Is Protein Phosphatase Inhibition Responsible for the Toxic Effects of Okadaic Acid in Animals?

Abstract
:1. Introduction
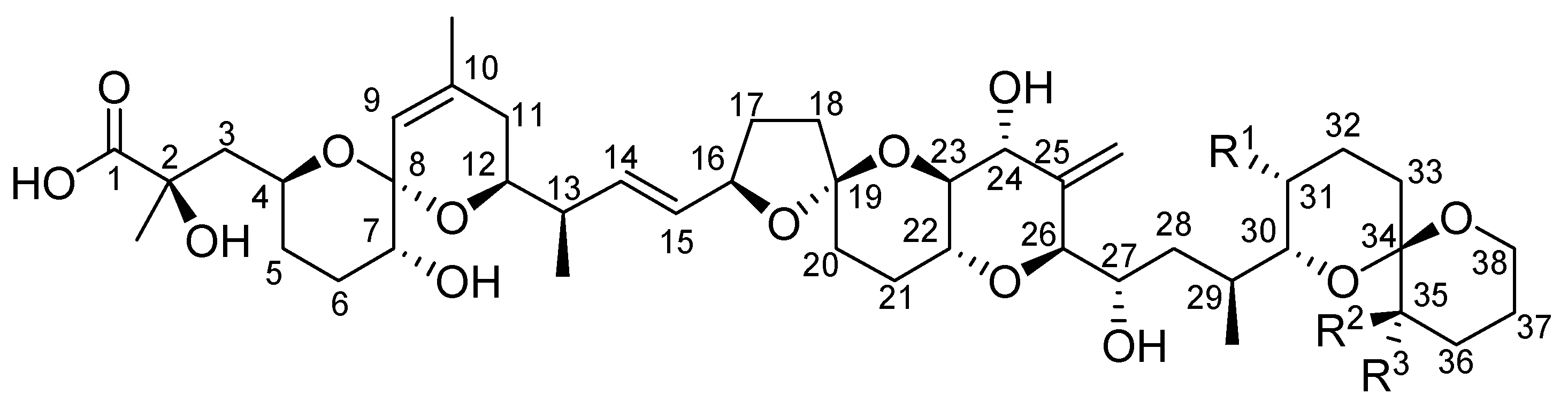
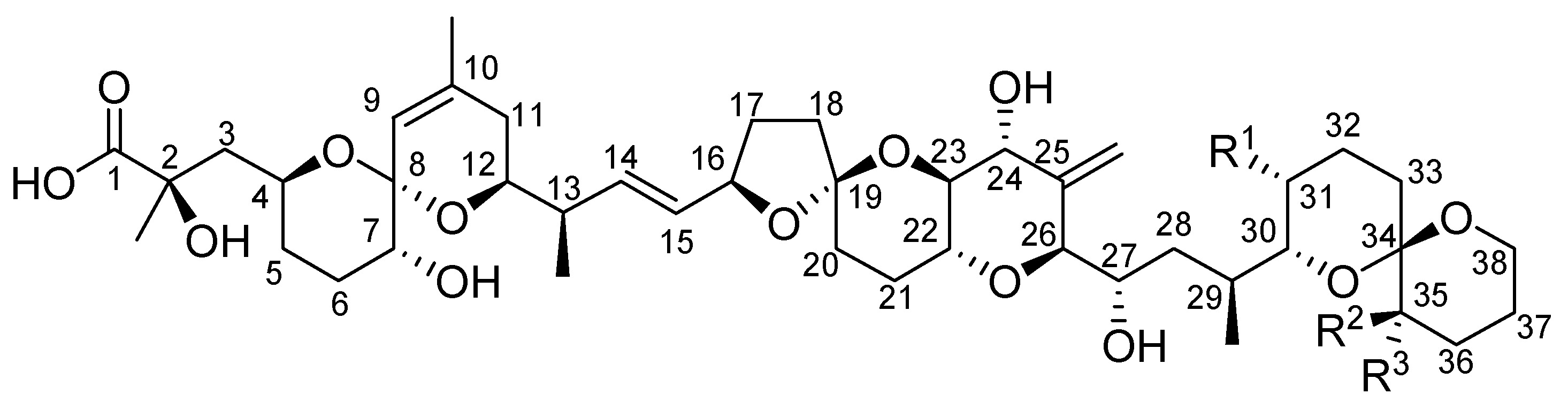
2. Toxicity of Okadaic Acid and Derivatives to Experimental Animals
2.1. Acute Toxicity of Okadaic Acid and Derivatives
2.2. Diarrhoeagenicity of Okadaic Acid
2.3. Toxicity of Okadaic Acid and Derivatives through Dermal Application
2.4. Tumour Promotion by Okadaic Acid and Derivatives
2.5. Neurotoxicity of Okadaic Acid after Intra-Cerebral Injection
3. Inhibition of Protein Phosphatases by Okadaic Acid and Derivatives
4. Involvement of Protein Phosphatase Inhibition in the Toxic Effects of Okadaic Acid
- Demonstration of protein phosphatase inhibition in vivo, at the sites at which toxicity has been observed.
