Venom Peptides as a Rich Source of Cav2.2 Channel Blockers


Institute for Molecular Bioscience, The University of Queensland, St Lucia, Queensland, 4072, Australia
*
Author to whom correspondence should be addressed.
Toxins 2013, 5(2), 286-314; https://doi.org/10.3390/toxins5020286
Submission received: 2 November 2012
/
Revised: 7 January 2013
/
Accepted: 25 January 2013
/
Published: 4 February 2013
(This article belongs to the Special Issue Animal Toxins Targeting Ion Channels Involved in Pain)
Abstract
:Cav2.2 is a calcium channel subtype localized at nerve terminals, including nociceptive fibers, where it initiates neurotransmitter release. Cav2.2 is an important contributor to synaptic transmission in ascending pain pathways, and is up-regulated in the spinal cord in chronic pain states along with the auxiliary α2δ1 subunit. It is therefore not surprising that toxins that inhibit Cav2.2 are analgesic. Venomous animals, such as cone snails, spiders, snakes, assassin bugs, centipedes and scorpions are rich sources of remarkably potent and selective Cav2.2 inhibitors. However, side effects in humans currently limit their clinical use. Here we review Cav2.2 inhibitors from venoms and their potential as drug leads.
1. Introduction
A wide diversity of venomous animals has evolved a large range of peptide toxins that target ion channels expressed in the neuronal and neuromuscular systems of prey and predators as part of efficient prey immobilization and deterrent strategies. Accordingly, many of the most selective ion channel modulators known originate from venoms (reviewed by [1]). These peptide toxins have evolved from a relatively small number of structural frameworks that are particularly well suited to address crucial issues such as, potency and stability [1]. While venoms from some spiders, such as Phoneutria nigriventer, are dominated by Nav inhibitors [2]; in general, Cav inhibition dominates the pharmacology of spider venom peptides (reviewed by [3]). However, activity of many spider toxins at Cav2.2 has not been characterized extensively, and many of these peptides preferentially target Cav channels other than Cav2.2, such as Cav2.1, Cav2.3 or invertebrate Cav [4,5,6,7].
In contrast, a range of disulfide rich peptides from cone snails (conotoxins) preferentially inhibit Cav2.2 (see Table 2; reviewed by: [8,9]). GVIA from Conus geographus has been used for many years as probe to discriminate Cav2.2 from other closely related Cav channel subtypes ([10,11,12,13], for review see: [14]). In addition, several cone snail toxins have direct diagnostic and therapeutic potential [8,13,15,16] (reviewed by: [9,17]). A synthetic version of a Cav2.2 channel blocker toxin ω-conotoxin MVIIA (ziconotide, Prialt®), from the venom of the cone snail Conus magus is currently in use clinically, validating Cav2.2 as an analgesic target in humans [15,18]. Unfortunately, intrathecal administration and undesirable side effects have limited the clinical use of ziconitide [15,19]. Here we review Cav2.2 channel inhibitor toxins from venoms, their pharmacological and structural properties as well as their therapeutic potential.
2. Cav Channels
Calcium (Ca2+) currents in mammalian excitable cells have diverse pharmacological properties, and control essential physiological functions, including muscle contraction, hormone secretion, neurotransmitter release and nociceptive transmission. Voltage-gated calcium channels (Cav), are multi-subunit complexes composed of different pore-forming/voltage-sensing α1 subunit types, and several α2δ, β and γ regulatory subunit isoforms. Genes encoding 10 pore-forming α1 (α1A–α1I and α1s) as well as several splice variants have been identified and characterized. Cav superfamilies 1 and 2 require higher voltage steps to be activated, and are thus classified as high-threshold calcium channels, while superfamily 3 has a lower threshold for activation. High threshold currents include L-type (encoded by Cav1.1–1.4 genes), N-type (Cav2.2), P/Q-type (Cav2.1) and R-type (Cav2.3) channels, while T-type (Cav3.1–3.3) calcium channels are low-threshold channels (Table 1) (for review see [14,20]).
The primary structure of the Cav family has been determined by a combination of protein chemistry, cDNA cloning and sequencing [14,21,22]. Their hetero-oligomeric nature was established from biochemical glycosylation and hydrophobicity analyses. At least three auxiliary subunits, which regulate Cav2.2 expression and function, have been defined. The ~190 kDa pore-forming transmembrane α1 subunit (~2000 amino acids), is organized in four homologous domains (I–IV), comprising six transmembrane α helices (S1–S6) and the pore-forming P loop between S5 and S6 (Figure 1) [14]. Studies on the structure and function of the related pore-forming subunits of Na+ and K+ channels have resulted in the identification of their principal functional domains [23]. The S4 segments form a key part of the voltage sensor module (Figure 1), moving outward and rotating under the influence of the electric field and initiating a conformational change that opens the pore. The external entrance to the ion conducting pore of the channel is lined by the P loop, which contains a pair of glutamate residues in each domain, required for Ca2+ ion selectivity. The inner pore is lined by the S6 segments (Figure 1), which forms the receptor site for the pore-blocking Cav1 antagonist drugs (for review see: [22]). While no high-resolution crystal structure of the Cav is available to date, structures from related ion channels, in particular the recently determined bacterial voltage-gated sodium channel [24,25], promise to provide significant insight into the structure and function of Cav channels.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
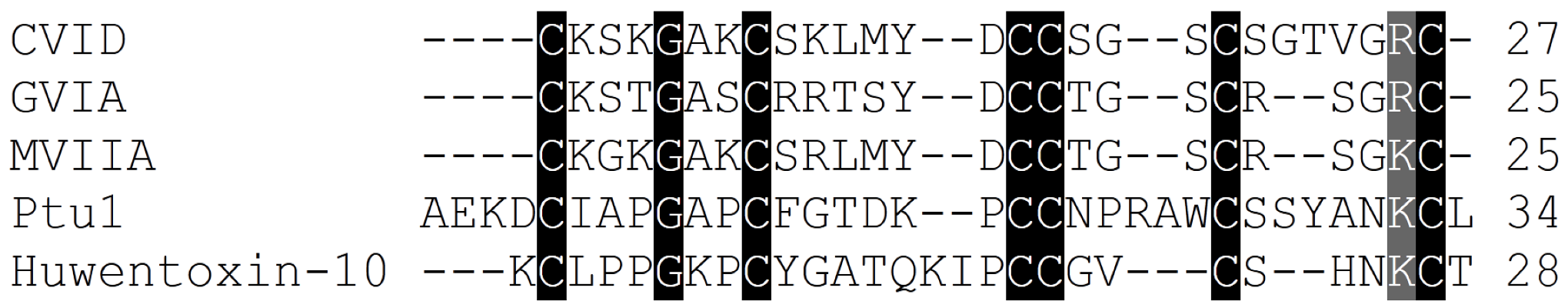{kind=link}
| Cav subtype | Current type | Localization | Antagonist class/Name | Physiological function |
|---|---|---|---|---|
| Cav1.1 | L | Skeletal muscle, transverse tubules | DHP, PHA, BTZ | Excitation-contraction coupling, gene regulation |
| Cav1.2 | L | Cardiac myocytes, smooth muscle myocytes, endocrine cells, neuronal cell bodies, proximal dendrites | DHP, PHA, BTZ | Excitation-contraction coupling, hormone secretion, gene regulation |
| Cav1.3 | L | Endocrine cells, neuronal cell bodies and dendrites, cardiac atrial myocytes and pacemarker cells, cochlear hair cells | DHP, PHA, BTZ | Hormone secretion, gene regulation, tonic transmitter release |
| Cav1.4 | L | Retinal rod and bipolar cells, spinal cord, adrenal gland, mast cells | DHP, PHA, BTZ | Tonic neurotransmitter release |
| Cav2.1 | P/Q | Nerve terminals and dendrites, neuroendocrine cells | ω-agatoxin IVA | Neurotransmitter release, dendritic Ca2+ transient currents |
| Cav2.2 | N | Nerve terminals and dendrites, neuroendocrine cells | ω-conotoxin CVID, GVIA MVIIA | Neurotransmitter release, Ca2+-dependent action potentials |
| Cav2.3 | R | Neuronal cell bodies and dendrites | ω-theraphotoxin-Hg1a(SNX-482) | NeurotransmitterRelease |
| Cav3.1 | T | Neuronal cell bodies and dendrites, cerebellum and thalamus, cardiac and smooth muscles | Pimozide, mibefradil,TTA-P2, Ni2+, Zn2+ | Pacemaking, repetitive firing |
| Cav3.2 | T | CNS: neuronal cell bodiesand dendrites, heart, liver, kidney, lung, skeletal muscle, pancreas | Kurtoxin, pimopzide, mibefradil, Z123212, TTA-P2, Ni2+, Zn2+ | Pacemaking, repetitive firing |
| Cav3.3 | T | CNS: neuronal cell bodies and dendrites | Pimozide, TTA-P2, Zn2+ Ni2+, mibefradil | Pacemaking, repetitive firing |
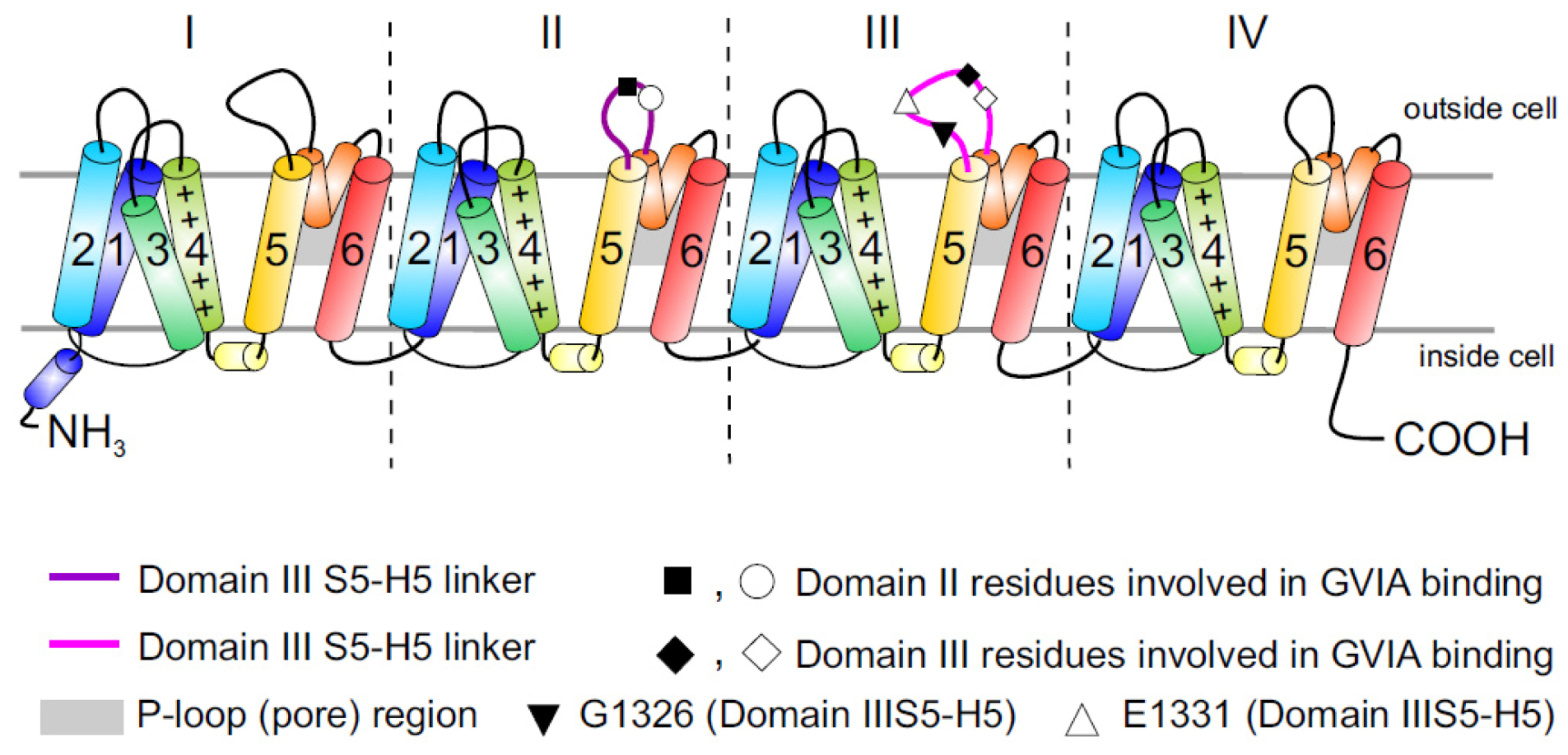
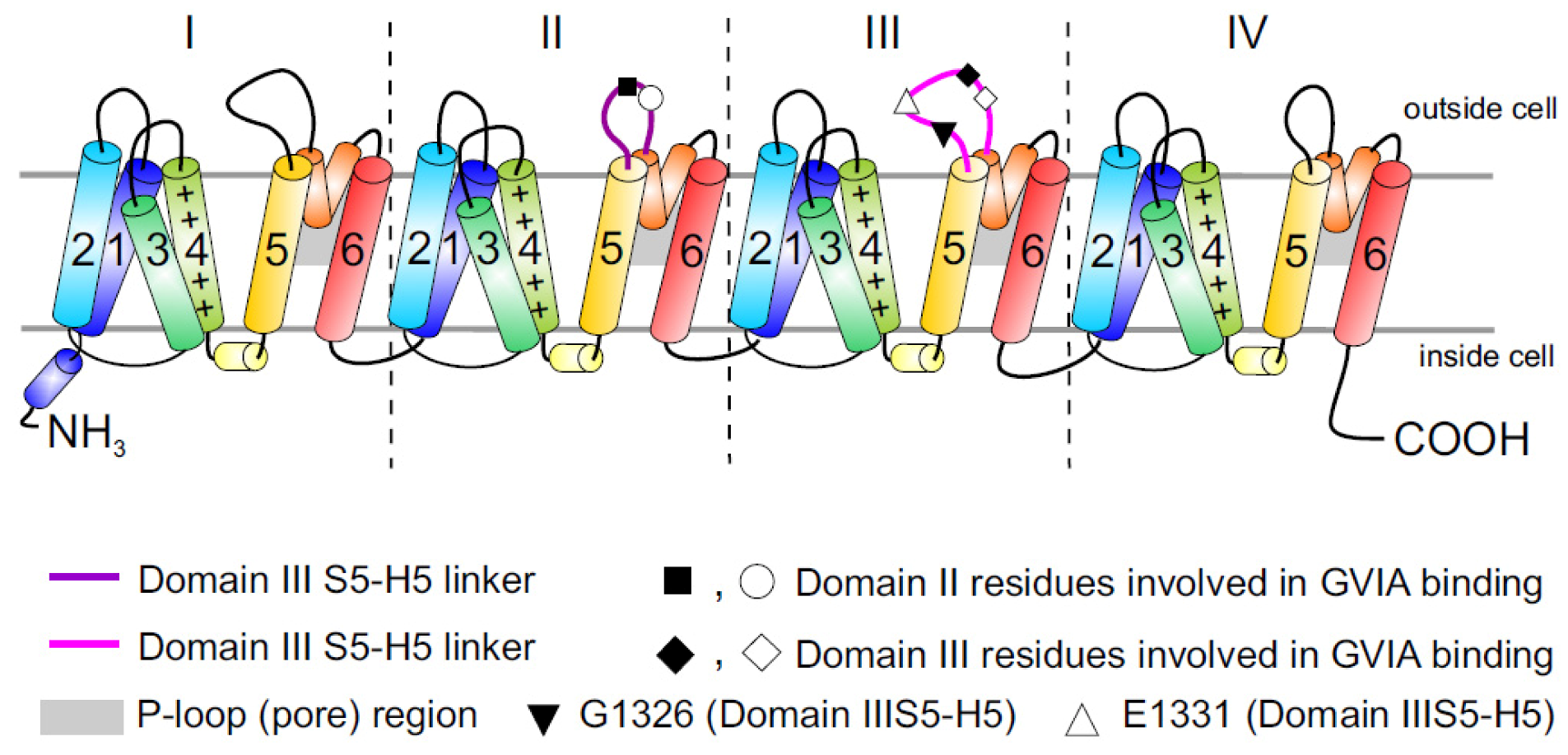
Figure 1.
