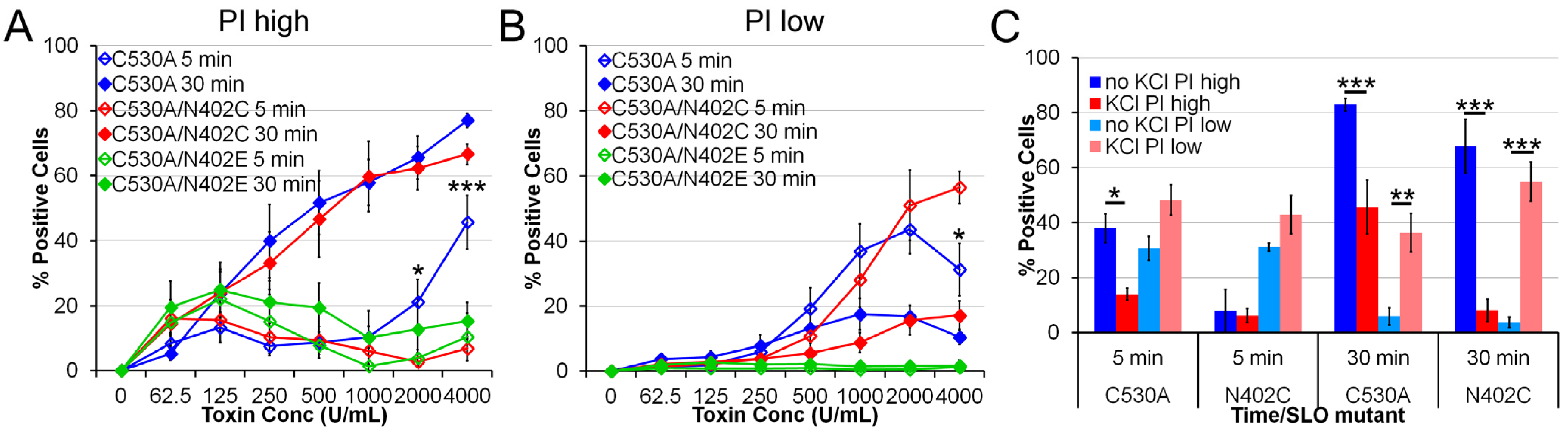
2.3. SLO C530A/N402C Has Similarly Reduced Toxicity
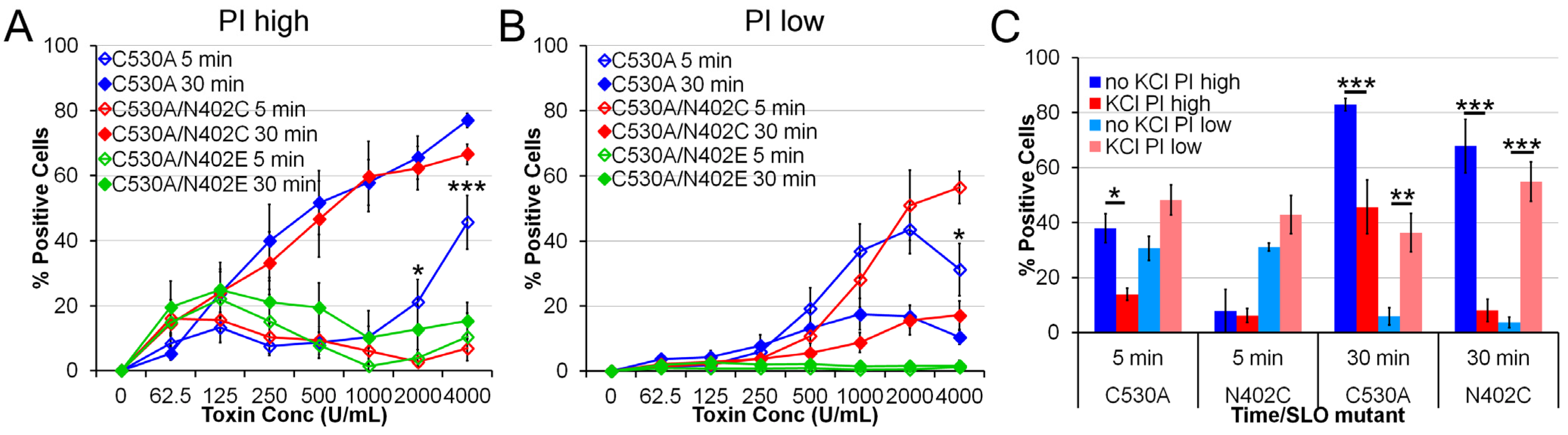
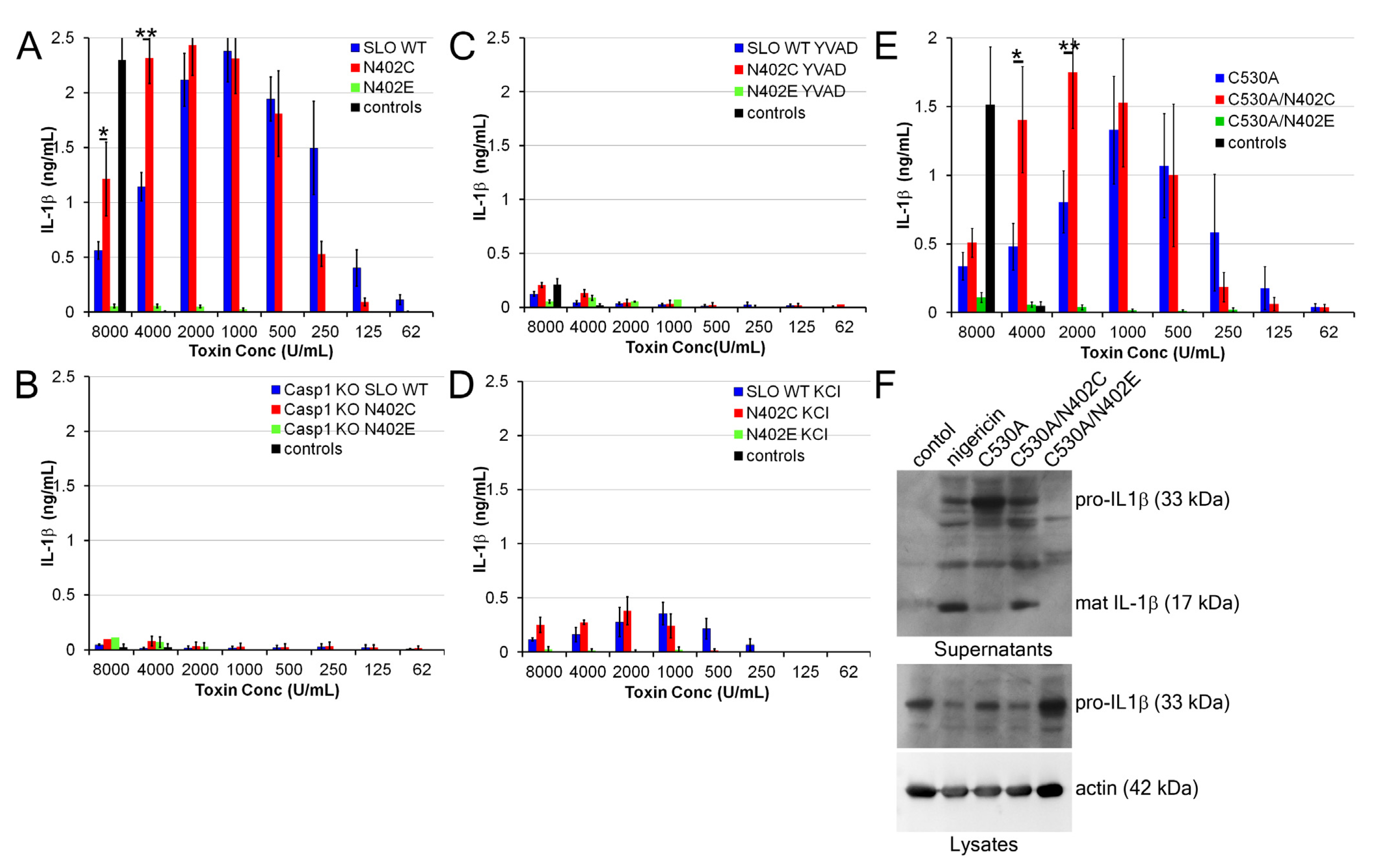
2.4. Increased IL-1β Production by SLO N402C
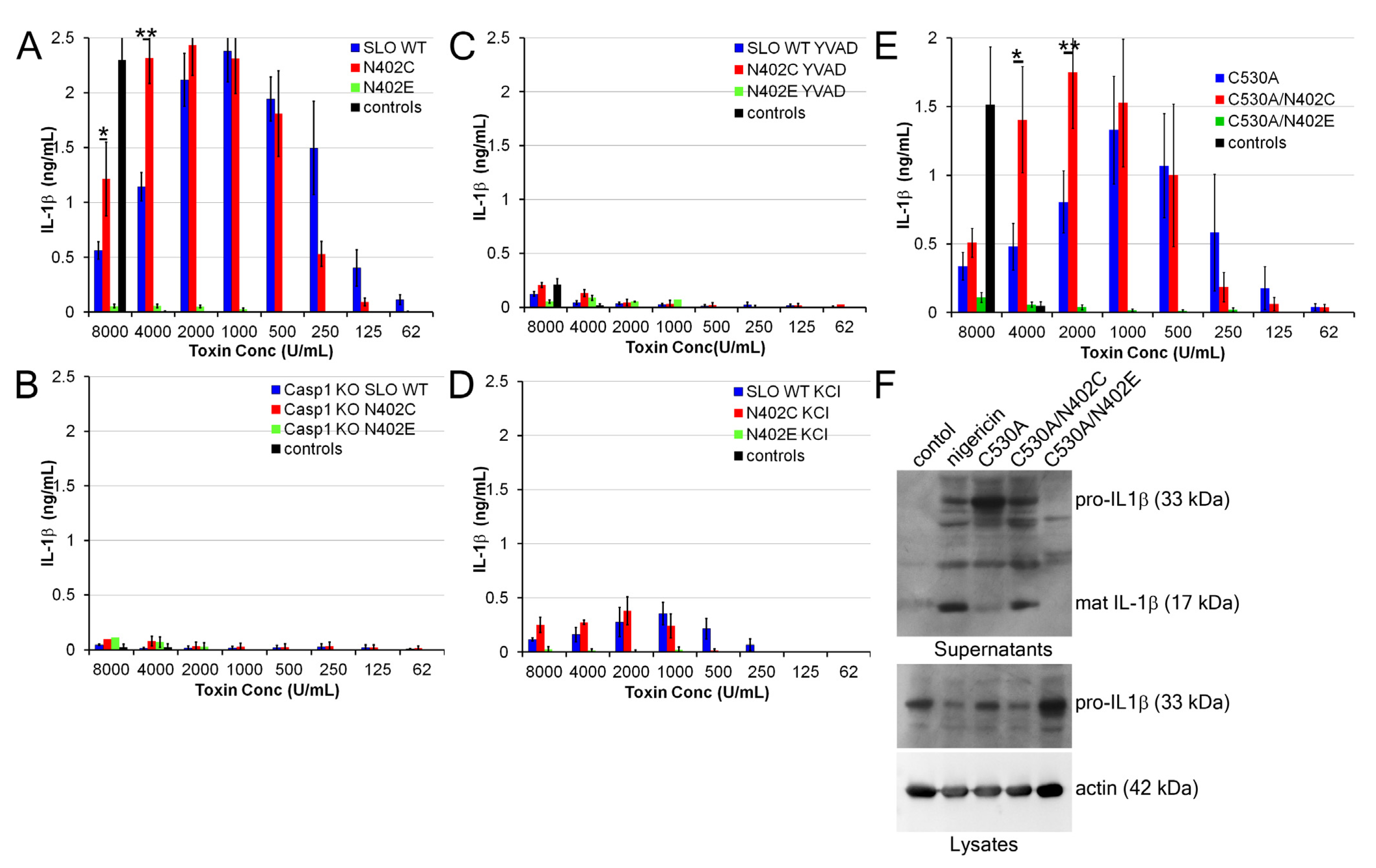
3. Discussion
4. Experimental Section
4.1. Reagents
4.2. Cell Culture
4.3. Electron Microscopy
4.4. Toxicity Assay
4.5. IL-1β Release
4.6. Statistics
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Limbago, B.; Penumalli, V.; Weinrick, B.; Scott, J.R. Role of streptolysin o in a mouse model of invasive group a streptococcal disease. Infect. Immun. 2000, 68, 6384–6390. [Google Scholar] [CrossRef]
- Bricker, A.L.; Cywes, C.; Ashbaugh, C.D.; Wessels, M.R. Nad+-glycohydrolase acts as an intracellular toxin to enhance the extracellular survival of group a streptococci. Mol. Microbiol. 2002, 44, 257–269. [Google Scholar] [CrossRef]
- Madden, J.C.; Ruiz, N.; Caparon, M. Cytolysin-mediated translocation (cmt): A functional equivalent of type iii secretion in gram-positive bacteria. Cell 2001, 104, 143–152. [Google Scholar] [CrossRef]
- Gilbert, R.J.; Mikelj, M.; Dalla Serra, M.; Froelich, C.J.; Anderluh, G. Effects of macpf/cdc proteins on lipid membranes. Cell. Mol. Life Sci. 2012, 70, 2083–2098. [Google Scholar]
- Rossjohn, J.; Polekhina, G.; Feil, S.C.; Morton, C.J.; Tweten, R.K.; Parker, M.W. Structures of perfringolysin o suggest a pathway for activation of cholesterol-dependent cytolysins. J. Mol. Biol. 2007, 367, 1227–1236. [Google Scholar]
- Tweten, R.K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 2005, 73, 6199–6209. [Google Scholar] [CrossRef]
- Tilley, S.J.; Orlova, E.V.; Gilbert, R.J.; Andrew, P.W.; Saibil, H.R. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 2005, 121, 247–256. [Google Scholar]