- Proportionality between the efficacy of OA derivatives in inhibition of protein phosphatases and the severity of the toxic effects that they induce.
- The induction of toxic effects similar to those observed with OA by other inhibitors of protein phosphatases.
- A defined pathway from protein phosphatase inhibition to the observed toxic effects.
4.1. Protein Phosphatase Inhibition and Acute Toxicity of Okadaic Acid

{kind=link}
| Compound | Relative toxicity to mice (i.p. injection) | Relative inhibition of PP2A | Relative inhibition of PP1 |
|---|---|---|---|
| OA | 1.0 | 1.0 | 1.0 |
| DTX-1 | 1.0 [11] | 1.6–2.4 [45,48,49] | 0.4–0.9 [45,48] |
| DTX-2 | 0.6 [7] | 0.5 [7] | - |
| DTX-4 | 0.3 [13] | 0.002 [51] | - |
| 7-O-palmitoyl-OA | 0.05 [14] | <0.0003 [45] | <0.00005 [45] |
| 7-O-docosahexaenoyl-OA | 0.5 [14] | <0.0003 [45,50] | - |
4.2. Protein Phosphatase Inhibition and Diarrhoeagenicity of Okadaic Acid
4.3. Protein Phosphatase Inhibition and Tumour Promotion by Okadaic Acid
4.4. Protein Phosphatase Inhibition and Neurotoxicity of Okadaic Acid
5. Conclusion
Acknowledgment
Conflict of Interest
References
- Reguera, B.; Velo-Suárez, L.; Raine, R.; Park, M.G. Harmful Dinophysis species: a review. Harmful Algae 2012, 14, 87–106. [Google Scholar] [CrossRef]
- Hu, W.; Xu, J.; Sinkkonen, J.; Wu, J. Polyketides from marine dinoflagellates of the genus Prorocentrum, biosynthetic origin and bioactivity of their okadaic acid analogues. Mini-Rev. Med. Chem. 2010, 10, 51–61. [Google Scholar] [CrossRef]
- Suzuki, T.; Ota, H.; Yamasaki, M. Direct evidence of transformation of dinophysistoxin-1 to 7-O-acyl-dinophysistoxin-1 (dinophysistoxin-3) in the scallop Patinopecten yessoensis. Toxicon 1999, 37, 187–198. [Google Scholar] [CrossRef]
- Yasumoto, T.; Oshima, Y.; Yamaguchi, M. Occurrence of a new type of shellfish poisoning in the Tohoku district. Bull. Jpn. Soc. Sci. Fish. 1978, 44, 1249–1255. [Google Scholar] [CrossRef]
- James, K.J.; Carey, B.; O'Halloran, J.O.; van Pelt, F.N.A.M.; Škrabáková, Z. Shellfish toxicity: human health implications of marine algal toxins. Epidemiol. Infect. 2010, 138, 927–940. [Google Scholar]
- Picot, C.; Nguyen, T.A.; Roudot, A.C.; Parent-Massin, D. A preliminary risk assessment of human exposure to phycotoxins in shellfish: a review. Hum. Ecol. Risk Assess. 2011, 17, 328–366. [Google Scholar] [CrossRef]
- Aune, T.; Larsen, S.; Aasen, J.A.B.; Rehmann, N.; Satake, M.; Hess, P. Relative toxicity of dinophysistoxin-2 (DTX-2) compared with okadaic acid, based on acute intraperitoneal toxicity in mice. Toxicon 2007, 49, 1–7. [Google Scholar] [CrossRef]
- Dickey, R.W.; Bobzin, S.C.; Faulkner, D.J.; Bencsath, F.A.; Andrzejewski, D. Identification of okadaic acid from a Caribbean dinoflagellate, Prorocentrum concavum. Toxicon 1990, 28, 371–377. [Google Scholar] [CrossRef]
- Tubaro, A.; Sosa, S.; Carbonatto, M.; Altinier, G.; Vita, F.; Melato, M.; Satake, M.; Yasumoto, T. Oral and intraperitoneal acute toxicity studies of yessotoxin and homoyessotoxins in mice. Toxicon 2003, 41, 783–792. [Google Scholar] [CrossRef]
- Tachibana, K.; Scheuer, P.J.; Tsukitani, Y.; Kikuchi, H.; Van Engen, D.; Clardy, J.; Gopichand, Y.; Schmitz, F.J. Okadaic acid, a cytotoxic polyether from two marine sponges of the genus Halichondria. J. Am. Chem. Soc. 1981, 103, 2469–2471. [Google Scholar]
- Murata, M.; Shimatani, M.; Sugitani, H.; Oshima, Y.; Yasumoto, T. Isolation and structural elucidation of the causative toxin of the diarrhetic shellfish poisoning. Bull. Jpn. Soc. Sci. Fish. 1982, 48, 549–552. [Google Scholar]
- Yasumoto, T.; Murata, M.; Oshima, Y.; Sano, M.; Matsumoto, G.K.; Clardy, J. Diarrhetic shellfish toxins. Tetrahedron 1985, 41, 1019–1025. [Google Scholar]