Topology of Cav channels: Represented is the pore-forming α1 subunit of the Cav2.2 channels. This large protein consists of four homologous transmembrane domains (I–IV) and each domain contains six segments (S1–S6) and a membrane-associated P loop between S5 and S6 (represented in orange/grey) where the binding site of ω-conotoxins is localized. Circles, triangles and rectangles represent the localization of specific residues described to be important for binding of Cav2.2 to the ω-conotoxin GVIA [11].
Figure 1.
Topology of Cav channels: Represented is the pore-forming α1 subunit of the Cav2.2 channels. This large protein consists of four homologous transmembrane domains (I–IV) and each domain contains six segments (S1–S6) and a membrane-associated P loop between S5 and S6 (represented in orange/grey) where the binding site of ω-conotoxins is localized. Circles, triangles and rectangles represent the localization of specific residues described to be important for binding of Cav2.2 to the ω-conotoxin GVIA [11].
2.1. Cav2.2 Channels
The N-type voltage-gated Ca2+ channel Cav2.2 is expressed predominantly at presynaptic neuronal terminals throughout the central and peripheral nervous systems, where it is critical for neurotransmitter release. Like the other members of the Cav family, Cav2.2 is a hetero-oligomeric channel comprising the core pore-forming α1B subunit, which determines the main biophysical and pharmacological properties of the channel, and typically three auxiliary subunits.
2.2. Cav2.2 Splice Variants
Alternative splicing is an essential mechanism used extensively in the mammalian nervous system to increase the level of diversity that can be achieved by a set of genes [26]. Two Cav2.2 splice variants have been reported to occur predominantly in the central and peripheral nervous system [27]. The Cav2.2 splice variant 37a is of particular interest, as it replaces the usual variant 37b in a specific subset of rat nociceptive neurons, and may thus represent a potential therapeutic target [28,29,30]. Additional splice variants, named Δ1 and Δ2, lack large parts of the domain II–III linker region including the synaptic protein interaction site, have been isolated from human neuroblastoma cells and brain cDNA libraries [31]. Clinically important, the Δ1 channel was less sensitive to inhibition by both ω-conotoxin MVIIA and GVIA than either the Δ2 variant or the full-length construct [31]. However, since a human splice variant that is only expressed in pathological pain states or in nociceptive pathways has not been identified to date, targeting of Cav splice variants as an analgesic strategy remains to be validated in humans.
2.3. Cav2.2 Toxin Binding Sites
While it is now appreciated that ω-conotoxins represent some of the most selective known inhibitors of the neuronal Cav2.2 isoform, and many of the key residues involved in binding have been identified, the precise peptide binding determinants and binding sites on Cav channels are not clearly identified. It is generally accepted that ω-conotoxins act as pore blockers [11,32], although the binding site has been mapped primarily to the external vestibule of the channel in the domain III pore-forming S5–S6 region (see [9] for docking model). In addition, inhibition of Cav2.2 by ω-conotoxins can be notably more complex than would be expected from simple pore blockers sharing a homologous binding site. While some of the best-characterized ω-conotoxins, such as GVIA and MVIIA, bind nearly irreversibly to Cav2.2, reversibility can be induced by voltage protocols which take advantage of the preferential binding of ω-conotoxin to the inactivated rather than resting state of Cav2.2 [33].
The large putative extracellular loop between IIIS5 and IIIH5 has been shown to be critically important for the block of Cav2.2 by the extensively studied peptide inhibitor of Cav2.2, ω-conotoxin GVIA (Figure 1). In particular, residues Gln1327, Glu1334, Glu1337, Gln1339 [11] and Gly1326 and Glu1332 of this region were identified as being important for blockage by GVIA [34]. The latter group proposed that Gly1326 may form a barrier that controls access of peptide toxins to the outer vestibule of the channel pore and stabilizes the toxin-channel interaction [34].In addition, the complex between MVIIA or GVIA toxins and Cav2.2 has been proposed to involve a central aromatic residue: tyrosine in the peptide, which is critical for high affinity interactions (Figure 3), plus lateral basic residues that form salt bridges with Glu1332 and perhaps Glu1334 and Glu1337 on the channel [34].
However, voltage-dependent reversibility varies significantly between ω-conotoxins, with CVIE and CVIF showing voltage-dependent reversal particularly in the presence of α2δ1 and β subunits, while reversibility of block by GVIA and MVIIA are only weakly influenced by co-expression with the subunits α2δ1 and β2a or β3 subunit [16,35]. Residues identified to contribute to this reversibility of ω-conotoxin block include in particular Gly1326, as well as intracellular domain II-III linker regions (Figure 1) [31,32].
2.4. Auxiliary Subunits of Cav Channels
While the pore-forming α1 subunit determines the main electrophysiological and pharmacological properties of Cav channels, auxiliary α2δ and β-subunits can modify channel gating properties and thus have a significant influence on calcium channel function [35,36]. To date, four auxiliary α2δ1–4 subunits, consisting of extracellular disulfide-linked α2δ dimers of 170 kDa, and four auxiliary β1–4 subunits [37] forming a 55 kDa cytoplasmic complex with the α1 subunit, have been identified. In addition, a 33 kDa γ subunit comprising four transmembrane segments was first found as a component of skeletal muscle Cav channels [38], and its related isoforms are expressed in heart and brain (for review see [14,22]). The presence or absence of the auxiliary subunits modulate the α1 subunit function and play an important functional role, modifying and regulating the kinetic as well as pharmacological properties of Cav channels [16,35,39].
2.4.1. α2δ Subunit
The α2δ proteins are auxiliary subunits of Cav2.2 that enhance Cav2.2 trafficking and insertion in the plasma membrane [39], but also influence the biophysical and pharmacological properties of the channel (for review see: [40]). A single gene product translates the α2δ subunit, which is post-translationally cleaved into the α2 and δ parts that remain associated via disulphide bridges. The α2 protein (~950 amino acids) is entirely extracellular, while the δ part has a small extracellular part that is attached to α2, and a transmembrane domain with a very short cytoplasmic tail [41]. The α2δ protein was originally isolated from skeletal muscle as a non-essential subunit of the L-type calcium channel complex [39]. Later it was found to be expressed in many tissues, specifically; the α2δ isoforms 1 and 2 are highly expressed by many CNS neurons [42]. Importantly, the isoform 1 is involved in neuropathic pain and is overexpressed after peripheral sensory nerve injury [43,44]. α2δ1 and α2δ2 are the targets for the gabapentinoid drugs (gabapentin and pregabalin), which are drugs currently used in the treatment of neuropathic pain [44,45,46].
The α2δ subunits increase the Cav2.2 inactivation rate to different extents [47]. Specifically, co-expression of α2δ subunits has been reported to cause hyperpolarization of the steady-state inactivation as well as an increase in the voltage-dependence [41,47]. Importantly, co-expression of α2δ subunit decreases the potency of ω-conotoxins [16,35], which has implications for the therapeutic potential of these peptides.
Both the physiological functions of α2δ subunits and the mechanisms by which binding of gabapentinoid drugs such as gabapentin and pregabalin to α2δ subunit translates into therapeutic action are not fully understood. Intriguingly, despite binding to α2δ subunits, gabapentin and pregabalin produce little acute inhibition of calcium channel currents. Inhibition of Cav2.2 currents after chronic treatment is generally attributed to down-regulation of Cav2.2 trafficking (for review see [41,47,48]).
Although most of the role of α2δ1 in the regulation of pain has been related to regulation of Cav2.2function and trafficking, an alternative analgesic mechanism was proposed recently [49]. The presence of a large extracellular region containing a protein-protein interaction fold, the Von Willebrand Factor A (VWF-A) domain, suggests that the α2δ1 subunit could serve as a receptor for extracellular ligands itself [41,49]. Indeed, it has been reported that the VWF-A domain of the α2δ1 subunit binds to proteins of the thrombospondin family [49]. Thus, α2δ1 is proposed to be the neuronal thrombospondin receptor which is involved in CNS synaptogenesis (synapse formation) and synaptic maturation [49]. The α2δ1 inhibitor gabapentin was also found to disrupt the interaction of the α2δ1 subunit with proteins of the thrombospondin family, thus leading to inhibition of synapse formation [49]. This occurred both in vitro and in vivo when neonatal mice were treated with gabapentin [47,49], and it has been proposed that inhibited synapse formation represent an additional mechanism by which α2δ1 inhibitors produce analgesia [49].
2.4.2. β Subunit
Four different genes encode the β subunits (β1–β4) and numerous splice variants are known [39]. The β subunit is the only subunit of the channel that is entirely cytosolic. It has been proposed that these subunits associate with the α1 subunit predominantly through a highly conserved high affinity interaction that is mediated by the Alpha Interaction Domain (AID) in the α1 subunit [50] and a corresponding Beta Interaction Domain (BID) in the β subunit [51]. The β subunit aids the trafficking of α1 to the plasma membrane. This was initially thought to occur due to its ability to mask an endoplasmic reticulum retention signal in the α1 subunit domain I-II linker [51,52]. However, several recent studies have reported data which is inconsistent with this hypothesis (for review see [53]). Transplanting the I–II linker of the Cav2.2 α1 subunit into Cav3.1, which does not require the β subunit for its function, caused current up-regulation rather than down-regulation [54]. Similarly, domain I–II linkers from several different Cavα1 subunits do not cause ER retention of CD8 or CD4 reporter protein [55,56]. In addition, several studies have implicated regions other than the I–II linker in Cavα1 trafficking [56,57,58], and another study suggested that the mechanism of β subunit-mediated Cav trafficking involves proteosomal degradation [59].
β subunit co-expression has a large effect on the level of expression, voltage dependence and kinetics of gating of cardiac and neuronal Cav channels. In general, the level of Cavexpression is increased, the voltage dependence of activation and inactivation is shifted to more negative membrane potentials, and the rate of inactivation is increased. However, these effects are β subunit specific [22], with the β2 subunit slowing channel inactivation in combination with α1B-b/α2δ1, while the β3 induces faster inactivation in combination with α1B-b/α2δ1 [16]. Recovery from ω-conotoxins block is also influenced to varying degrees by the different β subunit isoforms co-expressed with α1B-b/α2δ subunits [8,16].
2.4.3. γ Subunit
The γ subunit was originally known to only be associated with the skeletal muscle voltage-gated channel complex. However, recently, expression of γ isoforms 2 and 3 were established in the brain [60,61] and genetic studies revealed the existence of a γ subunit isoform in the brain whose lack of expression is responsible for the epileptic and ataxic phenotype of the stargazer mouse [61]. In addition, the γ subunit has been found as part of a neuronal membrane complex with Cav1.2 [62]. The γ subunits share a conserved four transmembrane domain topology, with predicted intracellular amino and carboxy termini, and a consensus site for cAMP/cGMP phosphorylation [39]. Although the effects of auxiliary γ subunits on the pharmacology of Cav channels have not been extensively studied, a γ isoform-dependent negative effect on Cav3.1 low voltage-activated current density has been described [63]. In addition, patch-clamp recordings showed that transient transfection of γ1 drastically inhibited macroscopic currents through recombinant N-type calcium channels (CaV2.2/α2δ–1/β3) expressed in HEK-293 cells [64].
2.5. Cav2.2 Channels as Analgesic Target
Cav2.2 is expressed in a common pathway downstream from the large variety of receptors that mediate pain responses, thus, inhibition of this Cav subtype can mediate analgesia [65,66]. Although different types of calcium channels are found in nociceptive pathways, Cav2.2 is particularly important in controlling signaling in nociceptive pathways such as the ventral and dorsal horn of the spinal cord and dorsal root ganglion (DRG) neurons, especially along the dendrites and at presynaptic terminals where it contributes critically to neurotransmitter release [67]. As a consequence of the change in membrane potential that occurs in response to a painful peripheral stimulus, Cav2.2 channels open, resulting in an increase in intracellular calcium. This in turn triggers synaptic vesicle release of neurotransmitters such as glutamate, substance P and CGRP (Calcitonin Gene Related Peptide), which activate post-synaptic receptors in the membrane of spinothalamic neurons and nerve terminals localized in the dorsal horn of the spinal cord [67,68] (Figure 2), allowing the propagation of pain signals.