- Shatursky, O.; Heuck, A.P.; Shepard, L.A.; Rossjohn, J.; Parker, M.W.; Johnson, A.E.; Tweten, R.K. The mechanism of membrane insertion for a cholesterol-dependent cytolysin: A novel paradigm for pore-forming toxins. Cell 1999, 99, 293–299. [Google Scholar] [CrossRef]
- Abdel Ghani, E.M.; Weis, S.; Walev, I.; Kehoe, M.; Bhakdi, S.; Palmer, M. Streptolysin o: Inhibition of the conformational change during membrane binding of the monomer prevents oligomerization and pore formation. Biochemistry 1999, 38, 15204–15211. [Google Scholar]
- Palmer, M.; Saweljew, P.; Vulicevic, I.; Valeva, A.; Kehoe, M.; Bhakdi, S. Membrane-penetrating domain of streptolysin o identified by cysteine scanning mutagenesis. J. Biol. Chem. 1996, 271, 26664–26667. [Google Scholar]
- Palmer, M.; Harris, R.; Freytag, C.; Kehoe, M.; Tranum-Jensen, J.; Bhakdi, S. Assembly mechanism of the oligomeric streptolysin o pore: The early membrane lesion is lined by a free edge of the lipid membrane and is extended gradually during oligomerization. EMBO J. 1998, 17, 1598–1605. [Google Scholar] [CrossRef]
- Pinkney, M.; Beachey, E.; Kehoe, M. The thiol-activated toxin streptolysin o does not require a thiol group for cytolytic activity. Infect. Immun. 1989, 57, 2553–2558. [Google Scholar]
- Harris, J.R.; Adrian, M.; Bhakdi, S.; Palmer, M. Cholesterol-streptolysin o interaction: An em study of wild-type and mutant streptolysin o. J. Struct. Biol. 1998, 121, 343–355. [Google Scholar] [CrossRef]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell. Biol. 2005, 6, 499–505. [Google Scholar] [CrossRef]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin o clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell. Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef]