- Hu, T.; Curtis, J.M.; Walter, J.A.; Wright, J.L.C. Identification of DTX-4, a new water-soluble phosphatase inhibitor from the toxic dinoflagellate Prorocentrum lima. J. Chem. Soc. Chem. Comm. 1995, 597–599. [Google Scholar]
- Yanagi, T.; Murata, M.; Torigoe, K.; Yasumoto, T. Biological activities of semisynthetic analogs of dinophysistoxin-3, the major diarrhetic shellfish toxin. Agric. Biol. Chem. 1989, 53, 525–529. [Google Scholar] [CrossRef]
- Ito, E.; Yasumoto, T.; Takai, A.; Imanishi, S.; Harada, K. Investigation of the distribution and excretion of okadaic acid in mice using immunostaining method. Toxicon 2002, 40, 159–165. [Google Scholar] [CrossRef]
- Aune, T.; Espenes, A.; Aasen, J.A.B.; Quilliam, M.A.; Hess, P.; Larsen, S. Study of possible combined toxic effects of azaspiracid-1 and okadaic acid in mice via the oral route. Toxicon 2012, 60, 895–906. [Google Scholar] [CrossRef]
- Le Hégarat, L.; Jacquin, A.-G.; Bazin, E.; Fessard, V. Genotoxicity of the marine toxin okadaic acid, in human Caco-2 cells and in mice gut cells. Environ. Toxicol. 2006, 21, 55–64. [Google Scholar]
- Ito, E.; Terao, K. Injury and recovery process of intestine caused by okadaic acid and related compounds. Nat. Toxins 1994, 2, 371–377. [Google Scholar]
- Ogino, H.; Kumagai, M.; Yasumoto, T. Toxicologic evaluation of yessotoxin. Nat. Toxins 1997, 5, 255–259. [Google Scholar] [CrossRef]
- Terao, K.; Ito, E.; Yanagi, T.; Yasumoto, T. Histopathological studies on experimental marine toxin poisoning. I. Ultrastructural changes in the small intestine and liver of suckling mice induced by dinophysistoxin-1 and pectenotoxin-1. Toxicon 1986, 24, 1141–1151. [Google Scholar] [CrossRef]
- Terao, K.; Ito, E.; Ohkusu, M.; Yasumoto, T. A comparative study of the effects of DSP-toxins on mice and rats. In Toxic Phytoplankton Blooms in the Sea; Smayda, T.J., Shimizu, Y., Eds.; Elsevier: New York, NY, USA, 1993; pp. 581–586. [Google Scholar]
- Yuasa, H.; Yoshida, K.; Iwata, H.; Nakanishi, H.; Suganuma, M.; Tatematsu, M. Increase of labeling indices in gastrointestinal mucosae of mice and rats by compounds of the okadaic acid type. J. Cancer Res. Clin. Oncol. 1994, 120, 208–212. [Google Scholar] [CrossRef]
- Berven, G.; Sætre, F.; Halvorsen, K.; Seglen, P.O. Effects of the diarrhetic shellfish toxin, okadaic acid, on cytoskeletal elements, viability and functionality of rat liver and intestinal cells. Toxicon 2001, 39, 349–362. [Google Scholar] [CrossRef]
- Tubaro, A.; Sosa, S.; Altinier, G.; Soranzo, M.R.; Satake, M.; Della Loggia, R.; Yasumoto, T. Short-term oral toxicity of homoyessotoxins, yessotoxin and okadaic acid in mice. Toxicon 2004, 43, 439–445. [Google Scholar] [CrossRef]
- Fujiki, H.; Suganuma, M.; Suguri, H.; Yoshizawa, S.; Takagi, K.; Uda, N.; Wakamatsu, K.; Yamada, K.; Murata, M.; Yasumoto, T.; Sugimura, T. Diarrhetic shellfish toxin, dinophysistoxin-1, is a potent tumor promoter on mouse skin. Jpn. J. Cancer Res. 1988, 79, 1089–1093. [Google Scholar]
- Fujiki, H.; Suganuma, M.; Suguri, H.; Yoshizawa, S.; Ojika, M.; Wakamatsu, K.; Yamada, K.; Sugimura, T. Induction of ornithine decarboxylase activity in mouse skin by a possible tumor promoter, okadaic acid. Proc. Jpn. Acad. Ser. B 1987, 63, 51–53. [Google Scholar] [CrossRef]
- Suganuma, M.; Fujiki, H.; Suguri, H.; Yoshizawa, S.; Hirota, M.; Nakayasu, M.; Ojika, M.; Wakamatsu, K.; Yamada, K.; Sugimura, T. Okadaic acid: an additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc. Natl Acad. Sci. USA 1988, 85, 1768–1771. [Google Scholar] [CrossRef]
- Suganuma, M.; Tatematsu, M.; Yatsunami, J.; Yoshizawa, S.; Okabe, S.; Uemura, D.; Fujiki, H. An alternative theory of tissue specificity by tumor promotion of okadaic acid in glandular stomach of SD rats. Carcinogenesis 1992, 13, 1841–1845. [Google Scholar] [CrossRef]