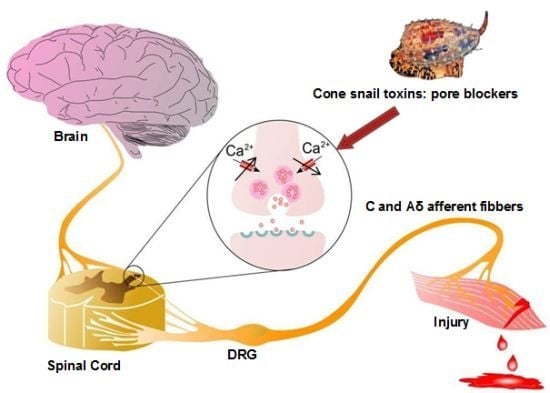
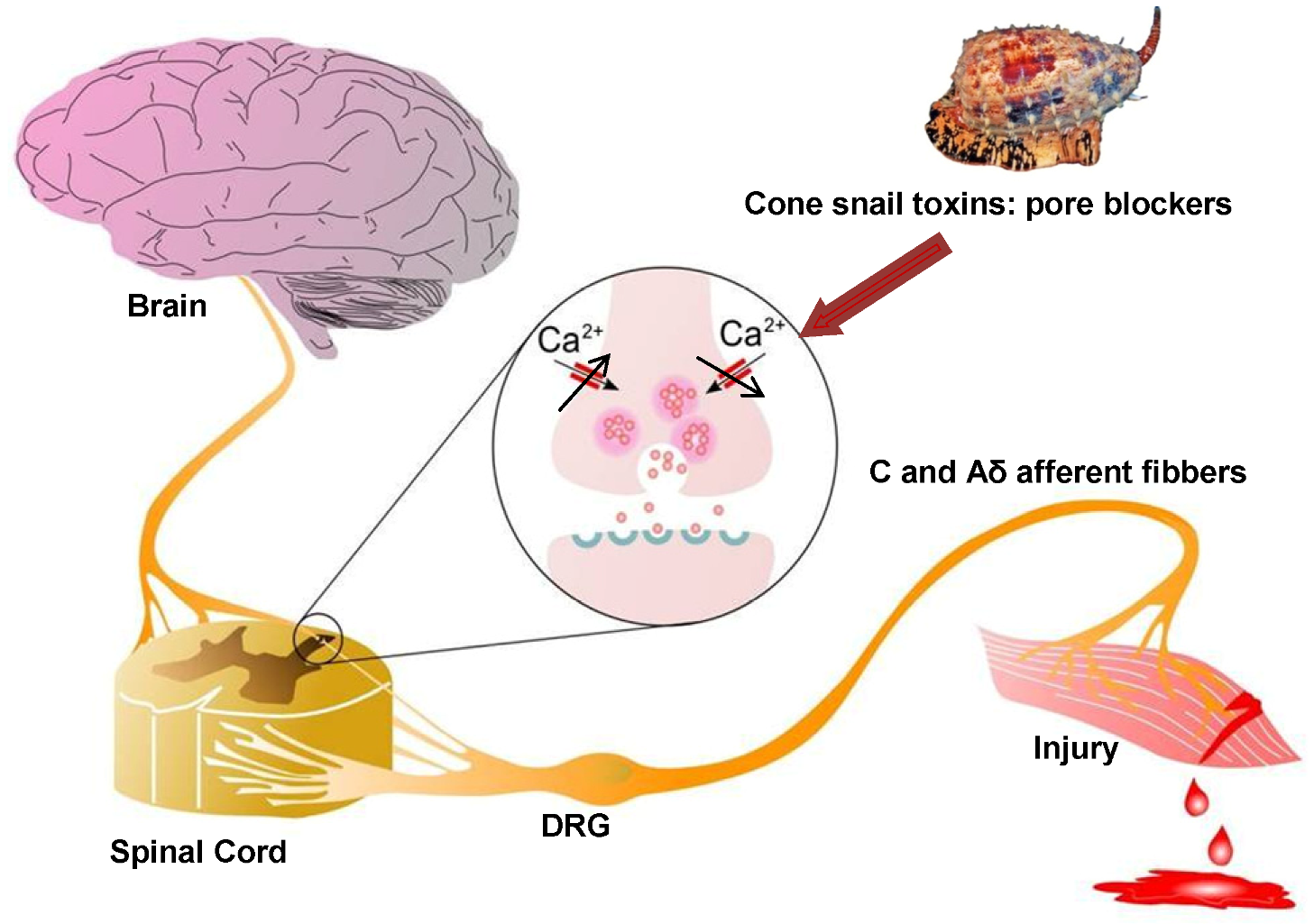
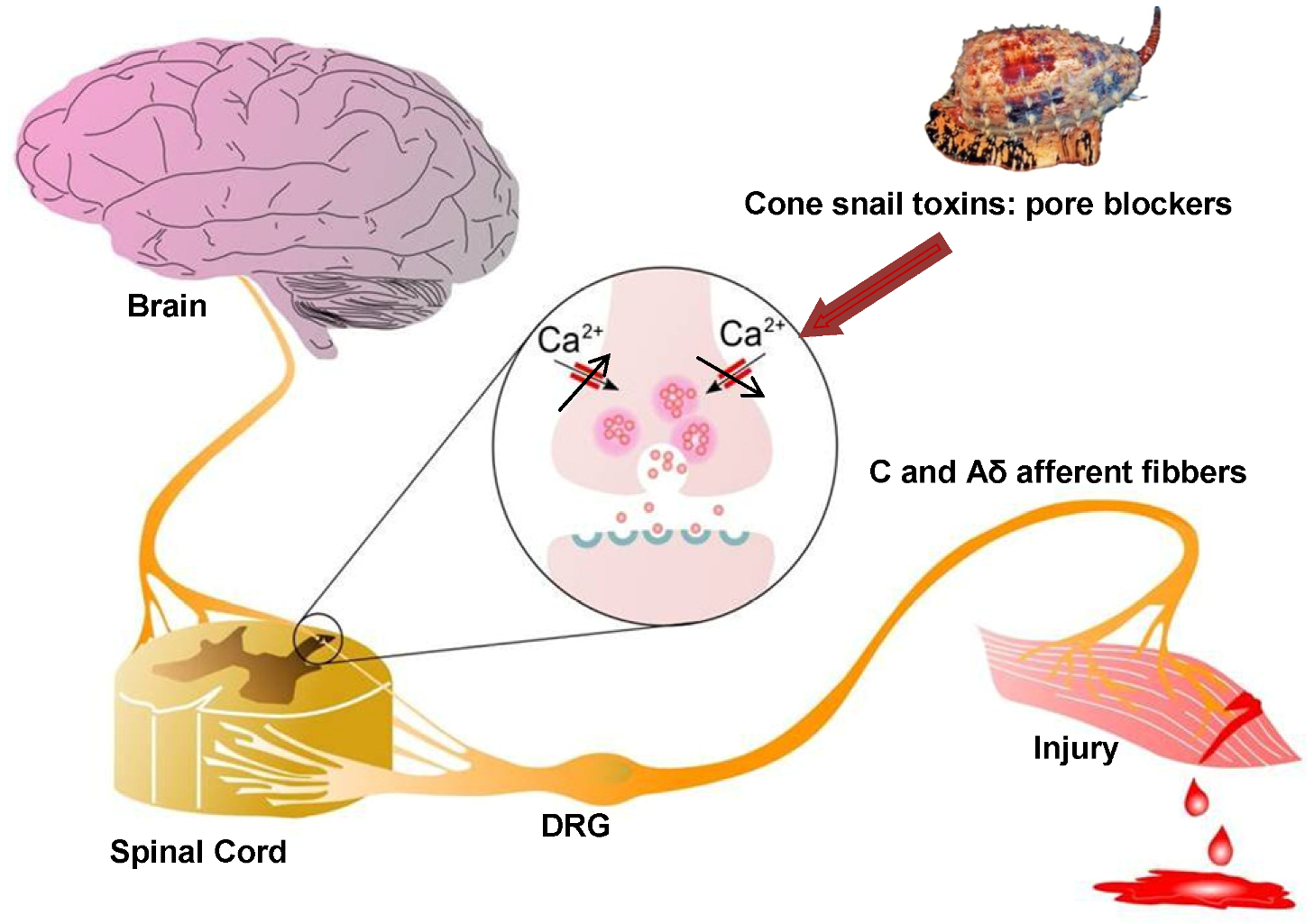
Figure 2.
Role of Cav2.2 in ascending pain pathway: Pain signals originating from peripheral C and Aδ afferent fibers evoke Cav2.2-mediated synaptic vesicle release of neurotransmitters such as glutamate, substance P, and CGRP which activate spinal neurons, altering sensory excitability and leading to pain sensations. Direct block of Cav2.2 channels by ω-conotoxins from cone snail venoms stops the link between the origin of pain and the transmission of pain sensation to the brain, because it decreases excessive calcium signalling during hyperactive excitation. Figure adapted from Zamponi et al. [67].
Figure 2.
Role of Cav2.2 in ascending pain pathway: Pain signals originating from peripheral C and Aδ afferent fibers evoke Cav2.2-mediated synaptic vesicle release of neurotransmitters such as glutamate, substance P, and CGRP which activate spinal neurons, altering sensory excitability and leading to pain sensations. Direct block of Cav2.2 channels by ω-conotoxins from cone snail venoms stops the link between the origin of pain and the transmission of pain sensation to the brain, because it decreases excessive calcium signalling during hyperactive excitation. Figure adapted from Zamponi et al. [67].
Several lines of evidence support Cav2.2 as an important pain target. Studies of Cav2.2 knock-out mice have shown that these animals, in contrast to Cav2.1 knock-out mice, had normal CNS (central nerve system) and motor function, but were resistant to development of neuropathic pain in a spinal nerve ligation model, and were insensitive to formalin-induced or visceral pain [69,70]. Furthermore, morphine, an opioid analgesic used for many years as the first option to treat severe pain, indirectly modulates Cav2.2 channels. Binding of morphine to μ-opioid receptors leads to inhibition of Cav2.2 through Gβγ-mediated signaling that reduces the ability of DRG sensory neurons to propagate pain signals centrally [67,71].
In addition, the α2δ1 auxiliary subunit of the Cav2.2 channels has been reported to be up-regulated in pain states in the dorsal root ganglion (DRG) and spinal dorsal horn after nerve injury [43,44,72,73], suggesting an involvement of this subunit with pathophysiological mechanisms of pain [73]. Although as discussed above, other mechanisms may underlie the involvement of the α2δ subunit with pain; studies using transgenic mice have found that the pro-algesic effects of α2δ subunits are mediated at least partially by enhancing Cav2.2 activity in sensory neurons [73]. In this study the author suggested it occurred possibly through enhanced formation of the functional Cav complex in lipid raft micro domains, as well as through hyper-excitability in dorsal horn neurons in response to peripheral stimulation [73].
Lastly, in 2004 the Cav2.2 blocker peptide ω-conotoxin MVIIA or ziconotide (Prialt®), was approved for the treatment of severe chronic pain associated with cancer, AIDS and neuropathies. Intrathecal injection (delivered directly to the spinal cord) of this synthetic peptide has proved effective against both neuropathic and inflammatory pain in laboratory animals and man [15,18,69,74,75], although associated side effects limit its application. Importantly, ziconotide acts synergistically with opioid analgesics without inducing tolerance or addiction [19].
2.6. Venoms as a Rich Source of Cav2.2 Channel Blockers
Cone snails comprise over 500 species of marine predatory gastropods that are mostly found on or near coral reefs in tropical and subtropical waters, including the coastal waters of Australia. They produce a highly complex mixture of venom peptides which have evolved for prey capture and defense (for review see [1,9]). This includes the ω-conotoxins, small disulfide-rich peptides with defined activity at mammalian Cav isoforms.
The ω-conotoxins belong to the O-superfamily, which also includes the δ-conotoxins (inhibit the fast inactivation of the voltage gated Na+ channels), µO-conotoxins (inhibit voltage-gated Na+ currents) and κ-conotoxins (interact with K+ channels). To date, ω-conotoxins targeting mammalian Cav isoforms have only been isolated from piscivorous cone snails, where they are likely to have evolved as part of the “motor cabal” leading to rapid flaccid paralysis of their fish prey [76]. In contrast, the few ω-conotoxins isolated from mollusc-hunting species to date (PnVIA and PnVIB) have been found to be inactive at mammalian Cav channels, suggesting distinct phylum-selective pharmacology, consistent with their sequence diversity (for review see: [9]).
The ω-conotoxin family comprises peptides ranging from 24 to 30 amino acids in length. As seen for the snake and spider venom toxins, the relatively low sequence homology among ω-conotoxins suggests that the overall three-dimensional structure and charge distribution underpin their interaction with Cav2.2 channels (Table 2, Figure 3). Although the remainder of the amino acids shows no absolute sequence conservation, positions 2 and 25 are always occupied by either a lysine (K) or an arginine (R), and the most active forms have a tyrosine (Y) at position 13. Moreover, a high proportion of residues contain hydroxyl moieties, which is accentuated in many of the ω-conotoxins by the substitution of γ-hydroxyproline for proline [77]. Disulphide bonds fold the peptide into a highly conserved cysteine framework pattern (C-C-CC-C-C) (Table 2, Figure 3) that contributes to tertiary structure stabilization. This configuration defines the canonical ω-conotoxin fold, which comprises a triple-stranded β-sheet/inhibitory cysteine-knot framework (Figure 3) [78]. A unique feature of conotoxins is their high degree of post-translational modification [79], and several ω-conotoxin have stability enhanced naturally through the use of these post-translational modifications (PTMs) [1,80]. This mechanism may limit potential degradation by carboxylases, which would otherwise rend the peptide biologically inactive. In addition, a C-terminal amide and the relative abundance of basic residues within ω-conotoxins class gives them an overall net positive charge, which presumably assists in their complementary binding to Cav2.2 channels [81].
The binding determinants for the high affinity interaction of ω-conotoxins with Cav2.2 have been proposed to rely on a two-point pharmacophore formed by the highly conserved Y13 (tyrosine) and K2 (lysine) [8,85,86,93,94]. However, Y13 and K2 are also often conserved in ω-conotoxins with activity at Cav2.1. Thus, residues contributing to selectivity at Cav2.2 over Cav2.1 are less clear. Intriguingly, the two ω-conotoxins that display most sequence homology, MVIIA and MVIIC, target quite different Cav subtypes (Cav2.2 and Cav2.1, respectively), whereas conversely, ω-conotoxins GVIA and MVIIA inhibit the same Cav subtype (Cav2.2), despite significantly lower sequence homology (Table 2, Figure 4). An extensive study assessing loop splice hybrids found that selectivity of MVIIA and MVIIC for Cav2.2 and Cav2.1 respectively, is controlled in a concerted manner by residues of loop 2 and 4 (Figure 3) [81]. Peptides with homogeneous combinations of loop 2 and 4 display clear selectivity while those with heterogeneous combinations of loops 2 and 4, are less discriminatory (Figure 3A−B) [81]. CVID is notable for its high potency at Cav2.2 and low potency at Cav2.1, making this peptide the most Cav2.2-selective peptide described to date [8,90].
Table 2.
ω-ConotoxinCav2.2 blockers: Sequence, indicating conserved cysteine residues in bold face type and potency at 125I-GVIA or MVIIA binding assays.
| ω-conotoxin name | ω-conotoxin Sequence | 125I-Ctx binding assays to rat brain IC50/Kd (nM) | Reference |
|---|---|---|---|
| CnVIIA | CKGKGAOCTRLMYDCCHGSCSSSKGRC* | 0.4 (2.2 > 2.1) | [82] |
| CVIA | CKSTGASCRRTSYDCCTGSCRSGRC | 0.6 (2.2 > 1.2) | [8] |
| CVIB | CKGKGASCRKTMYDCCRGSCRSGRC | 7.7 (2.2~2.1 > 2.3) | [8] |
| CVIC | CKGKGQSCSKLMYDCCTGSC-SRRGKC | 7.6 (2.1~2.2) | [8] |
| CVID | CKSKGAKCSKLMYDCCSGSCSGTVGRC | 0.07 (2.2 > 2.1) | [8] |
| CVIE | CKGKGASCRRTSYDCCTGSCRSGRC | 0.025 (2.2 > 2.1 > 1.2~1.3~2.3 | [16] |
| CVIF | CKGKGASCRRTSYDCCTGSCRLGRC | 0.098 (2.2 > 2.1 > 1.2~1.3~2.3) | [16] |
| FVIA | CKGTGKSCSRIAYNCCTGSCRSGKC | ND (2.2 > 2.1 > 3.2) | [83] |
| GVIA | CKSOGSSCSOTSYNCCRSCNOYTKRCY* | 0.04 (2.2 > 2.1) | [84,85,86,87,88] |
| GVIB | CKSOGSSCSOTSYNCCR-SCNOYTKRCYG* | ND | [88,89] |
| GVIIA | CKSOGTOCSRGMRDCCTSCLLYSNKCRRY* | 3.7 (ND) | [88,89,90] |
| GVIIB | CKSOGTOCSRGMRDCCTSCLSYSNKCRRY* | ND | [88] |
| MVIIA | CKGKGAKCSRLMYDCCTGSCRSGKC | 0.055 (2.2 > 2.1) | [13,81,87] |
| RVIA | CKPPGSPCRVSSYNCCSSCKSYNKKCG | 0.25 (2.2) | [10] |
| TVIA | CLSXGSSCSXTSYNCCRSCNXYSRKCR | ND (2.2 > 2.1) | [91,92] |
Source: Conoserver database: www.conoserver.org. 125I-Ctx = 125I-GVIA or 125I-MVIIA displacement assays to define ω- conotoxins binding to Cav2.2 expressed in different brain preparations including rat, chicken and mouse brain. ND= Not determined; in brackets the order of Cav type selectivity for each ω-conotoxin is described.* O=hydoxyproline (PTM: post-translational modification).
Importantly, it has been reported that the affinity of ω-conotoxins is often profoundly affected by the presence of auxiliary subunits, in particular α2δ and β subunits [16,35,95,96,97,98]. However, the degree to which co-expression of auxiliary subunits affects ω-conotoxin potency can vary significantly, with MVIIA, GVIA, CVID and CVIF being particularly susceptible to affinity reductions in the presence of α2δ1 subunits, while the potency of CVIE is affected to a lesser degree by co-expression with auxiliary subunits [16,35]. Such pharmacological effects can have profound implications for the therapeutic potential of ω-conotoxins, as α2δ1 subunit expression is increased in dorsal root ganglion and spinal cord in several animal models of neuropathic pain [1,16,99].