- Braun, J.S.; Novak, R.; Gao, G.; Murray, P.J.; Shenep, J.L. Pneumolysin, a protein toxin of streptococcus pneumoniae, induces nitric oxide production from macrophages. Infect. Immun. 1999, 67, 3750–3756. [Google Scholar]
- Ito, Y.; Kawamura, I.; Kohda, C.; Tsuchiya, K.; Nomura, T.; Mitsuyama, M. Seeligeriolysin o, a protein toxin of listeria seeligeri, stimulates macrophage cytokine production via toll-like receptors in a profile different from that induced by other bacterial ligands. Int. Immunol. 2005, 17, 1597–1606. [Google Scholar] [CrossRef]
- Walev, I.; Vollmer, P.; Palmer, M.; Bhakdi, S.; Rose-John, S. Pore-forming toxins trigger shedding of receptors for interleukin 6 and lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1996, 93, 7882–7887. [Google Scholar] [CrossRef]
- Keyel, P.A.; Heid, M.E.; Salter, R.D. Macrophage responses to bacterial toxins: A balance between activation and suppression. Immunol. Res. 2011, 50, 118–123. [Google Scholar] [CrossRef]
- Park, J.M.; Ng, V.H.; Maeda, S.; Rest, R.F.; Karin, M. Anthrolysin o and other gram-positive cytolysins are toll-like receptor 4 agonists. J. Exp. Med. 2004, 200, 1647–1655. [Google Scholar] [CrossRef]
- Harder, J.; Franchi, L.; Munoz-Planillo, R.; Park, J.H.; Reimer, T.; Nunez, G. Activation of the nlrp3 inflammasome by streptococcus pyogenes requires streptolysin o and nf-kappa b activation but proceeds independently of tlr signaling and p2×7 receptor. J. Immunol. 2009, 183, 5823–5829. [Google Scholar] [CrossRef]
- Chu, J.; Thomas, L.M.; Watkins, S.C.; Franchi, L.; Nunez, G.; Salter, R.D. Cholesterol-dependent cytolysins induce rapid release of mature il-1beta from murine macrophages in a nlrp3 inflammasome and cathepsin b-dependent manner. J. Leukoc. Biol. 2009, 86, 1227–1238. [Google Scholar] [CrossRef]