- Mudher, A.K.; Perry, V.H. Using okadaic acid as a tool for the in vivo induction of hyperphosphorylated tau. Neuroscience 1998, 85, 1329–1332. [Google Scholar] [CrossRef]
- He, J.; Yamada, K.; Zou, L.-B.; Nabeshima, T. Spatial memory deficit and neurodegeneration induced by the direct injection of okadaic acid into the hippocampus in rats. J. Neural Transm. 2001, 108, 1435–1443. [Google Scholar]
- Arias, C.; Becerra-García, F.; Arrieta, I.; Tapia, R. The protein phosphatase inhibitor okadaic acid induces heat shock protein expression and neurodegeneration in rat hippocampus in vivo. Exp. Neurol. 1998, 153, 242–254. [Google Scholar]
- Arendt, T.; Hanisch, F.; Holzer, M.; Brückner, M.K. In vivo phosphorylation in the rat basal nucleus induces PHF-like and APP immunoreactivity. NeuroReport 1994, 5, 1397–1400. [Google Scholar]
- Cummings, B.J.; Hayward, N.; Stoltzner, S.; Molineaux, S.; Nixon, R.A. Intraventricular infusion of okadaic acid induces mild changes in tau and APP, but fails to produce AD-like neuropathology in adult rats. Soc. Neurosci. Abstracts 1997, 232, 1639. [Google Scholar]
- Van Dam, A.M.; Bol, J.G.J.M.; Binnekade, R.; van Muiswinkel, F.L. Acute or chronic administration of okadaic acid to rats induces brain damage rather than Alzheimer-like neuropathology. Neuroscience 1998, 85, 1333–1335. [Google Scholar]
- Zhang, Z.; Simpkins, J.W. An okadaic acid-induced model of tauopathy and cognitive deficiency. Brain Res. 2010, 1359, 233–246. [Google Scholar]
- He, J.; Yang, Y.; Xu, H.; Zhang, X.; Li, X.-M. Olanzapine attenuates the okadaic acid-induced spatial memory impairment and hippocampal cell death in rats. Neuropsychopharmacology 2005, 30, 1511–1520. [Google Scholar] [CrossRef]
- Kamat, P.K.; Tota, S.; Rai, S.; Swarnkar, S.; Shukla, R.; Nath, C. A study on neuroinflammatory marker in brain areas of okadaic acid (ICV) induced memory impaired rats. Life Sci. 2012, 90, 713–720. [Google Scholar] [CrossRef]
- Yin, Y.-Y.; Liu, H.; Cong, X.-B.; Liu, Z.; Wang, Q.; Wang, J.-Z.; Zhu, L.-Q. Acetyl-L-carnitine attenuates okadaic acid induced tau hyperphosphorylation and spatial memory impairment in rats. J. Alzheimer's Dis. 2010, 19, 735–746. [Google Scholar]
- Costa, A.P.; Tramontina, A.C.; Biasibetti, R.; Batassini, C.; Lopes, M.W.; Wartchow, K.M.; Bernardi, C.; Tortorelli, L.S.; Leal, R.B.; Gonçalves, C.-A. Neuroglial alterations in rats submitted to the okadaic acid-induced model of dementia. Behav. Brain Res. 2012, 226, 420–427. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Fruth, R.; Brückner, M.K.; Gärtner, U. Paired helical filament-like phosphorylation of tau, deposition of β/A4-amyloid and memory impairment in rat induced by chronic inhibition of phosphatase 1 and 2A. Neuroscience 1995, 69, 691–698. [Google Scholar] [CrossRef]
- Lee, J.-H.; Hong, H.-N.; Im, J.-O.; Byun, H.-S.; Kim, D. The formation of PHF-1 and SMI-31 positive dystrophic neurites in rat hippocampus following acute injection of okadaic acid. Neurosci. Lett. 2000, 282, 49–52. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Fruth, R.; Brückner, M.K.; Gärtner, U. Phosphorylation of tau, Aβ-formation, and apoptosis after in vivo inhibition of PP-1 and PP-2A. Neurobiol. Aging 1998, 19, 3–13. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Brückner, M.K.; Janke, C.; Gärtner, U. The use of okadaic acid in vivo and the induction of molecular changes typical for Alzheimer's disease. Neuroscience 1998, 85, 1337–1340. [Google Scholar] [CrossRef]
- Bialojan, C.; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 1988, 256, 283–290. [Google Scholar]
- Takai, A.; Murata, M.; Torigoe, K.; Isobe, M.; Mieskes, G.; Yasumoto, T. Inhibitory effect of okadaic acid derivatives on protein phosphatases. A study on structure-affinity relationship. Biochem. J. 1992, 284, 539–544. [Google Scholar]