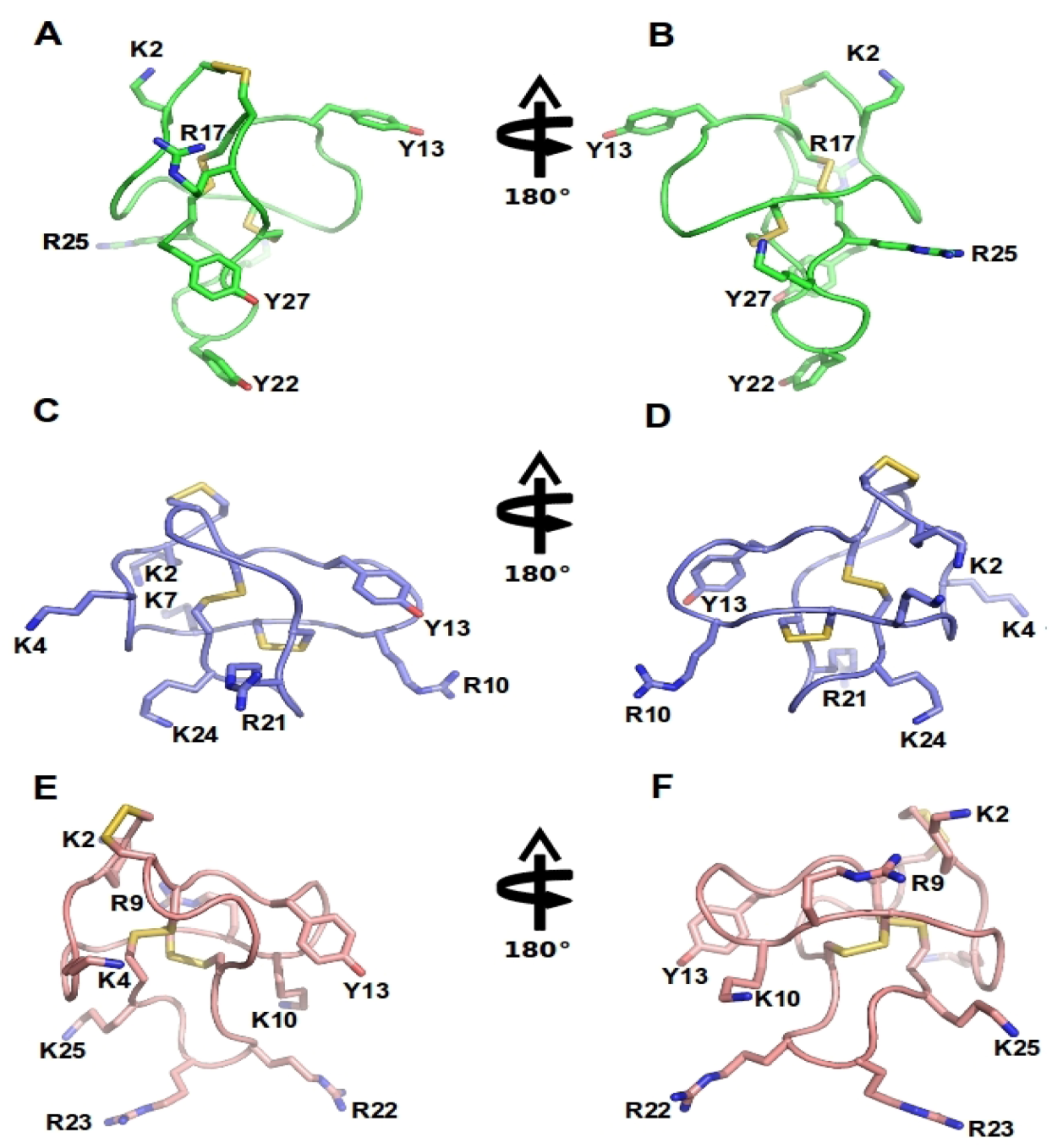
Figure 3.
ω-Conotoxins structure: NMR structure of GVIA (PDB 1TTL, green A-B), MVIIA (PDB 1 TTK, blue C-D) and MVIIC (PDB 1CNN, pink D-E). Represented are two different orientations. Disulfide bridges are shown in yellow and important amino acid residues, including Y13 (tyrosine13) and K2 (lysine2) and several positively charged residues exposed in the side chain are labeled.
Figure 3.
ω-Conotoxins structure: NMR structure of GVIA (PDB 1TTL, green A-B), MVIIA (PDB 1 TTK, blue C-D) and MVIIC (PDB 1CNN, pink D-E). Represented are two different orientations. Disulfide bridges are shown in yellow and important amino acid residues, including Y13 (tyrosine13) and K2 (lysine2) and several positively charged residues exposed in the side chain are labeled.
2.6.1. Cone Snail Venom Peptide Cav2.2 Inhibitors for the Treatment of Pain
While both direct and indirect inhibition of Cav2.2 by toxins and small molecules is a clinically validated analgesic strategy, currently available peptide Cav2.2 inhibitors suffer from limitations that restrict their more widespread use. These limitations include the need for intrathecal administration for effective delivery to spinal sites of action. In addition, dose-limiting side effects including dizziness, nystagmus, somnolence, abnormal gait and ataxia lead to a narrow therapeutic index. Thus, since its approval in 2004, ziconotide remains the only peptidic Cav2.2 inhibitor approved for the treatment of severe refractory pain. The precise mechanisms underlying this unfavorable side effect profile is not entirely clear. Contributing factors most likely include a lack of in vivo selectivity over other Cav subtypes (although the in vitro binding selectivity is exceptional), inhibition of Cav2.2 at supraspinal sites [15], inhibition at inhibitory interneurons or descending inhibitory synapses [57], or pharmacodynamic effects such as a slow off-rate and poor affinity for Cav2.2 co-expressed with auxiliary subunits [100].
2.6.2. Effect of Selectivity on Side Effect Profile
While Cav2.2 is important in mediating synaptic transmission at nociceptive synapses, contributions from other Cav channel subtypes, most notably Cav2.1 and Cav2.3, also mediate significant neurotransmitter release at neuronal synapses [101,102]. Thus, non-selective block of these Cav isoforms can contribute to severe side effects arising from inhibition of neurotransmitter release in non-nociceptive neurons. Accordingly, a significant challenge in targeting Cav2.2 for therapeutic drug discovery is likely to be selectivity over other Cav subtypes, especially Cav2.1, which is highly homologous to Cav2.2. However, the residues lining the pore in all S5 and S6 segments, which are proposed to contain the binding sites for most of the therapeutically useful drugs that block voltage-gated calcium channels, are nearly identical [103]. Thus, many venom peptides with activity at Cav2.2 also inhibit Cav2.1 to varying degrees, and vice versa. Systemically administered small molecule or peptide inhibitors of Cav2.2, while efficacious, lead to additional side effects resulting predominantly from action on the cardiovascular system [104]. However, recent data from animal models provide some evidence that a better therapeutic margin may be achievable with the ω-conotoxins. For example, intravenously administered leconotide (CVID, AM336), the most selective N-type blocker described to date [8], has shown efficacy in animal models of bone cancer pain [8]. Accordingly, additional ω-conotoxins are currently undergoing pre-clinical and clinical trials, and novel ω-contoxins with improved safety margin will hopefully reach the clinic in the future.
2.7. Cav2.2 Inhibitor Toxins from Spiders
Spiders, the most species-rich family of terrestrial venomous predators, have evolved highly complex venoms to assist with prey capture ([105], for review see [106]). Like cone snails, spiders have evolved a myriad of peptide venom components with activity at voltage-gated ion channels including Cav2.2 (for review see [106,107]).
Interestingly, spider toxins have provided some of the most subtype-selective Cav2.1 and Cav2.3 inhibitors known to date [108,109]. However, in contrast to conotoxins, which are notable for their selectivity for Cav2.2 in particular (for review see [9]), relatively few spider venom peptides are active at Cav2.2, and even fewer show selectivity for Cav2.2 over other Cav isoforms (see Table 2 and Table 3) [4,5].
| Toxin name/Synonym | Functional (IC50)/Binding (Kd) at Cav2.2 | Amino acid sequence | Reference |
|---|---|---|---|
| μ/ω-theraphotoxin-Hh1a/Huwentoxin-1 | 100 nM (ND) | ACKGVFGACTPGKNECCPNRVCSDKHKWCKWKL | [110,111] |
| μ/ω-theraphotoxin-Hh1b/Huwentoxin1a3 | (ND) | ACKGVFGACTPGKNECCPNRVCSDKHKWCKWKL | [112] |
| μ/ω-theraphotoxin Hh1c/Huwentoxin1a10 | (ND) | ACKGVFDACTPGKNECCSNRVCSDKHKWCKWKL | [112,113] |
| μ/ω-theraphotoxin-Hh1d/Huwentoxin-1a6 | (ND) | ACKGVFDACTPGKNECCPNRVCSDEHKWCKWKL | [112] |
| ω-agatoxin-Aa2a/ω-agatoxin IIA | 10 nM (Y) | GCIEIGGDCDGYQEKSYCQCCRNNGFCS | [114,115] |
| ω-agatoxin-Aa3a/ω-agatoxin IIIA | 1.4 nM/170 pM (N) | SCIDIGGDCDGEKDDCQCCRRNGYCSCYSLFGYLKSGCKCVVGTSAEFQGICRRKARQCYNSDPDKCESHNKPKRR | [116,133] |
| ω-agatoxin-Aa3b/ω-agatoxin IIIB | 140 nM/2.4 nM (N) | SCIDFGGDCDGEKDDCQCCRSNGYCSCYNLFGYLKSGCKCEVGTSAEFRRICRRKAKQCYNSDPDKCVSVYKPKRR | [116,117] |
| ω-agatoxin-Aa3d/ω-agatoxin IIID | 35 nM (N) | SCIKIGEDCDGDKDDCQCCRTNGYCSXYXLFGYLKSG | [116] |
| ω-agatoxin-Aa3f/ω-agatoxin IIIA (58T) | 1.4 nM (N) | SCIDIGGDCDGEKDDCQCCRRNGYCSCYSLFGYLKSGCKCVVGTSAEFQGICRRKARTCYNSDPDKCESHNKPKRR | [116] |
| ω-agatoxin-Aa3g/ω-agatoxin IIIB (35R) | 2.4 nM (N) | SCIDFGGDCDGEKDDCQCCRSNGYCSCYNLFGYLRSGCKCEVGTSAEFRRICRRKAKQCYNSDPDKCVSVYKPKRR | [116] |
| ω-agatoxin-Aa3h/ω-agatoxin IIIB (29S) | 2.4 nM (N) | SCIDFGGDCDGEKDDCQCCRSNGYCSCYSLFGYLKSGCKCEVGTSAEFRRICRRKAKQCYNSDPDKCVSVYKPKRR | [116] |
| ω-ctenitoxin-Pn2a/Neurotoxin Tx3–3 | >320nM/50 pM (N) | GCANAYKSCNGPHTCCWGYNGYKKACICSGXNWK | [7,118,119] |
| ω-ctenitoxin-Pn3a/Neurotoxin Tx3–4 | 50 pM (N) | SCINVGDFCDGKKDDCQCCRDNAFCSCSVIFGYKTNCRCEVGTTATSYGICMAKHKCGRQTTCTKPCLSKRCKKNH | [119] |
| ω-ctenitoxin-Pn4a/Neurotoxin Tx3–6 PnTx3–6/Phα1β | 122 nM (N) | ACIPRGEICTDDCECCGCDNQCYCPPGSSLGIFKCSCAHANKYFCNRKKEKCKKA | [119,120] |
| ω-ctenitoxin-Pr1a/Neurotoxin PRTx3–7 | >1000 nM (N) | ACAGLYKKCGKGVNTCCENRPCKCDLAMGNCICKKKFVEFFGG | [121,122] |
| ω-segestritoxin-Sf1a/SNX-325 | ~10 nM (Y) | GSCIESGKSCTHSRSMKNGLCCPKSRCNCRQIQHRHDYLGKRKYSCRCS | [123] |
| ω-theraphotoxin-Hh1a/Huwentoxin-10 | 40 nM (Y) | KCLPPGKPCYGATQKIPCCGVCSHNKCT | [113] |
Source: Arachnoserver spider venom database: http://www.arachnoserver.org [4,5]. Selective for Cav2.2 channel? (Y) = yes, (N) = no, ND = Not determined; Binding: 125I-Ctx performed in different brain preparations, including rat, chicken and mouse brain.
Spider venom peptides with activity at Cav2.2 have to date only been described from Haplopelma huwenum, Agelenopsis aperta, Phoneutria nigriventer, Phoneutria reidyi and Segestria florentina [7,110,111,112,113,114,115,116,117,118,119,120,121,122,123]. These Cav2.2 inhibitors are structurally diverse and can share common structural motifs with the ω-conotoxins (Figure 3, Figure 4, Figure 5), with 3 disulfide bonds forming an inhibitory cysteine knot, or have as many as 7 disulfide bonds, as is the case for ω-ctenitoxin-Pn3a [113,119]. The majority of these peptides have little selectivity for Cav2.2 and displays activity at Cav1, Cav2.1 and Cav2.3 isoforms. For example, ω-agatoxin-Aa3a is equipotent at Cav1 and Cav2.2, and ω-ctenitoxin-Pn2a from Phoneutria nigriventer blocks voltage-gated calcium channels Cav2.1, Cav2.3, Cav1 and Cav2.2 in decreasing order of potency [119]. Some spider venom peptides, such as the theraphotoxins Hh1a-d, even inhibit the related Nav channels and are designated as ω-/µ-toxins based on this pharmacological profile [110]. In contrast, ω-segestritoxin-Sf1a, ω-agatoxin-Aa2a and ω-theraphotoxin-Hh1a or huwentoxin-10 preferably inhibit vertebrate Cav2.2, although it is unclear whether their selectivity can match the more than 10,000-fold preference for Cav2.2 over Cav2.1 exhibited by some conotoxins [8,113,115,123]. Given the similar overall structure shared by cone snail and spider venom peptides, these divergent selectivity profiles are surprising. However, as illustrated by conopeptides, small differences in structure may result in profound effects on Cav selectivity. Specifically, conotoxins MVIIA, MVIIC, GVIA and CVID share a high degree of sequence similarity and are structurally closely related. Nonetheless, MVIIC is a selective Cav2.1 inhibitor, while MVIIA, GIVA and CVID are highly selective Cav2.2 blockers [8].
