Towards Systems Biology of Mycotoxin Regulation

{kind=link}
Abstract
:1. Biological Networks in the Context of Mycotoxin Biosynthesis
2. Saccharomyces cerevisiae as a Model System to Identify Components in Networks in Fusarium
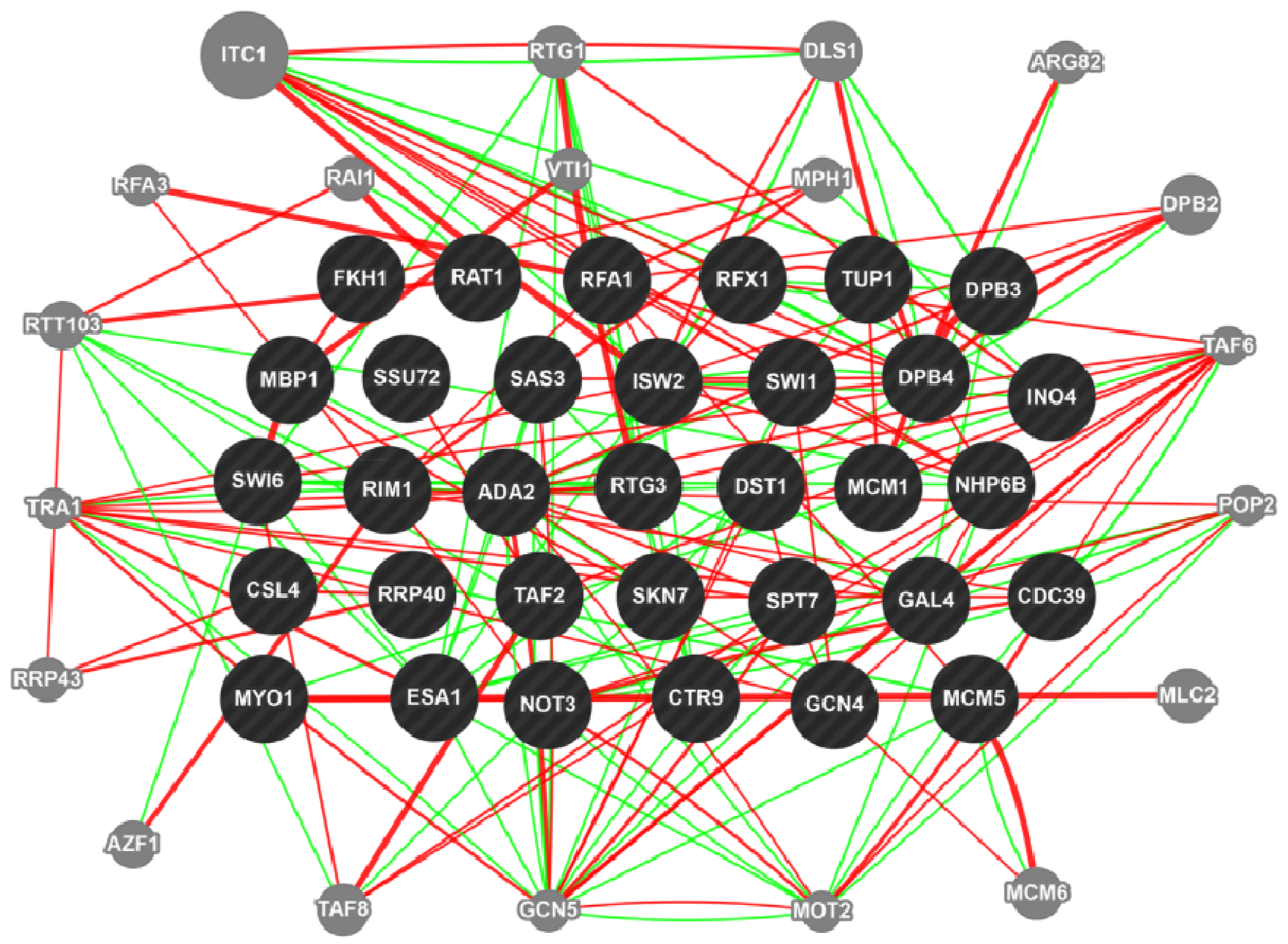
3. Chemogenomics to Decipher Mycotoxin Pathway in Fusarium
4. Proteomics to Identify Components of a Cellular Network
5. Concluding Remarks
References
- Vidal, M. A unifying view of 21st century systems biology. FEBS Lett. 2009, 583, 3891–3894. [Google Scholar]
- Westerhoff, H.V.; Winder, C.; Messiha, H.; Simeonidis, E.; Adamczyk, M.; Verma, M.; Bruggeman, F.J.; Dunn, W. Systems biology: The elements and principles of life. FEBS Lett. 2009, 583, 3882–3890. [Google Scholar] [CrossRef]
- Yu, J.-H.; Keller, N. Regulation of secondary metabolism in filamentous fungi. Annu. Rev. Phytopathol. 2005, 43, 437–458. [Google Scholar] [CrossRef]
- Alexander, N.J.; Proctor, R.H.; McCormick, S.P. Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusariu. Toxin Rev. 2009, 28, 198–215. [Google Scholar] [CrossRef]
- Ponts, N.; Pinson-Gadais, L.; Barreau, C.; Richard-Forget, F.; Ouellet, T. Exogenous H2O2 and catalase treatments interfere with Tri genes expression in liquid cultures of Fusarium graminearum. FEBS Lett. 2007, 581, 443–447. [Google Scholar] [CrossRef]
- Gardiner, D.M.; Osborne, S.; Kazan, K.; Manners, J.M. Low pH regulates the production of deoxynivalenol by Fusarium graminearum. Microbiology 2009, 155, 3149–3156. [Google Scholar] [CrossRef]
- Gardiner, D.M.; Kazan, K.; Manners, J.M. Nutrient profiling reveals potent inducers of trichothecene biosynthesis in Fusarium graminearum. Fungal Genet. Biol. 2009, 46, 604–613. [Google Scholar] [CrossRef]
- Jiao, F.; Kawakami, A.; Nakajima, T. Effects of different carbon sources on trichothecene production and Tri gene expression by Fusarium graminearum in liquid culture. FEMS Microbiol. Lett. 2008, 285, 212–219. [Google Scholar]
- Merhej, J.; Richard-Forget, F.; Barreau, C. The pH regulatory factor PaC1 regulates Tri gene expression and trichothecene production in Fusarium graminearum. Fungal Genet. Biol. 2011, 48, 275–284. [Google Scholar] [CrossRef]
- Merhej, J.; Urban, M.; Dufresne, M.; Hammond-Kosack, K.E.; Richard-Forget, F.; Barreau, C. The velvet gene FgVe1, affects fungal development and positively regulates trichothecene biosynthesis and pathogenicity in Fusarium graminearum. Mol. Plant Pathol. 2012, 13, 363–374. [Google Scholar] [CrossRef]