- Swingle, M.; Ni, L.; Honkanen, R.E. Small-molecule inhibitors of Ser/Thr protein phosphatases. In Methods in Molecular Biology, Volume 365: Protein Phosphatase Protocols; Moorhead, G., Ed.; Springer: New York, NY, USA, 2007; pp. 23–38. [Google Scholar]
- Sheppeck, J.E.; Gauss, C.-M.; Chamberlin, A.R. Inhibition of the Ser-Thr phosphatases PP1 and PP2A by naturally occurring toxins. Bioorg. Med. Chem. 1997, 5, 1739–1750. [Google Scholar] [CrossRef]
- Holmes, C.F.B.; Luu, H.A.; Carrier, F.; Schmitz, F.J. Inhibition of protein phosphatases-1 and -2A with acanthifolicin. Comparison with diarrhetic shellfish toxins and identification of a region on okadaic acid important for phosphatase inhibition. FEBS Lett. 1990, 270, 216–218. [Google Scholar]
- Rivas, M.; García, C.; Liberona, J.L.; Lagos, N. Biochemical characterization and inhibitory effects of dinophysistoxin-1, okadaic acid and microcystine l-r on protein phosphatase 2a purified from the mussel Mytilus chilensis. Biol. Res. 2000, 33, 197–206. [Google Scholar]
- Nishiwaki, S.; Fujiki, H.; Suganuma, M.; Furuya-Suguri, H.; Matsushima, R.; Iida, Y.; Ojika, M.; Yamada, K.; Uemura, D.; Yasumoto, T.; Schmitz, F.J.; Sugimura, T. Structure-activity relationship within a series of okadaic acid derivatives. Carcinogenesis 1990, 11, 1837–1841. [Google Scholar] [CrossRef]
- Hu, T.; Curtis, J.M.; Walter, J.A.; McLachlan, J.L.; Wright, J.L.C. Two new water-soluble DSP toxin derivatives from the dinoflagellate Prorocentrum maculosum: possible storage and excretion products. Tetrahedron Lett. 1995, 36, 9273–9276. [Google Scholar] [CrossRef]
- Albano, C.; Ronzitti, G.; Rossini, A.M.; Callegari, F.; Rossini, G.P. The total activity of a mixture of okadaic acid-group compounds can be calculated by those of individual analogues in a phosphoprotein phosphatase 2A assay. Toxicon 2009, 53, 631–637. [Google Scholar] [CrossRef]
- Garibo, D.; Dàmaso, E.; Eixarch, H.; de la Iglesia, P.; Fernández-Tejedor, M.; Diogène, J.; Pazos, Y.; Campàs, M. Protein phosphatase inhibition assays for okadaic acid detection in shellfish: matrix effects, applicability and comparison with LC-MS/MS analysis. Harmful Algae 2012, 19, 68–75. [Google Scholar] [CrossRef]
- Ikehara, T.; Imamura, S.; Yoshino, A.; Yasumoto, T. PP2A inhibition assay using recombinant enzyme for rapid detection of okadaic acid and its analogs in shellfish. Toxins 2010, 2, 195–204. [Google Scholar]
- Vilariño, N.; Louzao, M.C.; Vieytes, M.R.; Botana, L.M. Biological methods for marine toxin detection. Anal. Bioanal. Chem. 2010, 397, 1673–1681. [Google Scholar] [CrossRef]
- Cohen, P.; Holmes, C.F.B.; Tsukitani, Y. Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem. Sci. 1990, 15, 98–102. [Google Scholar]
- Kikuchi, K.; Shima, H.; Mitsuhashi, S.; Suzuki, M.; Oikawa, H. The apoptosis-inducing activity of the two protein phosphatase inhibitors, tautomycin and thyrsiferyl 23-acetate, is not due to the inhibition of protein phosphatases PP1 and PP2A. Int. J. Mol. Med. 1999, 4, 395–401. [Google Scholar]
- Espiña, B.; Louzao, M.; Cagide, E.; Alfonso, A.; Vieytes, M.R.; Yasumoto, T.; Botana, L.M. The methyl ester of okadaic acid is more potent than okadaic acid in disrupting the actin cytoskeleton and metabolism of primary cultured hepatocytes. Br. J. Pharmacol. 2010, 159, 337–344. [Google Scholar] [CrossRef]
- Doucet, E.; Ross, N.; Quilliam, M. Enzymatic hydrolysis of esterified diarrhetic shellfish poisoning toxins and pectenotoxins. Anal. Bioanal. Chem. 2007, 389, 335–342. [Google Scholar] [CrossRef]
- Runnegar, M.T.; Kong, S.; Berndt, N. Protein phosphatase inhibition and in vivo hepatotoxicity of microcystins. Am. J. Physiol. 1993, 265, G224–G230. [Google Scholar]