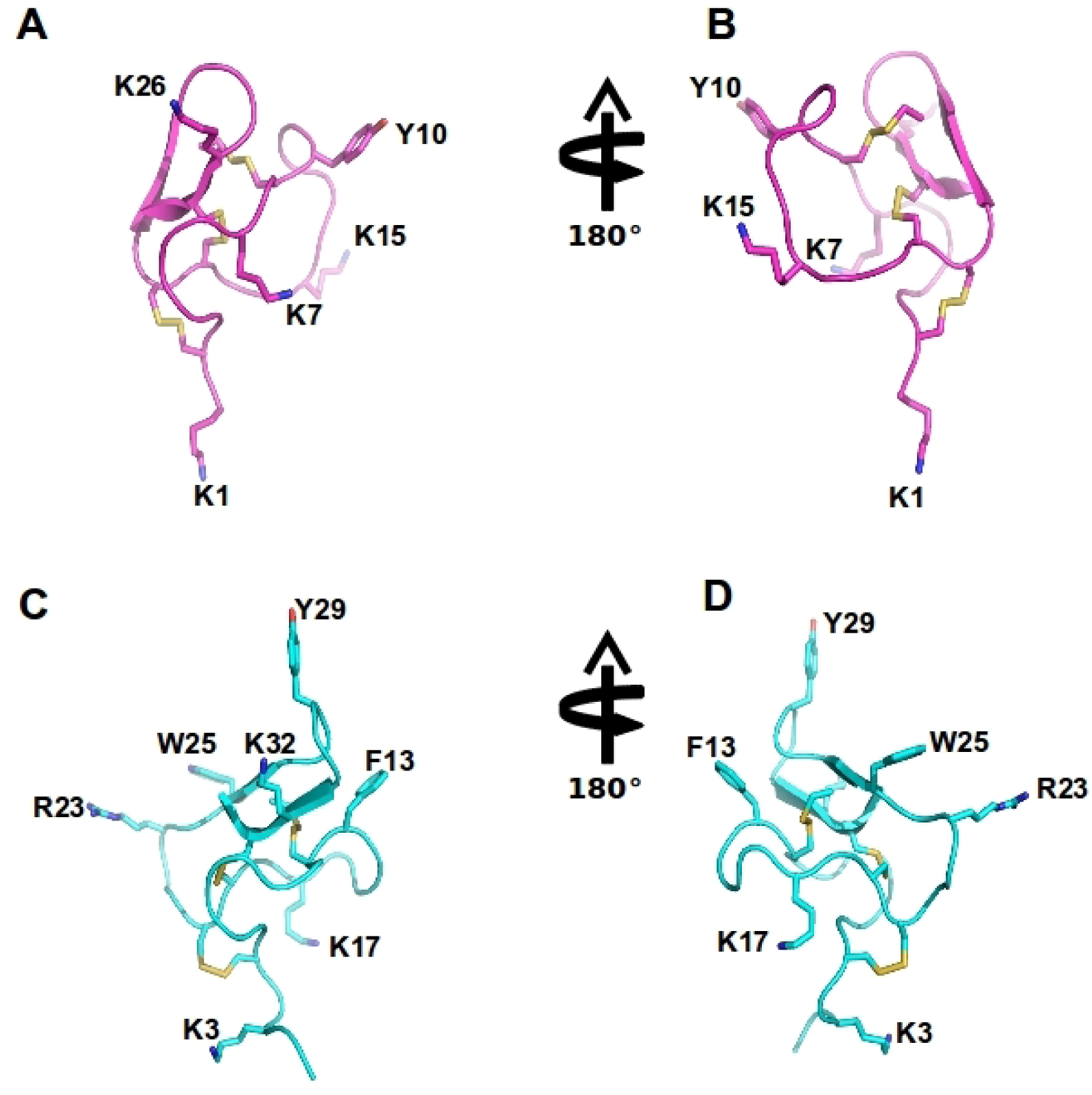
Figure 4.
Spider ω-theraphotoxin-Hh1a (HWTX-X) and Ptu1 structure: The structure of huwentoxin 10 (HWTX-X) (pink A–B) and Ptu1 (blue C–D) showing two different orientations. The position of the four loops is indicated, and disulfide bridges are shown in yellow. Important amino acid residues described to have similar function to tyrosine13 and lysine 2 in the ω-conotoxins are represented, including Y10 and K7 in Ptu1, as well as F13 (phenylalanine13) and K17 (lysine17) and several positively charged residues exposed in the side chain are labeled.
Figure 4.
Spider ω-theraphotoxin-Hh1a (HWTX-X) and Ptu1 structure: The structure of huwentoxin 10 (HWTX-X) (pink A–B) and Ptu1 (blue C–D) showing two different orientations. The position of the four loops is indicated, and disulfide bridges are shown in yellow. Important amino acid residues described to have similar function to tyrosine13 and lysine 2 in the ω-conotoxins are represented, including Y10 and K7 in Ptu1, as well as F13 (phenylalanine13) and K17 (lysine17) and several positively charged residues exposed in the side chain are labeled.
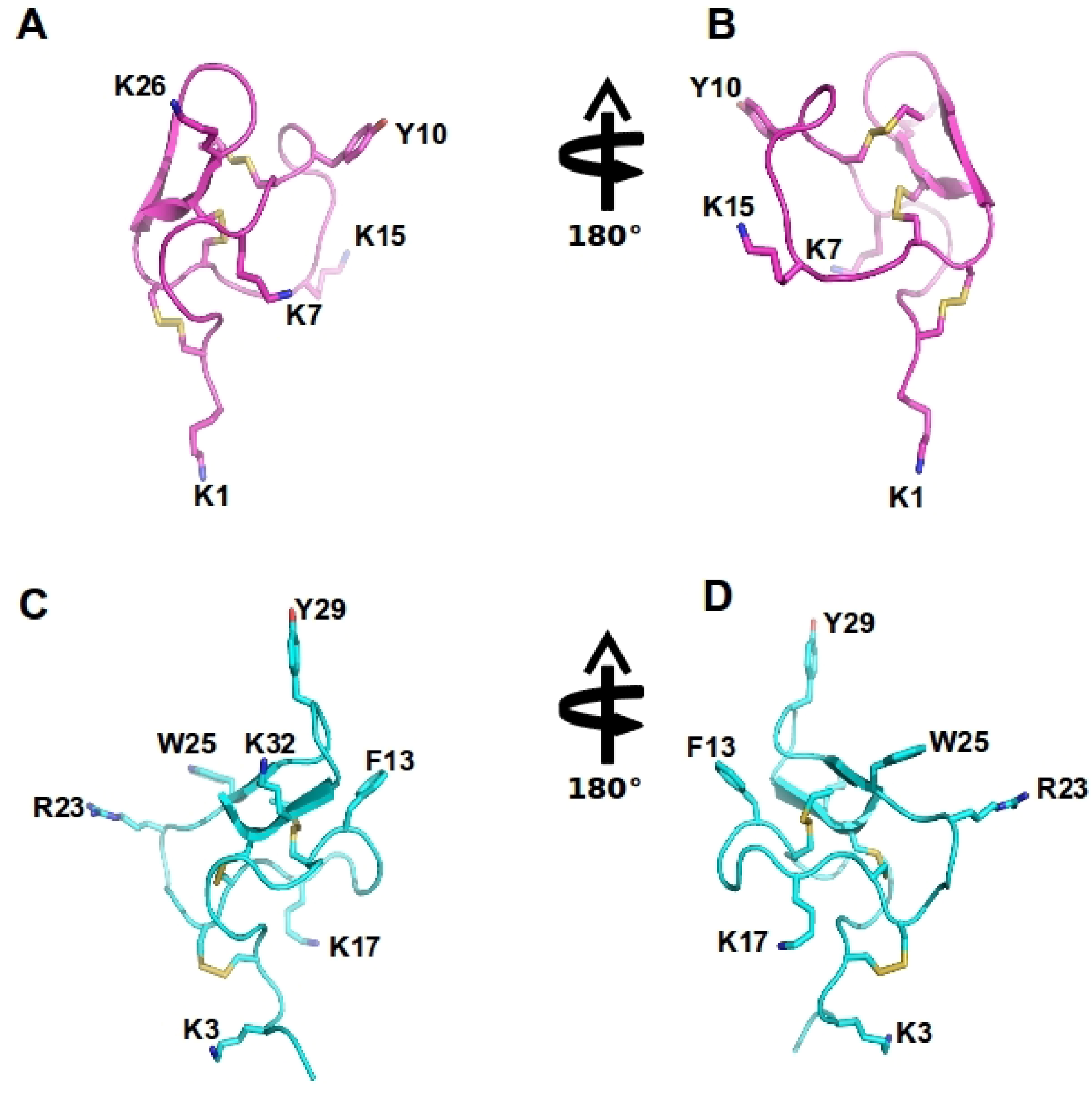
Figure 5.
Amino acid sequence alignment of the Cav2.2 inhibitor toxins, ω-conotoxins from cone snails, spiders and the assassin bug Peirates turpis. Cysteines common to all toxins which are important for these peptides extraordinary stability are highlighted, in addition to positively charged amino acids suggested to be important for binding of these toxins to Cav2.2 channels.
Figure 5.
Amino acid sequence alignment of the Cav2.2 inhibitor toxins, ω-conotoxins from cone snails, spiders and the assassin bug Peirates turpis. Cysteines common to all toxins which are important for these peptides extraordinary stability are highlighted, in addition to positively charged amino acids suggested to be important for binding of these toxins to Cav2.2 channels.
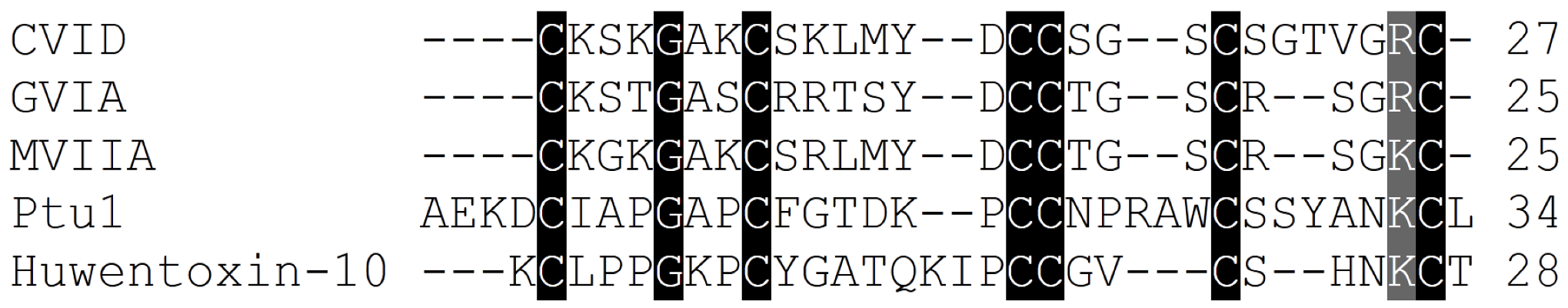
Though spider toxins are generally accepted to act as gating modifiers at Cav channels, relatively little is known about the mechanism of action of Cav2.2 block. For example, ω-ctenitoxin-Pn4a was shown to decrease peak Cav2.2 current with little effect on voltage-dependence of activation [120], and Cav2.2-specific inhibitors from spider venoms such as ω-segestritoxin-Sf1a were able to displace radiolabelled ω-conotoxins (Table 3) [123], suggesting that these peptides may act like pore blockers rather than gating modifiers at Cav2.2. Importantly, little is known about the influence of Cav auxiliary subunits on inhibition by spider venom peptides. Given that the pharmacology of cone snail toxins is known to be affected by auxiliary subunits [16,35,124], future studies should include characterising the effect of auxiliary subunits on inhibition of Cav2.2 by spider venom peptides.
2.7.1. ω-Agatoxin-Aa2a
The venom of Agelenopsis aperta provided the first source of Cav inhibitors, making the agatoxins some of the best-studied spider Cav channel antagonists. Based on their structural homology and pharmacological properties, agatoxins have been classified into four distinct groups (agatoxins I –IV). While type I and III agatoxins are selective for Cav1 and Cav2.1, respectively, type II and III agatoxins display activity at Cav2.2. However, while type III agatoxins such as ω-agatoxin-Aa3a are active at all high-threshold Cav channel isoforms, including Cav2.1, Cav2.2, Cav2.3 and Cav1, type II agatoxins target Cav2.2 over other Cav isoforms [114,115]. ω-Agatoxin-Aa2a, an 11 kDa mature toxin comprised of 92 residues, displaced ω-conotoxin GVIA binding and synergistically blocked neurotransmitter release with the unrelated L-type toxin ω-AGTX-Aa1a [114]. While more detailed selectivity studies have not been carried out, this suggests that the toxin targets primarily Cav2.2 channels [115].
The structural requirements for high affinity inhibition of Cav2.1 by type IV agatoxins such as ω-agatoxin-Aa4a have been relatively well defined, and are proposed to involve a positively charged area, formed by several basic amino acid residues near the hydrophobic C-terminus [125], as well as a crucial tryptophan residue in position 14. In contrast, nothing is known about the structure-activity of ω-agatoxin-Aa2a. Thus, future studies are necessary to improve our understanding of the molecular interaction between ω-agatoxin-Aa2a and Cav2.2.
2.7.2. ω-Theraphotoxin-Hh1a
ω-Theraphotoxin-Hh1a (huwentoxin 10 or HWTX-X) was isolated from the venom of the Chinese bird spider and shares several properties with the ω-conotoxins [113]. In contrast to ω-conotoxins, the C-terminus of HWTX-X is not amidated (Table 3, Figure 5) [113].This relatively small 28 residue peptide is stabilized by three disulfide bonds, and shares a functional motif, defined by a critical aromatic residue and several basic residues, with the ω-conotoxin GVIA (Figure 4). Intriguingly, huwentoxin 10 was unable to inhibit twitch responses of electrically stimulated rat vas deferens, suggesting selectivity for different Cav2.2 or splice variants [113]. While the analgesic potential of ω-theraphotoxin-Hh1a has not been assessed to date, intraperitoneal injection in mice produced no toxic effects [113], suggesting that this peptide could be a promising therapeutic lead for the treatment of pain.
2.7.3. ω-Segestritoxin-Sf1a
ω-Segestritoxin-Sf1a (SNX-325) has been described as a selective Cav2.2 inhibitor, based on its ability to inhibit Cav2.2 responses in oocytes as well as KCl-mediated neurotransmitter release in hippocampal slices [123]. Interestingly, although ω-segestritoxin-Sf1a shares little structural homology with the ω-conotoxins and is a large peptide stabilized by 4 disulfide bonds, it shares a common binding site with the ω-conotoxins and was able to displace radiolabelled MVIIA from a rat brain synaptosome preparation [123].
2.8. Spider Venom Cav2.2 Inhibitors in Pain
While the analgesic potential of Cav2.2-selective peptides from spider venom has not been assessed extensively, several studies report efficacy with little side effects in various animal models of pain. ω-ctenitoxin-Pn4a (PnTx3–6, Phα1β, neurotoxin 3–6), a toxin from the spider Phoneutria nigriventer which non-selectively inhibits neuronal Cav with a rank order of potency of Cav1.2 > 2.2 > 2.1 > 2.3 [119,120,126,127], elicited prolonged analgesia after intrathecal administration in an animal model of incisional pain [128,129]. ω-ctenitoxin-Pn4a and had no effect on mean arterial blood pressure, heart rate or gross neuronal performance, suggesting that Cav inhibitors from spider venoms could also find therapeutic application for the treatment of pain [128,129]. Another non-selective toxin from Phoneutria nigriventer, ω-ctenitoxin-Pn2a (rank order of potency: Cav2.1 > 2.3 > 1 > 2.2) [118], showed prevalent antinociceptive effects in neuropathic pain models and did not cause adverse motor effects efficacious doses [130]. However, as Cav2.1 and Cav3 have been proposed as analgesic targets in their own right [131,132], the contribution of non-Cav2.2 channels to these observed in vivo effects remains to be determined.
3. Cav2.2 Inhibitors from Other Venomous Animals
Cav modulators are also found in the venom of other venomous species, including snakes, scorpions, and centipedes. However, these peptides, including calcicludine from the green mamba [134] and kurtoxin from the venom of the scorpion Parabuthus transvaalicus [135], show no selectivity for Cav2.2, or in the case of glycerotoxin [136] from the marine blood worm Glycera convulata, are Cav2.2 enhancers and are thus included here only for completeness.
3.1. Scorpion Venom Peptides
Kurtoxin, a 63-residue peptide isolated from the venom of the scorpion Parabuthus transvaalicus is related to the α-scorpion toxins, a family of toxins that slow inactivation of Nav channels. While kurtoxin has shown selectivity for heterologous expressed Cav3 or T-type calcium channels [137], activity at Cav2.2 has been described in rat sympathetic and thalamic neurons [135]. In contrast to ω-conotoxins with activity at Cav2.2 channels, scorpion toxins with activity at Cav channels, including kurtoxin, act as gating modifiers. Thus, selectivity differences observed in overexpression systems compared to neurons may be due to the influence of auxiliary subunits, although this has not been assessed to date.
3.2. Snake Venom Peptides
Calcicludine (60 residues, 3 disulfide bonds) and calciseptine (60 residues, 4 disulfide bonds) were isolated from the venom of the black mamba, Dendroaspis polyepsis, and the green mamba, Dendroaspis angusticeps, respectively. While calciseptine inhibits L-type calcium channels and has no activity at Cav2.2 [138], calcicludine is less selective and potently inhibits all high-voltage-activated calcium channels, including Cav2.2 [139].