- Kanneganti, T.D. Central roles of nlrs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar] [CrossRef]
- Broz, P.; von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell. Host Microbe 2010, 8, 471–483. [Google Scholar] [CrossRef]
- Keller, M.; Ruegg, A.; Werner, S.; Beer, H.D. Active caspase-1 is a regulator of unconventional protein secretion. Cell 2008, 132, 818–831. [Google Scholar] [CrossRef]
- Lamkanfi, M. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 2011, 11, 213–220. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Jin, C.; Flavell, R.A. Molecular mechanism of nlrp3 inflammasome activation. J. Clin. Immunol. 2010, 30, 628–631. [Google Scholar] [CrossRef]
- Leemans, J.C.; Cassel, S.L.; Sutterwala, F.S. Sensing damage by the nlrp3 inflammasome. Immunol. Rev. 2011, 243, 152–162. [Google Scholar] [CrossRef]
- Scott, R.E. Plasma membrane vesiculation: A new technique for isolation of plasma membranes. Science 1976, 194, 743–745. [Google Scholar]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 increases susceptibility to salmonella infection in the absence of caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar]
- Metkar, S.S.; Wang, B.; Catalan, E.; Anderluh, G.; Gilbert, R.J.; Pardo, J.; Froelich, C.J. Perforin rapidly induces plasma membrane phospholipid flip-flop. PLoS One 2011, 6, e24286. [Google Scholar]
- Keefe, D.; Shi, L.; Feske, S.; Massol, R.; Navarro, F.; Kirchhausen, T.; Lieberman, J. Perforin triggers a plasma membrane-repair response that facilitates ctl induction of apoptosis. Immunity 2005, 23, 249–262. [Google Scholar]
- Li, H.; Willingham, S.B.; Ting, J.P.; Re, F. Cutting edge: Inflammasome activation by alum and alum's adjuvant effect are mediated by nlrp3. J. Immunol. 2008, 181, 17–21. [Google Scholar]