- Falconer, I.R.; Jackson, A.R.B.; Langley, J.; Runnegar, M.T. Liver pathology in mice in poisoning by the blue-green alga Microcystis aeruginosa. Aust. J. Biol. Sci. 1981, 34, 179–188. [Google Scholar]
- Seawright, A.A.; Cain, K.; Nolan, C.C.; Dinsdale, D.; Codd, G.A. The lesion caused by microcystin-LR in the liver of the rat. Is it a model for apoptosis in vivo in the liver? Phycologia 1996, 35 (Suppl. 6), 172–176. [Google Scholar]
- Graziano, M.J.; Casida, J.E. Comparison of the acute toxicity of endothal and cantharidic acid on mouse liver in vivo. Toxicol. Lett. 1987, 37, 143–148. [Google Scholar]
- Bagatell, F.K.; Dugan, K.; Wilgram, G.F. Structural and biochemical changes in tissues isolated from the cantharidin-poisoned rat with special emphasis upon hepatic subcellular particles. Toxicol. Appl. Pharmacol. 1969, 15, 249–261. [Google Scholar] [CrossRef]
- Ito, Y.; Abril, E.R.; Bethea, N.W.; McCuskey, R.S. Inhibition of matrix metalloproteinases minimizes hepatic microvascular injury in response to acetaminophen in mice. Toxicol. Sci. 2005, 83, 190–196. [Google Scholar]
- DeLeve, L.D.; Wang, X.; Kaplowitz, N.; Shulman, H.M.; Bart, J.A.; van der Hoek, A. Sinusoidal endothelial cells as a target for acetaminophen toxicity: direct action versus requirement for hepatocyte activation in different mouse strains. Biochem. Pharmacol. 1997, 53, 1339–1345. [Google Scholar] [CrossRef]
- Wieland, T.; Faulstich, H.; Fiume, L. Amatoxins, phallotoxins, phallolysin, and antamanide: the biologically active components of poisonous Amanita mushroo. CRC Crit. Rev. Biochem. Mol. Biol. 1978, 5, 185–260. [Google Scholar] [CrossRef]
- Nakamura, S.-I.; Mori, K.J. Effects of reticuloendothelial blockade on acute saponin poisoning in mice. Toxicology 1984, 29, 235–242. [Google Scholar] [CrossRef]
- Hong, S.J. Inhibition of mouse neuromuscular transmission and contractile function by okadaic acid and cantharidin. Br. J. Pharmacol. 2000, 130, 1211–1218. [Google Scholar] [CrossRef]
- Munday, R.; Selwood, A.I.; Rhodes, L. Acute toxicity of pinnatoxins E, F and G to mice. Toxicon 2012, 60, 995–999. [Google Scholar]
- Valdiglesias, V.; Fernández-Tajes, J.; Pásaro, E.; Méndez, J.; Laffon, B. Identification of differentially expressed genes in SHSY5Y cells exposed to okadaic acid by suppression subtractive hybridization. BMC Genomics 2012, 13:46. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.-Y.; Lin, L.; Gao, Y.; Hong, H.-S.; Wang, D.-Z. Quantitative proteomic analysis of okadaic acid treated mouse small intestines reveals differentially expressed proteins involved in diarrhetic shellfish poisoning. J. Proteomics 2012, 75, 2038–2052. [Google Scholar]
- Tripuraneni, J.; Koutsouris, A.; Pestic, L.; De Lanerolle, P.; Hecht, G. The toxin of diarrheic shellfish poisoning, okadaic acid, increases intestinal epithelial paracellular permeability. Gastroenterology 1997, 112, 100–108. [Google Scholar] [CrossRef]
- Okada, T.; Narai, A.; Matsunaga, S.; Fusetani, N.; Shimizu, M. Assessment of the marine toxins by monitoring the integrity of human intestinal Caco-2 cell monolayers. Toxicol. in Vitro 2000, 14, 219–226. [Google Scholar] [CrossRef]
- Hosokawa, M.; Tsukada, H.; Saitou, T.; Kodama, M.; Onomura, M.; Nakamura, H.; Fukuda, K.; Seino, Y. Effects of okadaic acid on rat colon. Dig. Dis. Sci. 1998, 43, 2526–2535. [Google Scholar] [CrossRef]
- Rao, R.K.; Basuroy, S.; Rao, V.U.; Karnaky, K.J.; Gupta, A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-β-catenin complexes from the cytoskeleton by oxidative stress. Biochem. J. 2002, 368, 471–481. [Google Scholar]
- Anderson, J.M.; van Itallie, C.M. Tight junctions and the molecular basis for regulation of paracellular permeability. Am. J. Physiol. 1995, 269, G467–G475. [Google Scholar]