3.3. Centipede Venom Peptides
Recently, two peptides with activity at neuronal Cav channels were isolated from the venom of the centipede Scolopendra subspinipes mutilans. ω-SLPTX-Ssm1a, an 83 residues peptide containing 7 cysteines was found to act as an activator of Cav channels [140], while a smaller peptide, ω-SLPTX-Ssm2a (54 residues) was shown to inhibit Cav channels expressed in DRG neurons [140]. However, the Cav subtype selectivity of these peptides is currently unknown but it may include Cav2.2, which is expressed in peripheral sensory neurons.
3.4. Assassin Bug Toxins
The predatory assassin bugs (Hemiptera: Reduviidae) contain a complex mixture of small and large peptides in its toxic saliva which is used to immobilize and pre-digest their prey, and for defense against predators [141]. Three novel peptide toxins, named Ado1, Ptu1 and Iob1, isolated from three species of assassin bugs (P. turpis, A. dohrni, and I. obscurus), were biologically active in electrophysiological assays using BHK-N101 cells stably expressing rabbit Cav2.2, β1A subunit, and α2δ subunit [141]. Ptu1 binds reversibly to Cav2.2 with lower affinity than ω-conotoxin MVIIA. Ptu1 lacks most of the residues shown to be important for ω-conotoxin binding to the N-type calcium channel, including equivalents of Tyr13 or Lys2 (Figure 4 and Figure 5). This peptide belongs to the inhibitory cysteine knot structural family (ICK) that consists of a four-loop Cys scaffold forming a compact disulfide-bonded core [142]. Thus, as for the other venom peptides compared here, the structure of Ptu1 aligned with related Cav2.2 blockers MVIIA and GVIA, indicating that a common functional motif can be supported by a common three-dimensional structure despite the lack of sequence homology (Figure 5).
4. High Throughput Assays for Novel Cav2.2 Channel Inhibitors
Venoms represent complex natural compound libraries which have evolved over millions of years, and contain some of the most subtype-selective ion channel modulators known. Assay-guided fractionation, the process whereby bioactive components from venom are isolated based on sequential rounds of fractionation and activity testing, has been used as a successful strategy for the discovery and isolation of novel venom components for many years (for review see [143]). This process requires sensitive, accurate and robust assays which are able to detect activity at the biological target of interest.
Electrophysiological techniques are generally considered the “gold standard” technique to study ion channels, including Cav2.2 channels. However, classical electrophysiological recordings are labour-intensive and generally low-throughput, while electrophysiology in high throughput format for primary drug screening is difficult and/or costly to implement [98,144]. Therefore, incorporation of functional cell-based high-throughput screening (HTS) assays can significantly speed up the identification of novel candidates from large compound libraries [144,145], such as animal venoms.
A range of HTS assays to screen new Cav2.2 channel inhibitors have been described, including fluorescence-based Ca2+ assays and radioligand binding assays [124,144,145,146,147]. Radioligand binding assays are amenable to HTS, however, these assays cannot detect modulators acting at different sites from the ω-conotoxin site on Cav2.2. Given the narrow safety window of pore blocking peptides such as ziconotide, small molecule inhibitors of Cav2.2 channels with state-dependent blocking activity may provide improved therapeutic margins (for review see: [148]). In addition, the membrane potential as well as association of the pore-forming α subunit of Cav2.2 with auxiliary subunits is disrupted in these assays, which may influence binding affinity determination [149].
Functional HTS, fluorescent-cell based assays are advantageous, because they can address some of the problems above mentioned. For example, using heterologous expression systems, the Cav2.2 α subunit can be co-expressed with auxiliary α2δ and β subunit to produce biophysical properties more similar to the native channel [124]. In one study, Kir2.3, a potassium channel, was co-expressed together with Cav2.2 α and auxiliary subunits, to control membrane potential [145]. This mechanism allowed identification of novel state-dependent inhibitors [145]. In addition, cell lines expressing Cav2.2 and auxiliary subunits endogenously can be used in HTS to provide information on the mechanisms of novel toxins inhibition, in a native context [149]. Thus, functional HTS assays are expected to accelerate identification, pharmacological characterization and selectivity profile of novel Cav2.2 modulators.
5. Conclusions
Animal venoms are rich sources of Cav channel modulators. Cone snail venoms in particular have provided a diverse array of inhibitors [9], including the most subtype-selective Cav2.2 inhibitors known [8]. These peptide toxins are valuable drug leads, pharmacological tools and drugs in their own right [9,115,117]. In addition, Cav2.2 inhibitor toxins have served as templates for the development of peptidomimetic small molecules, which can then be engineered in an attempt to circumvent some of the disadvantages inherent to peptidic Cav2.2 inhibitors, especially the need for intrathecal use. Key strategies for improving the therapeutic potential of calcium channel include identification of inhibitors of Cav2.2 splice variants that are only expressed in pain states [28], inhibition of Cav2.2 in combination with specific auxiliary subunits [35], optimizing state and/or use-dependent inhibition, and targeting other Cav2.2 regulatory pathways [148].
Acknowledgments
We acknowledge Anderson Wang and Olivier Allart for graphic design support.
Conflict of Interest
The authors declare no conflict of interest.
References
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar]
- Gomez, M.V.; Kalapothakis, E.; Guatimosim, C.; Prado, M.A. Phoneutria nigriventer venom: A cocktail of toxins that affect ion channels. Cell Mol. Neurobiol. 2002, 22, 579–588. [Google Scholar]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef]
- Herzig, V.; Wood, D.L.; Newell, F.; Chaumeil, P.A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. Arachnoserver 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2011, 39, D653–D657. [Google Scholar] [CrossRef]
- Arachnoserver 2.0. Available online: www.arachnoserver.org (accessed on 12 December 2012).
- Adams, M.E.; Bindokas, V.P.; Hasegawa, L.; Venema, V.J. Omega-agatoxins: Novel calcium channel antagonists of two subtypes from funnel web spider (agelenopsis aperta) venom. J. Biol. Chem. 1990, 265, 861–867. [Google Scholar]
- Dos Santos, R.G.; Van Renterghem, C.; Martin-Moutot, N.; Mansuelle, P.; Cordeiro, M.N.; Diniz, C.R.; Mori, Y.; De Lima, M.E.; Seagar, M. Phoneutria nigriventer ω-phonetoxin iia blocks the cav2 family of calcium channels and interacts with ω-conotoxin-binding sites. J. Biol. Chem.. 2002, 277, 13856–13862. [Google Scholar]
- Lewis, R.J.; Nielsen, K.J.; Craik, D.J.; Loughnan, M.L.; Adams, D.A.; Sharpe, I.A.; Luchian, T.; Adams, D.J.; Bond, T.; Thomas, L. Novel ω-conotoxins from conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem. 2000, 275, 35335–35344. [Google Scholar]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef]
- Abbott, J.R.; Litzinger, M.J. Different ω-conotoxins mark the development of swiss webster mouse cortex suggesting N-type voltage sensitive calcium channel subtypes. Int. J. Dev. Neurosci. 1994, 12, 43–47. [Google Scholar] [CrossRef]
- Ellinor, P.T.; Zhang, J.F.; Horne, W.A.; Tsien, R.W. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature 1994, 372, 272–275. [Google Scholar] [CrossRef]
- Wagner, J.; Snowman, A.; Biswas, A.; Olivera, B.; Snyder, S. Ω-conotoxin gvia binding to a high affinity receptor in brain: Characterization, calcium sensitivity and solubilization. J. Neurosci. 1988, 9, 3354–3359. [Google Scholar]
- Olivera, B.M.; Cruz, L.J.; de Santos, V.; LeCheminant, G.W.; Griffin, D.; Zeikus, R.; McIntosh, J.M.; Galyean, R.; Varga, J.; Gray, W.R. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using ω-conotoxin from conus magus venom. Biochemistry 1987, 26, 2086–2090. [Google Scholar]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef]
- Berecki, G.; Motin, L.; Haythornthwaite, A.; Vink, S.; Bansal, P.; Drinkwater, R.; Wang, C.I.; Moretta, M.; Lewis, R.J.; Alewood, P.F.; et al. Analgesic ω-conotoxins cvie and cvif selectively and voltage-dependently block recombinant and native N-type calcium channels. Mol. Pharmacol. 2010, 77, 139–148. [Google Scholar]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- Wermeling, D.P. Ziconotide, an intrathecally administered N-type calcium channel antagonist for the treatment of chronic pain. Pharmacotherapy 2005, 25, 1084–1094. [Google Scholar] [CrossRef]
- Alicino, I.; Giglio, M.; Manca, F.; Bruno, F.; Puntillo, F. Intrathecal combination of ziconotide and morphine for refractory cancer pain: A rapidly acting and effective choice. Pain 2012, 153, 245–249. [Google Scholar] [CrossRef]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International union of pharmacology. Xlviii. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef]
- Williams, M.E.; Brust, P.F.; Feldman, D.H.; Patthi, S.; Simerson, S.; Maroufi, A.; McCue, A.F.; Velicelebi, G.; Ellis, S.B.; Harpold, M.M. Structure and functional expression of an ω-conotoxin-sensitive human N-type calcium channel. Science 1992, 257, 389–395. [Google Scholar]
- Catterall, W.A. Structure and regulation of voltage-gated ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef]
- Yu, F.H.; Catterall, W.A. The vgl-chanome: A protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. STKE 2004, 2004. [Google Scholar] [CrossRef]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar]
- Payandeh, J.; Gamal El-Din, T.M.; Scheuer, T.; Zheng, N.; Catterall, W.A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012, 486, 135–139. [Google Scholar]
- Lipscombe, D.; Pan, J.Q.; Gray, A.C. Functional diversity in neuronal voltage-gated calcium channels by alternative splicing of ca(v)α1. Mol. Neurobiol. 2002, 26, 21–44. [Google Scholar] [CrossRef]
- Lin, Z.; Haus, S.; Edgerton, J.; Lipscombe, D. Identification of functionally distinct isoforms of the N-type ca2+ channel in rat sympathetic ganglia and brain. Neuron 1997, 18, 153–166. [Google Scholar] [CrossRef]
- Bell, T.J.; Thaler, C.; Castiglioni, A.J.; Helton, T.D.; Lipscombe, D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 2004, 41, 127–138. [Google Scholar]
- Lipscombe, D.; Raingo, J. Alternative splicing matters: N-type calcium channels in nociceptors. Channels (Austin) 2007, 1, 225–227. [Google Scholar]
- Zamponi, G.W.; McCleskey, E.W. Splicing it up: A variant of the N-type calcium channel specific for pain. Neuron 2004, 41, 3–4. [Google Scholar] [CrossRef]
- Kaneko, S.; Cooper, C.B.; Nishioka, N.; Yamasaki, H.; Suzuki, A.; Jarvis, S.E.; Akaike, A.; Satoh, M.; Zamponi, G.W. Identification and characterization of novel human ca(v)2.2 (α1b) calcium channel variants lacking the synaptic protein interaction site. J. Neurosci. 2002, 22, 82–92. [Google Scholar]
- McDonough, S.I.; Boland, L.M.; Mintz, I.M.; Bean, B.P. Interactions among toxins that inhibit N-type and P-type calcium channels. J. Gen. Physiol. 2002, 119, 313–328. [Google Scholar] [CrossRef]
- Stocker, J.W.; Nadasdi, L.; Aldrich, R.W.; Tsien, R.W. Preferential interaction of ω-conotoxins with inactivated N-type Ca2+ channels. J. Neurosci. 1997, 17, 3002–3013. [Google Scholar]
- Feng, Z.P.; Hamid, J.; Doering, C.; Bosey, G.M.; Snutch, T.P.; Zamponi, G.W. Residue gly1326 of the N-type calcium channel α1b subunit controls reversibility of ω-conotoxin gvia and mviia block. J. Biol. Chem. 2001, 276, 15728–15735. [Google Scholar]
- Mould, J.; Yasuda, T.; Schroeder, C.I.; Beedle, A.M.; Doering, C.J.; Zamponi, G.W.; Adams, D.J.; Lewis, R.J. The α2δ auxiliary subunit reduces affinity of ω-conotoxins for recombinant N-type (Cav2.2) calcium channels. J. Biol. Chem. 2004, 279, 34705–34714. [Google Scholar]
- Dolphin, A.; Wyatt, C.; Richards, J.; Beattie, R.; Craig, P.; Lee, J.H.; Cribbs, L.; Volsen, S.; Perez‐Reyes, E. The effect of α2δ and other accessory subunits on expression and properties of the calcium channel α1g. J. Physiol. 1999, 519, 35–45. [Google Scholar]
- Birnbaumer, L.; Qin, N.; Olcese, R.; Tareilus, E.; Platano, D.; Costantin, J.; Stefani, E. Structures and functions of calcium channel β subunits. J. Bioenerg. Biomembr. 1998, 30, 357–375. [Google Scholar] [CrossRef]
- Takahashi, M.; Seagar, M.J.; Jones, J.F.; Reber, B.F.; Catterall, W.A. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 5478–5482. [Google Scholar] [CrossRef]
- Arikkath, J.; Campbell, K.P. Auxiliary subunits: Essential components of the voltage-gated calcium channel complex. Curr. Opin. Neurobiol. 2003, 13, 298–307. [Google Scholar] [CrossRef]
- Dolphin, A.C. Calcium channel diversity: Multiple roles of calcium channel subunits. Curr. Opin. Neurobiol. 2009, 19, 237–244. [Google Scholar] [CrossRef]
- Davies, A.; Hendrich, J.; Van Minh, A.T.; Wratten, J.; Douglas, L.; Dolphin, A.C. Functional biology of the α2δ subunits of voltage-gated calcium channels. Trends. Pharmacol. Sci. 2007, 28, 220–228. [Google Scholar] [CrossRef]