- Boone, C.; Bussey, H.; Andrews, B.J. Exploring genetic interactions and networks with yeast. Nat. Rev. Genet. 2007, 8, 437–449. [Google Scholar] [CrossRef]
- Costanzo, M.; Giaever, G.; Nislow, C.; Andrews, B. Experimental approaches to identify genetic networks. Curr. Opin. Biotech. 2006, 17, 472–480. [Google Scholar] [CrossRef]
- Sopko, R.; Huang, D.; Preston, N.; Chua, G.; Papp, B.; Kafadar, K.; Snyder, M.; Oliver, S.G.; Cyert, M.; Hughes, T.R.; et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol. Cell. 2006, 21, 319–330. [Google Scholar] [CrossRef]
- Ma, L.; Zhai, Y.; Feng, D.; Chan, T.C.; Lu, Y.; Fu, X.; Wang, J.; Chen, Y.; Li, J.; Xu, K.; et al. Identification of novel factors involved in or regulating initiation of DNA replication by a genome-wide phenotypic screen in Saccharomyces cerevisiae. Cell Cycle 2010, 9, 4399–4410. [Google Scholar] [CrossRef]
- Measday, V.; Baetz, K.; Guzzo, J.; Yuen, K.; Kwok, T.; Sheikh, B.; Ding, H.; Ueta, R.; Hoac, T.; Cheng, B.; et al. Systematic yeast synthetic lethal and synthetic dosage lethal screens identify genes required for chromosome segregation. Proc. Natl. Acad. Sci.USA 2005, 39, 13956–13961. [Google Scholar]
- Liu, C.; van Dyk, D.; Li, Y.; Andrews, B.; Rao, H. A genome-wide synthetic dosage lethality screen reveals multiple pathways that require the functioning of ubiquitin-binding proteins Rad23 and Dsk2. BMC Biol. 2009, 7. [Google Scholar] [CrossRef]
- Hsiang, T.; Baillie, D.L. Comparison of the yeast proteome to other fungal genomes to find core fungal genes. J. Mol. Evol. 2005, 60, 475–483. [Google Scholar] [CrossRef]
- Son, H.; Seo, Y.-S.; Min, K.; Park, A.R.; Lee, J.; Jin, J.-M.; Lin, Y.; Cao, P.; Hong, S.-Y.; Kim, E.-K.; et al. A phenome-based functional analysis of transcription factors in the cereal head blight fungus, Fusarium graminearum. PLoS Pathog. 2011, 7, e1002310. [Google Scholar] [CrossRef]
- Grant, P.A.; Duggan, L.; Cote, J.; Roberts, S.M.; Brownell, J.E.; Candau, R.; Ohba, R.; Owen-Hughes, T.; Allis, C.D.; Winston, F.; et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: Characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 1997, 11, 1640–1650. [Google Scholar] [CrossRef]
- Tishgarten, T.; Yin, F.F.; Grant, T.R.; Lipscomb, L.A.; Faucher, K.M.; Dluhy, R.A.; Stevens, T.H.; Fischer, G.; Mollard, V. Structures of yeast vesicle trafficking proteins. Protein Sci. 1998, 8, 2465–2473. [Google Scholar]