- Andreeva, A.Y.; Piontek, J.; Blasig, I.E.; Utepbergenov, D.I. Assembly of tight junction is regulated by the antagonism of conventional and novel protein kinase C isoforms. Int. J. Biochem. Cell Biol. 2006, 38, 222–233. [Google Scholar]
- Dunagan, M.; Chaudhry, K.; Samak, G.; Rao, R.K. Acetaldehyde disrupts tight junctions in Caco-2 cell monolayers by a protein phosphatase 2A-dependent mechanism. Am. J. Physiol. 2012, 303, G1356–G1364. [Google Scholar]
- Sheth, P.; Seth, A.; Atkinson, K.J.; Gheyi, T.; Kale, G.; Giorgianni, F.; Desiderio, D.M.; Li, C.; Naren, A.; Rao, R.K. Acetaldehyde dissociates the PTP1B-E-cadherin-β-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem. J. 2007, 402, 291–300. [Google Scholar] [CrossRef]
- Turner, J.R. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am. J. Path. 2006, 169, 1901–1909. [Google Scholar] [CrossRef]
- Seth, A.; Sheth, P.; Elias, B.C.; Rao, R. Protein phosphatases 2A and 1 interact with occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. J. Biol. Chem. 2007, 282, 11487–11498. [Google Scholar] [CrossRef]
- Nunbhakdi-Craig, V.; Machleidt, T.; Ogris, E.; Bellotto, D.; White, C.L.; Sontag, E. Protein phosphatase 2A associates with and regulates atypical PKC and the epithelial tight junction complex. J. Cell Biol. 2002, 158, 967–978. [Google Scholar]
- Kemler, R. From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993, 9, 317–321. [Google Scholar] [CrossRef]
- Rao, R.K. Occludin phosphorylation in regulation of epithelial tight junctions. Ann. N.Y. Acad. Sci. 2009, 1165, 62–68. [Google Scholar] [CrossRef]
- Sheth, P.; Samak, G.; Shull, J.A.; Seth, A.; Rao, R. Protein phosphatase 2A plays a role in hydrogen peroxide-induced disruption of tight junctions in Caco-2 cell monolayers. Biochem. J. 2009, 421, 59–70. [Google Scholar]
- Shasby, D.M.; Kamath, J.M.; Moy, A.B.; Shasby, S.S. Ionomycin and PDBU increase MDCK monolayer permeability independently of myosin light chain phosphorylation. Am. J. Physiol. 1995, 269, L144–L150. [Google Scholar]
- Vale, C.; Botana, L.M. Marine toxins and the cytoskeleton: okadaic acid and dinophysistoxins. FEBS J. 2008, 275, 6060–6086. [Google Scholar] [CrossRef]
- Martín-López, A.; Gallardo-Rodríguez, J.J.; Sánchez-Mirón, A.; García-Camacho, F.; Molina-Grima, E. Cytotoxicity of yessotoxin and okadaic acid in mouse T lymphocyte cell line EL-4. Toxicon 2012, 60, 1049–1056. [Google Scholar] [CrossRef]
- Vilariño, N.; Ares, I.R.; Cagide, E.; Louzao, M.C.; Vieytes, M.R.; Yasumoto, T.; Botana, L.M. Induction of actin cytoskeleton rearrangement by methyl okadaate–comparison with okadaic acid. FEBS J. 2008, 275, 926–934. [Google Scholar]
- Rossini, G.P.; Hess, P. Phycotoxins: chemistry, mechanisms of action and shellfish poisoning. In Molecular, Clinical and Environmental Toxicology. Volume2: Clinical Toxicology; Luch, A., Ed.; Birkhäuser: Basel, Switzerland, 2010; pp. 65–122. [Google Scholar]
- Serres, M.; Grangeasse, C.; Haftek, M.; Durocher, Y.; Duclos, B.; Schmitt, D. Hyperphosphorylation of β-catenin on seine-threonine residues and loss of cell-cell contacts induced by calyculin A and okadaic acid in human epidermal cells. Exp. Cell Res. 1997, 231, 163–172. [Google Scholar] [CrossRef]
- Malaguti, C.; Rossini, G.P. Recovery of cellular E-cadherin precedes replenishment of estrogen receptor and estrogen-dependent proliferation of breast cancer cells rescued from a death stimulus. J. Cell. Physiol. 2002, 192, 171–181. [Google Scholar]
- Sontag, J.-M.; Sontag, E. Regulation of cell adhesion by PP2A and SV40 small tumor antigen: an important link to cell transformation. Cell. Mol. Life Sci. 2006, 63, 2979–2991. [Google Scholar] [CrossRef]
- Fujiki, H.; Sueoka, E.; Komori, A.; Suganuma, M. Tumor promotion and TNF-α gene expression by the okadaic acid class tumor promoters. Environ. Carcinogen. Ecotox. Rev. 1997, 15, 1–40. [Google Scholar]