- Cole, R.L.; Lechner, S.M.; Williams, M.E.; Prodanovich, P.; Bleicher, L.; Varney, M.A.; Gu, G. Differential distribution of voltage-gated calcium channel α2δ delta (alpha2delta) subunit mrna-containing cells in the rat central nervous system and the dorsal root ganglia. J. Comp. Neurol. 2005, 491, 246–269. [Google Scholar] [CrossRef]
- Luo, Z.D.; Chaplan, S.R.; Higuera, E.S.; Sorkin, L.S.; Stauderman, K.A.; Williams, M.E.; Yaksh, T.L. Upregulation of dorsal root ganglion α2δ calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J. Neurosci. 2001, 21, 1868–1875. [Google Scholar]
- Luo, Z.; Calcutt, N.; Higuera, E.; Valder, C.; Song, Y.H.; Svensson, C.; Myers, R. Injury type-specific calcium channel α2δ/1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J. Pharmacol. Exp. Ther. 2002, 303, 1199–1205. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Sindrup, S.H.; Jensen, T.S. Chronic neuropathic pain: Mechanisms, drug targets and measurement. Fundam. Clin. Pharmacol. 2007, 21, 129–136. [Google Scholar] [CrossRef]
- Hendrich, J.; Van Minh, A.T.; Heblich, F.; Nieto-Rostro, M.; Watschinger, K.; Striessnig, J.; Wratten, J.; Davies, A.; Dolphin, A.C. Pharmacological disruption of calcium channel trafficking by the α2δ ligand gabapentin. Proc. Natl. Acad. Sci. USA 2008, 105, 3628–3633. [Google Scholar]
- Dolphin, A.C. Calcium Channel α2δ Subunits in Epilepsy and as Targets for Antiepileptic Drugs. In Jasper's Basic Mechanisms Epilepsies, 4th; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; Oxford University Press: Bethesda, Maryland, MD, USA, 2012. [Google Scholar]
- Taylor, C.P.; Angelotti, T.; Fauman, E. Pharmacology and mechanism of action of pregabalin: The calcium channel α2/δ (alpha2-delta) subunit as a target for antiepileptic drug discovery. Epilepsy. Res. 2007, 73, 137–150. [Google Scholar]
- Eroglu, C.; Allen, N.J.; Susman, M.W.; O'Rourke, N.A.; Park, C.Y.; Ozkan, E.; Chakraborty, C.; Mulinyawe, S.B.; Annis, D.S.; Huberman, A.D.; et al. Gabapentin receptor α2δ/1 is a neuronal thrombospondin receptor responsible for excitatory cns synaptogenesis. Cell 2009, 139, 380–392. [Google Scholar]
- Pragnell, M.; De Waard, M.; Mori, Y.; Tanabe, T.; Snutch, T.P.; Campbell, K.P. Calcium channel β-subunit binds to a conserved motif in the i-ii cytoplasmic linker of the α1-subunit. Nature 1994, 368, 67–70. [Google Scholar] [CrossRef]
- De Waard, M.; Pragnell, M.; Campbell, K.P. Ca2+ channel regulation by a conserved β subunit domain. Neuron 1994, 13, 495–503. [Google Scholar] [CrossRef]
- Bichet, D.; Cornet, V.; Geib, S.; Carlier, E.; Volsen, S.; Hoshi, T.; Mori, Y.; De Waard, M. The i-ii loop of the ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron 2000, 25, 177–190. [Google Scholar] [CrossRef]
- Buraei, Z.; Yang, J. The ss subunit of voltage-gated Ca2+ channels. Physiol. Rev. 2010, 90, 1461–1506. [Google Scholar] [CrossRef]
- Arias, J.M.; Murbartian, J.; Vitko, I.; Lee, J.H.; Perez-Reyes, E. Transfer of beta subunit regulation from high to low voltage-gated Ca2+ channels. FEBS Lett. 2005, 579, 3907–3912. [Google Scholar]
- Altier, C.; Garcia-Caballero, A.; Simms, B.; Walcher, J.; Tedford, H.; Hermosilla, G.; Zamponi, G. The Cav β subunit prevents RFT2-mediated ubiquitination and proteasomal degradation l-type calcium channels via the derlin-1/p97 erad protein complex. Nat. Neurosci. 2011, 14, 173–180. [Google Scholar] [CrossRef]
- Cornet, V.; Bichet, D.; Sandoz, G.; Marty, I.; Brocard, J.; Bourinet, E.; Mori, Y.; Villaz, M.; De Waard, M. Multiple determinants in voltage-dependent p/q calcium channels control their retention in the endoplasmic reticulum. Eur. J. Neurosci. 2002, 16, 883–895. [Google Scholar] [CrossRef]
- Flucher, B.E.; Kasielke, N.; Grabner, M. The triad targeting signal of the skeletal muscle calcium channel is localized in the cooh terminus of the alpha(1s) subunit. J. Cell Biol. 2000, 151, 467–478. [Google Scholar]
- Kobrinsky, E.; Tiwari, S.; Maltsev, V.A.; Harry, J.B.; Lakatta, E.; Abernethy, D.R.; Soldatov, N.M. Differential role of the alpha1c subunit tails in regulation of the cav1.2 channel by membrane potential, beta subunits, and Ca2+ ions. J. Biol. Chem. 2005, 280, 12474–12485. [Google Scholar]
- Waithe, D.; Ferron, L.; Page, K.M.; Chaggar, K.; Dolphin, A.C. Beta-subunits promote the expression of Cav2.2 channels by reducing their proteasomal degradation. J. Biol. Chem. 2011, 286, 9598–9611. [Google Scholar]
- Letts, V.A.; Felix, R.; Biddlecome, G.H.; Arikkath, J.; Mahaffey, C.L.; Valenzuela, A.; Bartlett, F.S., II; Mori, Y.; Campbell, K.P.; Frankel, W.N. The mouse stargazer gene encodes a neuronal Ca2+-channel γ subunit. Nat. Genet. 1998, 19, 340–347. [Google Scholar] [CrossRef]
- Kang, M.G.; Chen, C.C.; Felix, R.; Letts, V.A.; Frankel, W.N.; Mori, Y.; Campbell, K.P. Biochemical and biophysical evidence for γ2 subunit association with neuronal voltage-activated Ca2+ channels. J. Biol. Chem. 2001, 276, 32917–32924. [Google Scholar]
- Yang, L.; Katchman, A.; Morrow, J.P.; Doshi, D.; Marx, S.O. Cardiac l-type calcium channel (Cav1.2) associates with γ subunits. FASEB J. 2011, 25, 928–936. [Google Scholar] [CrossRef]
- Hansen, J.P.; Chen, R.S.; Larsen, J.K.; Chu, P.J.; Janes, D.M.; Weis, K.E.; Best, P.M. Calcium channel γ6 subunits are unique modulators of low voltage-activated (Cav3.1) calcium current. J. Mol. Cell. Cardiol. 2004, 37, 1147–1158. [Google Scholar] [CrossRef]
- Sandoval, A.; Arikkath, J.; Monjaraz, E.; Campbell, K.P.; Felix, R. Γ1-dependent down-regulation of recombinant voltage-gated Ca2+ channels. Cell. Mol. Neurobiol. 2007, 27, 901–908. [Google Scholar]
- Wallace, M.S.; Rauck, R.L.; Deer, T. Ziconotide combination intrathecal therapy: Rationale and evidence. Clin. J. Pain 2010, 26, 635–644. [Google Scholar] [CrossRef]
- Cizkova, D.; Marsala, J.; Lukacova, N.; Marsala, M.; Jergova, S.; Orendacova, J.; Yaksh, T.L. Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp. Brain Res. 2002, 147, 456–463. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Lewis, R.J.; Todorovic, S.M.; Arneric, S.P.; Snutch, T.P. Role of voltage-gated calcium channels in ascending pain pathways. Brain Res. Rev. 2009, 60, 84–89. [Google Scholar] [CrossRef]
- Krarup, C. An update on electrophysiological studies in neuropathy. Curr. Opin. Neurol. 2003, 16, 603–612. [Google Scholar] [CrossRef]
- Saegusa, H.; Kurihara, T.; Zong, S.; Kazuno, A.; Matsuda, Y.; Nonaka, T.; Han, W.; Toriyama, H.; Tanabe, T. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J. 2001, 20, 2349–2356. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Wakamori, M.; Ino, M.; Miyamoto, N.; Takahashi, E.; Yoshinaga, T.; Sawada, K.; Imoto, K.; Tanaka, I.; Yoshizawa, T.; et al. Differential nociceptive responses in mice lacking the α1b subunit of N-type Ca(2+) channels. Neuroreport 2001, 12, 2423–2427. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Snutch, T.P. Modulation of voltage-dependent calcium channels by g proteins. Curr. Opin. Neurobiol. 1998, 8, 351–356. [Google Scholar] [CrossRef]
- Li, C.Y.; Song, Y.H.; Higuera, E.S.; Luo, Z.D. Spinal dorsal horn calcium channel α2δ/1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J. Neurosci. 2004, 24, 8494–8499. [Google Scholar] [CrossRef]
- Li, C.Y.; Zhang, X.L.; Matthews, E.A.; Li, K.W.; Kurwa, A.; Boroujerdi, A.; Gross, J.; Gold, M.S.; Dickenson, A.H.; Feng, G.; et al. Calcium channel α2δ1 subunit mediates spinal hyperexcitability in pain modulation. Pain 2006, 125, 20–34. [Google Scholar] [CrossRef]
- Jain, K.K. An evaluation of intrathecal ziconotide for the treatment of chronic pain. Expert. Opin. Investig. Drugs 2000, 9, 2403–2410. [Google Scholar] [CrossRef]
- Staats, P.S.; Yearwood, T.; Charapata, S.G.; Presley, R.W.; Wallace, M.S.; Byas-Smith, M.; Fisher, R.; Bryce, D.A.; Mangieri, E.A.; Luther, R.R.; et al. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or aids: A randomized controlled trial. JAMA 2004, 291, 63–70. [Google Scholar]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; de Santos, V.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar]
- Skalicky, J.J.; Metzler, W.J.; Ciesla, D.J.; Galdes, A.; Pardi, A. Solution structure of the calcium channel antagonist omega-conotoxin gvia. Protein. Sci. 1993, 2, 1591–1603. [Google Scholar]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar]
- Olivera, B.M. Conus peptides: Biodiversity-based discovery and exogenomics. J. Biol. Chem. 2006, 281, 31173–31177. [Google Scholar] [CrossRef]
- Jakubowski, J.A.; Kelley, W.P.; Sweedler, J.V. Screening for post-translational modifications in conotoxins using liquid chromatography/mass spectrometry: An important component of conotoxin discovery. Toxicon 2006, 47, 688–699. [Google Scholar] [CrossRef]
- Nielsen, K.J.; Adams, D.; Thomas, L.; Bond, T.; Alewood, P.F.; Craik, D.J.; Lewis, R.J. Structure-activity relationships of ω-conotoxins mviia, mviic and 14 loop splice hybrids at n and p/q-type calcium channels. J. Mol. Biol. 1999, 289, 1405–1421. [Google Scholar] [CrossRef]
- Favreau, P.; Gilles, N.; Lamthanh, H.; Bournaud, R.; Shimahara, T.; Bouet, F.; Laboute, P.; Letourneux, Y.; Menez, A.; Molgo, J.; et al. A new ω-conotoxin that targets N-type voltage-sensitive calcium channels with unusual specificity. Biochemistry 2001, 40, 14567–14575. [Google Scholar]
- Lee, S.; Kim, Y.; Back, S.K.; Choi, H.W.; Lee, J.Y.; Jung, H.H.; Ryu, J.H.; Suh, H.W.; Na, H.S.; Kim, H.J.; et al. Analgesic effect of highly reversible ω-conotoxin fvia on n type Ca2+ channels. Mol. Pain 2010, 6, 97. [Google Scholar] [CrossRef]
- Olivera, B.M.; McIntosh, J.M.; Cruz, L.J.; Luque, F.A.; Gray, W.R. Purification and sequence of a presynaptic peptide toxin from conus geographus venom. Biochemistry 1984, 23, 5087–5090. [Google Scholar]
- Sato, K.; Park, N.G.; Kohno, T.; Maeda, T.; Kim, J.I.; Kato, R.; Takahashi, M. Role of basic residues for the binding of ω-conotoxin gvia to N-type calcium channels. Biochem. Biophys. Res. Commun. 1993, 194, 1292–1296. [Google Scholar] [CrossRef]
- Kim, J.I.; Takahashi, M.; Ogura, A.; Kohno, T.; Kudo, Y.; Sato, K. Hydroxyl group of tyr13 is essential for the activity of ω-conotoxin gvia, a peptide toxin for N-type calcium channel. J. Biol. Chem. 1994, 269, 23876–23878. [Google Scholar]
- Kim, J.I.; Takahashi, M.; Ohtake, A.; Wakamiya, A.; Sato, K. Tyr13 is essential for the activity of ω-conotoxin mviia and gvia, specific N-type calcium channel blockers. Biochem. Biophys. Res. Commun. 1995, 206, 449–454. [Google Scholar] [CrossRef]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; Santos, V.D.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar]
- Bingham, J.-P.; Baker, M.R.; Chun, J.B. Analysis of a cone snail's killer cocktail - the milked venom of conus geographus. Toxicon 2012, 60, 1166–1170. [Google Scholar]
- Nielsen, K.J.; Schroeder, T.; Lewis, R. Structure-activity relationships of ω-conotoxins at N-type voltage-sensitive calcium channels. J. Mol. Recognit. 2000, 13, 55–70. [Google Scholar] [CrossRef]
- Wang, Y.X.; Bezprozvannaya, S.; Bowersox, S.S.; Nadasdi, L.; Miljanich, G.; Mezo, G.; Silva, D.; Tarczy-Hornoch, K.; Luther, R.R. Peripheral versus central potencies of N-type voltage-sensitive calcium channel blockers. Naunyn. Schmiedebergs. Arch. Pharmacol. 1998, 357, 159–168. [Google Scholar] [CrossRef]
- Chung, D.; Gaur, S.; Bell, J.R.; Ramachandran, J.; Nadasdi, L. Determination of disulfide bridge pattern in ω-conopeptides. Int. J. Pept. Protein. Res. 1995, 46, 320–325. [Google Scholar]
- Lew, M.J.; Flinn, J.P.; Pallaghy, P.K.; Murphy, R.; Whorlow, S.L.; Wright, C.E.; Norton, R.S.; Angus, J.A. Structure-function relationships of ω-conotoxin gvia. Synthesis, structure, calcium channel binding, and functional assay of alanine-substituted analogues. J. Biol. Chem. 1997, 272, 12014–12023. [Google Scholar]
- Flinn, J.P.; Pallaghy, P.K.; Lew, M.J.; Murphy, R.; Angus, J.A.; Norton, R.S. Roles of key functional groups in ω-conotoxin gvia synthesis, structure and functional assay of selected peptide analogues. Eur. J. Biochem. 1999, 262, 447–455. [Google Scholar] [CrossRef]
- Motin, L.; Yasuda, T.; Schroeder, C.I.; Lewis, R.J.; Adams, D.J. Ω-conotoxin inhibition of excitatory synaptic transmission evoked by dorsal root stimulation in rat superficial dorsal horn-conotoxin cvib differentially inhibits native and recombinant n- and p/q-type calcium channels. Eur. J. Neurosci. 2007, 25, 435–444. [Google Scholar] [CrossRef]
- Motin, L.; Adams, D.J. Ω-conotoxin inhibition of excitatory synaptic transmission evoked by dorsal root stimulation in rat superficial dorsal horn. Neuropharmacology 2008, 55, 860–864. [Google Scholar] [CrossRef]
- Schroeder, K.; Neagle, B.; Trezise, D.J.; Worley, J. Ionworks ht: A new high-throughput electrophysiology measurement platform. J. Biomol. Screen 2003, 8, 50–64. [Google Scholar] [CrossRef]
- Swensen, A.M.; Niforatos, W.; Vortherms, T.A.; Perner, R.J.; Li, T.; Schrimpf, M.R.; Scott, V.E.; Lee, L.; Jarvis, M.F.; McGaraughty, S. An automated electrophysiological assay for differentiating Cav2. 2 inhibitors based on state dependence and kinetics. Assay Drug Develop. Technol. 2012, 10, 542–550. [Google Scholar] [CrossRef]