- Ruiz, C.; Escribano, V.; Morgado, E.; Molina, M.; Mazon, M.J. Cell-type-dependent repression of yeast a-specific genes requires Itc1p, a subunit of the Isw2p–Itc1p chromatin remodelling complex. Microbiology 2003, 149, 341–351. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, S.; Hou, R.; Zhao, Z.; Zheng, Q.; Xu, Q.; Zheng, D.; Wang, G.; Liu, H.; Gao, X.; et al. Functional analysis of the kinome of the wheat scab fungus Fusarium graminearum. PLoS Pathog. 2011, 7, e1002460. [Google Scholar] [CrossRef]
- Spring, D.R. Chemical genetics to chemical genomics: Small molecules offer big insights. Chem. Soc. Rev. 2005, 34, 472–482. [Google Scholar] [CrossRef]
- Yamanishi, Y. Chemogenomic Approaches to Infer Drug–Target Interaction Networks. Meth. Mol. Biol. 2013, 939, 97–113. [Google Scholar]
- Laggner, C.; Kokel, D.; Setola, V.; Tolia, A.; Lin, H.; Irwin, J.J.; Keiser, M.J.; Cheung, C.Y.J.; Minor, D.L., Jr.; Roth, B.L.; et al. Chemical informatics and target identification in a zebrafish phenotypic screen. Nat. Chem. Biol. 2012, 8, 144–146. [Google Scholar]
- Calvo, A.M.; Wilson, R.A.; Bok, J.W.; Keller, N.P. Relationship between secondary metabolism and fungal development. Microbial. Mol. Biol. Rev. 2002, 66, 447–459. [Google Scholar] [CrossRef]
- Roze, L.V.; Beudry, R.M.; Keller, N.P.; Linz, J.E. Regulation of aflatoxin synthesis by FadA/cAMP/protein kinase A signaling in Aspergillus parasiticus. Mycopathologia 2004, 158, 219–232. [Google Scholar]
- Parsons, A.B.; Brost, R.L.; Ding, H.; Li, Z.; Zhang, C.; Sheikh, B.; Brown, G.B.; Kane, P.M.; Hughes, T.R.; Boone, C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotech. 2004, 22, 62–69. [Google Scholar]
- Schreiber, K.; Nasmith, C.; Allard, G.; Singh, J.; Subramaniam, R.; Desveaux, D. Found in translation: High-throughput chemical screening in Arabidopsis thaliana identifies small molecules that reduce Fusarium head blight disease in wheat. Mol. Plant Microbe Interact. 2011, 24, 640–648. [Google Scholar] [CrossRef]
- Ravasz, E.; Somera, A.L.; Mongru, D.A.; Oltvai, Z.N.; Barabasi, A.L. Hierarchical organization of modularity in metabolic networks. Science 2002, 297, 1551–1555. [Google Scholar] [CrossRef]
- Bar-Joseph, Z.; Gerber, G.K.; Lee, T.I.; Rinaldi, N.J.; Yoo, J.Y.; Robert, F.; Gordon, D.B.; Fraenkel, E.; Jaakkola, T.S.; Young, R.A.; et al. Computational discovery of gene modules and regulatory networks. Nat. Biotech. 2003, 21, 1337–1342. [Google Scholar] [CrossRef]
- Krogan, N.J.; Cagney, G.; Yu, H.; Zhong, G.; Guo, X.; Ignatchenko, A.; Li, J.; Pu, S.; Datta, N.; Tikuisis, A.P.; et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006, 440, 637–843. [Google Scholar]