- Franchi, L.; Nunez, G. The nlrp3 inflammasome is critical for aluminium hydroxide-mediated il-1beta secretion but dispensable for adjuvant activity. Eur. J. Immunol. 2008, 38, 2085–2089. [Google Scholar] [CrossRef]
- Eisenbarth, S.C.; Colegio, O.R.; O’Connor, W.; Sutterwala, F.S.; Flavell, R.A. Crucial role for the nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 2008, 453, 1122–1126. [Google Scholar] [CrossRef]
- Magassa, N.; Chandrasekaran, S.; Caparon, M.G. Streptococcus pyogenes cytolysin-mediated translocation does not require pore formation by streptolysin o. EMBO Rep. 2010, 11, 400–405. [Google Scholar] [CrossRef]
- Keyel, P.A.; Heid, M.E.; Watkins, S.C.; Salter, R.D. Visualization of bacterial toxin induced responses using live cell fluorescence microscopy. J. Vis. Exp. 2012, e4227. [Google Scholar] [CrossRef]
- Heuser, J. The production of “cell cortices” for light and electron microscopy. Traffic 2000, 1, 545–552. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Keyel, P.A.; Roth, R.; Yokoyama, W.M.; Heuser, J.E.; Salter, R.D. Reduction of Streptolysin O (SLO) Pore-Forming Activity Enhances Inflammasome Activation. Toxins 2013, 5, 1105-1118. https://doi.org/10.3390/toxins5061105
Keyel PA, Roth R, Yokoyama WM, Heuser JE, Salter RD. Reduction of Streptolysin O (SLO) Pore-Forming Activity Enhances Inflammasome Activation. Toxins. 2013; 5(6):1105-1118. https://doi.org/10.3390/toxins5061105
Chicago/Turabian StyleKeyel, Peter A., Robyn Roth, Wayne M. Yokoyama, John E. Heuser, and Russell D. Salter. 2013. "Reduction of Streptolysin O (SLO) Pore-Forming Activity Enhances Inflammasome Activation" Toxins 5, no. 6: 1105-1118. https://doi.org/10.3390/toxins5061105