- Bauer, C.S.; Rahman, W.; Tran-van-Minh, A.; Lujan, R.; Dickenson, A.H.; Dolphin, A.C. The anti-allodynic α2δ ligand pregabalin inhibits the trafficking of the calcium channel α1b subunit to presynaptic terminals in vivo. Biochem. Soc. Trans. 2010, 38, 525–528. [Google Scholar] [CrossRef]
- Perret, D.; Luo, Z.D. Targeting voltage-gated calcium channels for neuropathic pain management. Neurotherapeutics 2009, 6, 679–692. [Google Scholar] [CrossRef]
- Heinke, B.; Balzer, E.; Sandkuhler, J. Pre- and post-synaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina i neurons. Eur. J. Neurosci. 2004, 19, 103–111. [Google Scholar] [CrossRef]
- Rycroft, B.K.; Vikman, K.S.; Christie, M.J. Inflammation reduces the contribution of N-type calcium channels to primary afferent synaptic transmission onto nk1 receptor-positive lamina i neurons in the rat dorsal horn. J. Physiol. 2007, 580, 883–894. [Google Scholar] [CrossRef]
- Hockerman, G.H.; Johnson, B.D.; Abbott, M.R.; Scheuer, T.; Catterall, W.A. Molecular determinants of high affinity phenylalkylamine block of l-type calcium channels in transmembrane segment iiis6 and the pore region of the α1 subunit. J. Biol. Chem. 1997, 272, 18759–18765. [Google Scholar]
- McGuire, D.; Bowersox, S.; Fellmann, J.D.; Luther, R.R. Sympatholysis after neuron-specific, N-type, voltage-sensitive calcium channel blockade: First demonstration of n-channel function in humans. J. Cardiovasc. Pharmacol. 1997, 30, 400–403. [Google Scholar] [CrossRef]
- Escoubas, P.; Sollod, B.; King, G.F. Venom landscapes: Mining the complexity of spider venoms via a combined cdna and mass spectrometric approach. Toxicon 2006, 47, 650–663. [Google Scholar] [CrossRef]
- Estrada, G.; Villegas, E.; Corzo, G. Spider venoms: A rich source of acylpolyamines and peptides as new leads for cns drugs. Nat. Product Rep. 2007, 24, 145–161. [Google Scholar] [CrossRef]
- Norton, R.S.; McDonough, S.I. Peptides targeting voltage-gated calcium channels. Curr. Pharm. Des. 2008, 14, 2480–2491. [Google Scholar] [CrossRef]
- Mintz, I.M.; Venema, V.J.; Swiderek, K.M.; Lee, T.D.; Bean, B.P.; Adams, M.E. P-type calcium channels blocked by the spider toxin omega-aga-iva. Nature 1992, 355, 827–829. [Google Scholar] [CrossRef]
- Newcomb, R.; Szoke, B.; Palma, A.; Wang, G.; Chen, X.; Hopkins, W.; Cong, R.; Miller, J.; Urge, L.; Tarczy-Hornoch, K.; et al. Selective peptide antagonist of the class e calcium channel from the venom of the tarantula hysterocrates gigas. Biochemistry 1998, 37, 15353–15362. [Google Scholar]
- Wang, M.; Guan, X.; Liang, S. The cross channel activities of spider neurotoxin huwentoxin-i on rat dorsal root ganglion neurons. Biochem. Biophys. Res. Commun. 2007, 357, 579–583. [Google Scholar] [CrossRef]
- Liang, S.P.; Zhang, D.Y.; Pan, X.; Chen, Q.; Zhou, P.A. Properties and amino acid sequence of huwentoxin-i, a neurotoxin purified from the venom of the chinese bird spider selenocosmia huwena. Toxicon 1993, 31, 969–978. [Google Scholar]
- Jiang, L.; Peng, L.; Chen, J.; Zhang, Y.; Xiong, X.; Liang, S. Molecular diversification based on analysis of expressed sequence tags from the venom glands of the chinese bird spider ornithoctonus huwena. Toxicon 2008, 51, 1479–1489. [Google Scholar]
- Liu, Z.; Dai, J.; Dai, L.; Deng, M.; Hu, Z.; Hu, W.; Liang, S. Function and solution structure of huwentoxin-x, a specific blocker of N-type calcium channels, from the chinese bird spider ornithoctonus huwena. J. Biol. Chem. 2006, 281, 8628–8635. [Google Scholar]
- Bindokas, V.P.; Adams, M.E. Ω-aga-i: A presynaptic calcium channel antagonist from venom of the funnel web spider, agelenopsis aperta. J. Neurobiol. 1989, 20, 171–188. [Google Scholar] [CrossRef]
- Adams, M.E.; Bindokas, V.P.; Hasegawa, L.; Venema, V.J. Ω-agatoxins: Novel calcium channel antagonists of two subtypes from funnel web spider (agelenopsis aperta) venom. J. Biol. Chem. 1990, 265, 861–867. [Google Scholar]
- Ertel, E.A.; Warren, V.A.; Adams, M.E.; Griffin, P.R.; Cohen, C.J.; Smith, M.M. Ω-agatoxins: A family of probes for similar binding sites on l- and N-type calcium channels. Biochemistry 1994, 33, 5098–5108. [Google Scholar]
- Yan, L.; Adams, M.E. The spider toxin ω-aga iiia defines a high affinity site on neuronal high voltage-activated calcium channels. J. Biol. Chem. 2000, 275, 21309–21316. [Google Scholar] [CrossRef]
- Leao, R.M.; Cruz, J.S.; Diniz, C.R.; Cordeiro, M.N.; Beirao, P.S. Inhibition of neuronal high-voltage activated calcium channels by the omega-phoneutria nigriventer tx3–3peptide toxin. Neuropharmacology 2000, 39, 1756–1767. [Google Scholar]
- Cordeiro Mdo, N.; de Figueiredo, S.G.; Valentim Ado, C.; Diniz, C.R.; von Eickstedt, V.R.; Gilroy, J.; Richardson, M. Purification and amino acid sequences of six tx3 type neurotoxins from the venom of the brazilian 'armed' spider phoneutria nigriventer (keys). Toxicon 1993, 31, 35–42. [Google Scholar] [CrossRef]
- Vieira, L.B.; Kushmerick, C.; Hildebrand, M.E.; Garcia, E.; Stea, A.; Cordeiro, M.N.; Richardson, M.; Gomez, M.V.; Snutch, T.P. Inhibition of high voltage-activated calcium channels by spider toxin pntx3-6. J. Pharmacol. Exp. Ther. 2005, 314, 1370–1377. [Google Scholar] [CrossRef]
- Vieira, L.B.; Pimenta, A.M.; Richardson, M.; Bemquerer, M.P.; Reis, H.J.; Cruz, J.S.; Gomez, M.V.; Santoro, M.M.; Ferreira-de-Oliveira, R.; Figueiredo, S.G.; et al. Leftward shift in the voltage-dependence for Ca2+ currents activation induced by a new toxin from phoneutria reidyi (aranae, ctenidae) venom. Cell. Mol. Neurobiol. 2007, 27, 129–146. [Google Scholar] [CrossRef]
- Richardson, M.; Pimenta, A.M.; Bemquerer, M.P.; Santoro, M.M.; Beirao, P.S.; Lima, M.E.; Figueiredo, S.G.; Bloch, C., Jr.; Vasconcelos, E.A.; Campos, F.A.; et al. Comparison of the partial proteomes of the venoms of brazilian spiders of the genus phoneutria. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2006, 142, 173–187. [Google Scholar]
- Newcomb, R.; Palma, A.; Fox, J.; Gaur, S.; Lau, K.; Chung, D.; Cong, R.; Bell, J.R.; Horne, B.; Nadasdi, L.; et al. Snx-325, a novel calcium antagonist from the spider segestria florentina. Biochemistry 1995, 34, 8341–8347. [Google Scholar]
- Benjamin, E.R.; Pruthi, F.; Olanrewaju, S.; Shan, S.; Hanway, D.; Liu, X.; Cerne, R.; Lavery, D.; Valenzano, K.J.; Woodward, R.M.; et al. Pharmacological characterization of recombinant N-type calcium channel (Cav2.2) mediated Calcium mobilization using flipr. Biochem. Pharmacol. 2006, 72, 770–782. [Google Scholar] [CrossRef]
- Adams, M.E.; Mintz, I.M.; Reily, M.D.; Thanabal, V.; Bean, B.P. Structure and properties of ω-agatoxin ivb, a new antagonist of p-type calcium channels. Mol. Pharmacol. 1993, 44, 681–688. [Google Scholar]
- Cardoso, F.C.; Pacifico, L.G.; Carvalho, D.C.; Victoria, J.M.; Neves, A.L.; Chavez-Olortegui, C.; Gomez, M.V.; Kalapothakis, E. Molecular cloning and characterization of phoneutria nigriventer toxins active on calcium channels. Toxicon 2003, 41, 755–763. [Google Scholar] [CrossRef]
- Vieira, L.B.; Kushmerick, C.; Reis, H.J.; Diniz, C.R.; Cordeiro, M.N.; Prado, M.A.; Kalapothakis, E.; Romano-Silva, M.A.; Gomez, M.V. Pntx3–6a spider neurotoxin inhibits K+-evoked increase in [Ca2+](i) and Ca2+-dependent glutamate release in synaptosomes. Neurochem. Int. 2003, 42, 277–282. [Google Scholar] [CrossRef]
- Souza, A.H.; Ferreira, J.; Cordeiro Mdo, N.; Vieira, L.B.; De Castro, C.J.; Trevisan, G.; Reis, H.; Souza, I.A.; Richardson, M.; Prado, M.A.; et al. Analgesic effect in rodents of native and recombinant ph α1β toxin, a high-voltage-activated calcium channel blocker isolated from armed spider venom. Pain 2008, 140, 115–126. [Google Scholar] [CrossRef]
- de Souza, A.H.; Lima, M.C.; Drewes, C.C.; da Silva, J.F.; Torres, K.C.; Pereira, E.M.; de Castro Junior, C.J.; Vieira, L.B.; Cordeiro, M.N.; Richardson, M.; et al. Antiallodynic effect and side effects of pα1β, a neurotoxin from the spider phoneutria nigriventer: Comparison with ω-conotoxin mviia and morphine. Toxicon 2011, 58, 626–633. [Google Scholar] [CrossRef]
- Dalmolin, G.D.; Silva, C.R.; Rigo, F.K.; Gomes, G.M.; Cordeiro Mdo, N.; Richardson, M.; Silva, M.A.; Prado, M.A.; Gomez, M.V.; Ferreira, J. Antinociceptive effect of brazilian armed spider venom toxin tx3–3in animal models of neuropathic pain. Pain 2011, 152, 2224–2232. [Google Scholar] [CrossRef]
- Fukumoto, N.; Obama, Y.; Kitamura, N.; Niimi, K.; Takahashi, E.; Itakura, C.; Shibuya, I. Hypoalgesic behaviors of p/q-type voltage-gated Ca2+ channel mutant mouse, rolling mouse nagoya. Neuroscience 2009, 160, 165–173. [Google Scholar]
- Todorovic, S.M.; Jevtovic-Todorovic, V. T-type voltage-gated calcium channels as targets for the development of novel pain therapies. Br. J. Pharmacol. 2011, 163, 484–495. [Google Scholar] [CrossRef]
- Olivera, B.M.; Miljanich, G.P.; Ramachandran, J.; Adams, M.E. Calcium channel diversity and neurotransmitter release: The omega-conotoxins and omega-agatoxins. Annu. Rev. Biochem. 1994, 63, 823–867. [Google Scholar] [CrossRef]
- Stotz, S.C.; Spaetgens, R.L.; Zamponi, G.W. Block of voltage-dependent calcium channel by the green mamba toxin calcicludine. J. Membr. Biol. 2000, 174, 157–165. [Google Scholar] [CrossRef]
- Sidach, S.S.; Mintz, I.M. Kurtoxin, a gating modifier of neuronal high- and low-threshold Ca2+ channels. J. Neurosci. 2002, 22, 2023–2034. [Google Scholar]
- Meunier, F.A.; Feng, Z.P.; Molgo, J.; Zamponi, G.W.; Schiavo, G. Glycerotoxin from glycera convoluta stimulates neurosecretion by up-regulating N-type Ca2+ channel activity. EMBO J. 2002, 21, 6733–6743. [Google Scholar] [CrossRef]
- Lee, C.W.; Eu, Y.J.; Min, H.J.; Cho, E.M.; Lee, J.H.; Kim, H.H.; Nah, S.Y.; Swartz, K.J.; Kim, J.I. Expression and characterization of recombinant kurtoxin, an inhibitor of t-type voltage-gated calcium channels. Biochem. Biophys. Res. Commun. 2011, 416, 277–282. [Google Scholar]
- de Weille, J.R.; Schweitz, H.; Maes, P.; Tartar, A.; Lazdunski, M. Calciseptine, a peptide isolated from black mamba venom, is a specific blocker of the l-type calcium channel. Proc. Natl. Acad. Sci. USA 1991, 88, 2437–2440. [Google Scholar] [CrossRef]
- Schweitz, H.; Heurteaux, C.; Bois, P.; Moinier, D.; Romey, G.; Lazdunski, M. Calcicludine, a venom peptide of the kunitz-type protease inhibitor family, is a potent blocker of high-threshold Ca2+ channels with a high affinity for l-type channels in cerebellar granule neurons. Proc. Natl. Acad. Sci. USA 1994, 91, 878–882. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Z.; Xiao, Y.; Li, Y.; Rong, M.; Liang, S.; Zhang, Z.; Yu, H.; King, G.F.; Lai, R. Chemical punch packed in venoms makes centipedes excellent predators. Mol. Cell. Proteom. 2012, 11, 640–650. [Google Scholar] [CrossRef]
- Corzo, G.; Adachi-Akahane, S.; Nagao, T.; Kusui, Y.; Nakajima, T. Novel peptides from assassin bugs (hemiptera: Reduviidae): Isolation, chemical and biological characterization. FEBS Lett. 2001, 499, 256–261. [Google Scholar] [CrossRef]
- Bernard, C.; Corzo, G.; Mosbah, A.; Nakajima, T.; Darbon, H. Solution structure of ptu1, a toxin from the assassin bug peirates turpis that blocks the voltage-sensitive calcium channel N-type. Biochemistry 2001, 40, 12795–12800. [Google Scholar] [CrossRef]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar]
- Finley, M.F.; Lubin, M.L.; Neeper, M.P.; Beck, E.; Liu, Y.; Flores, C.M.; Qin, N. An integrated multiassay approach to the discovery of small-molecule N-type voltage-gated calcium channel antagonists. Assay Drug Dev. Technol. 2010, 8, 685–694. [Google Scholar] [CrossRef]
- Dai, G.; Haedo, R.J.; Warren, V.A.; Ratliff, K.S.; Bugianesi, R.M.; Rush, A.; Williams, M.E.; Herrington, J.; Smith, M.M.; McManus, O.B.; et al. A high-throughput assay for evaluating state dependence and subtype selectivity of Cav2 calcium channel inhibitors. Assay Drug Dev. Technol. 2008, 6, 195–212. [Google Scholar] [CrossRef]
- Dunlop, J.; Bowlby, M.; Peri, R.; Tawa, G.; LaRocque, J.; Soloveva, V.; Morin, J. Ion channel screening. Comb. Chem. High Throughput Screen. 2008, 11, 514–522. [Google Scholar] [CrossRef]
- Lubin, M.L.; Reitz, T.L.; Todd, M.J.; Flores, C.M.; Qin, N.; Xin, H. A nonadherent cell-based hts assay for N-type calcium channel using calcium 3 dye. Assay Drug Dev. Technol. 2006, 4, 689–694. [Google Scholar]
- Winquist, R.J.; Pan, J.Q.; Gribkoff, V.K. Use-dependent blockade of Cav2.2 voltage-gated calcium channels for neuropathic pain. Biochem. Pharmacol. 2005, 70, 489–499. [Google Scholar] [CrossRef]
- Sousa, S.R.; Ragnarsson, L.; Vetter, I.; Herzig, V.; King, G.F.; Lewis, R.J. Development of High Throughput Calcium Channel Assays to Accelerate the Discovery of Novel Toxins Targeting Human Cav2.2 Channels. In Proceedings of 17th World Congress the International Society on Toxinology & Venom Week 2012, Honolulu, Hawaii, USA, 8–13 July 2012; Volume 60, p. 111.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Sousa, S.R.; Vetter, I.; Lewis, R.J. Venom Peptides as a Rich Source of Cav2.2 Channel Blockers. Toxins 2013, 5, 286-314. https://doi.org/10.3390/toxins5020286
AMA Style
Sousa SR, Vetter I, Lewis RJ. Venom Peptides as a Rich Source of Cav2.2 Channel Blockers. Toxins. 2013; 5(2):286-314. https://doi.org/10.3390/toxins5020286
Chicago/Turabian StyleSousa, Silmara R., Irina Vetter, and Richard J. Lewis. 2013. "Venom Peptides as a Rich Source of Cav2.2 Channel Blockers" Toxins 5, no. 2: 286-314. https://doi.org/10.3390/toxins5020286