- Zhao, X.-M.; Zhang, X.-W.; Tang, W.-H.; Chen, L. FPPI: Fusarium graminearum Protein-Protein Interaction Database. J. Proteome Res. 2009, 8, 4714–4721. [Google Scholar] [CrossRef]
- stlund, G.; Schmitt, T.; Forslund, K.; Köstler, T.; Messina, D.N.; Roopra, S.; Frings, O.; Sonnhammer, E.L.L. InParanoid 7: New algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010, 38, D196–D203. [Google Scholar] [CrossRef]
- Jørgensen, C.; Locard-Paulet, M. Analysing signalling networks by mass spectrometry. Amino Acids 2012, 43, 1061–1074. [Google Scholar] [CrossRef]
- Tichy, A.; Salovska, B.; Rehulka, P.; Klimentova, J.; Vavrova, J.; Stulik, J.; Hernychova, L. Phosphoproteomics: Searching for a needle in a haystack. J. Proteomics 2011, 74, 2786–2797. [Google Scholar]
- Amoutzias, G.D.; He, Y.; Lilley, K.S.; van de Peer, Y.; Oliver, S.G. Evaluation and properties of the budding yeast phosphoproteome. Mol. Cell. Proteomics 2012, 11, 1–13. [Google Scholar] [CrossRef]
- Yachie, N.; Saito, R.; Sugiyama, N.; Tomita, M.; Ishihama, Y. Integrative features of the yeast phosphoproteome and protein–protein interaction map. PLoS Comput. Biol. 2011, 7, e1001064. [Google Scholar] [CrossRef]
- Bodenmiller, B.; Wanka, S.; Kraft, C.; Urban, J.; Campbell, D.; Pedrioli, P.G.; Gerrits, B.; Picotti, P.; Lam, H.; Vitek, O.; et al. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef]
- Rampitsch, C.; Tinker, N.A.; Subramaniam, R.; Barkow-Österreicher, S.; Laczko, E. Phosphoproteome profile of Fusarium graminearum grown in vitro under non-limiting conditions. Proteomics 2012, 12, 1002–1005. [Google Scholar] [CrossRef]
- Takemoto, D.; Tanaka, A.; Scott, B. NADPH oxidases in fungi: Diverse roles of reactive oxygen species in fungal cellular differentiation. Fungal Genet. Biol. 2007, 44, 1065–1076. [Google Scholar] [CrossRef]
- Bykova, N.V.; Hoehn, B.; Rampitsch, C.; Banks, T.; Stebbing, Jo-Ann.; Fan, T.; Knox, R. Redox-sensitive proteome and antioxidant strategies in wheat seed dormancy control. Proteomics 2011, 11, 865–882. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Subramaniam, R.; Rampitsch, C. Towards Systems Biology of Mycotoxin Regulation. Toxins 2013, 5, 675-682. https://doi.org/10.3390/toxins5040675
Subramaniam R, Rampitsch C. Towards Systems Biology of Mycotoxin Regulation. Toxins. 2013; 5(4):675-682. https://doi.org/10.3390/toxins5040675
Chicago/Turabian StyleSubramaniam, Rajagopal, and Christof Rampitsch. 2013. "Towards Systems Biology of Mycotoxin Regulation" Toxins 5, no. 4: 675-682. https://doi.org/10.3390/toxins5040675