- Marano, C.W.; Lewis, S.A.; Garulacan, L.A.; Peralta Soler, A.; Mullin, J.M. Tumor necrosis factor-α increases sodium and chloride conductance across the tight junction of CACO-2 BNE, a human intestinal cell line. J. Membrane Biol. 1998, 161, 263–274. [Google Scholar] [CrossRef]
- Kamat, P.K.; Tota, S.; Shukla, R.; Ali, S.; Najmi, A.K.; Nath, C. Mitochondrial dysfunction: a crucial event in okadaic acid (ICV) induced memory impairment and apoptotic cell death in rat brain. Pharmacol. Biochem. Behav. 2011, 100, 311–319. [Google Scholar] [CrossRef]
- Schmidt, K.N.; Traenckner, E.B.-M.; Meier, B.; Baeuerle, P.A. Induction of oxidative stress by okadaic acid is required for activation of transcription factor NF-κB. J. Biol. Chem. 1995, 270, 27136–27142. [Google Scholar]
- Rao, R.K.; Baker, R.D.; Baker, S.S.; Gupta, A.; Holycross, M. Oxidant-induced disruption of intestinal barrier function: role of protein tyrosine phosphorylation. Am. J. Physiol. 1997, 273, G812–G823. [Google Scholar]
- Ewe, K. Intestinal transport in constipation and diarrhoea. Pharmacology 1988, 36 (Suppl. 1), 73–84. [Google Scholar] [CrossRef]
- Gaginella, T.S.; Bass, P. Laxatives: an update on mechanism of action. Life Sci. 1978, 23, 1001–1009. [Google Scholar]
- Cline, W.S.; Lorenzsonn, V.; Benz, L.; Bass, P.; Olsen, W. The effects of sodium ricinoleate on small intestinal function and structure. J. Clin. Invest. 1976, 58, 380–390. [Google Scholar] [CrossRef]
- Reynell, P.C.; Spray, G.H. Chemical gastroenteritis in the rat. Gastroenterology 1958, 34, 867–873. [Google Scholar]
- Saunders, D.R.; Sillery, J.; Rachmilewitz, D. Effect of dioctyl sodium sulfosuccinate on structure and function of rodent and human intestine. Gastroenterology 1975, 69, 380–386. [Google Scholar]
- Saunders, D.R.; Sillery, J.; Rachmilewitz, D.; Rubin, C.; Tytgat, G. Effect of bisacodyl on the structure and function of rodent and human intestine. Gastroenterology 1977, 72, 849–856. [Google Scholar]
- Surawicz, C.; Saunders, D.R.; Rubin, C.E.; Tytgat, G.N. Pharmacology of laxatives: effects of phenolphthalein (PHE) on structure and function of intestinal mucosa. Gastroenterology 1977, 72, A-114. [Google Scholar]
- Van Gorkom, B.A.P.; de Vries, E.G.E.; Karrenbeld, A.; Kleibeuker, J.H. Review article: anthranoid laxatives and their potential carcinogenic effects. Aliment. Pharmacol. Ther. 1999, 13, 443–452. [Google Scholar] [CrossRef]
- Van Gorkom, B.A.P.; Karrenbeld, A.; van der Sluis, T.; Koudstaal, J.; de Vries, E.G.E.; Kleibeuker, J.H. Influence of a highly purified senna extract on colonic epithelium. Digestion 2000, 61, 113–120. [Google Scholar] [CrossRef]
- Suganuma, M.; Fujiki, H.; Furuya-Suguri, H.; Yoshizawa, S.; Yasumoto, S.; Kato, Y.; Fusetani, N.; Sugimura, T. Calyculin A, an inhibitor of protein phosphatases, a potent tumor promoter on CD-1 mouse skin. Cancer Res. 1990, 50, 3521–3525. [Google Scholar]
- Ohta, T.; Sueoka, E.; Iida, N.; Komori, A.; Suganuma, M.; Nishiwaki, R.; Tatematsu, M.; Kim, S.J.; Carmichael, W.W.; Fujiki, H. Nodularin, a potent inhibitor of protein phosphatases 1 and 2A, is a new environmental carcinogen in male F344 rat liver. Cancer Res. 1994, 54, 6402–6406. [Google Scholar]
- Nishiwaki-Matsushima, R.; Ohta, T.; Nishiwaki, S.; Suganuma, M.; Kohyama, K.; Ishikawa, T.; Carmichael, W.W.; Fujiki, H. Liver tumor promotion by the cyanobacterial cyclic peptide toxin microcystin-LR. J. Cancer Res. Clin. Oncol. 1992, 118, 420–424. [Google Scholar] [CrossRef]
- Suganuma, M.; Okabe, S.; Sueoka, E.; Nishiwaki, R.; Komori, A.; Uda, N.; Isono, K.; Fujiki, H. Tautomycin: an inhibitor of protein phosphatases 1 and 2A but not a tumor promoter on mouse skin and in rat glandular stomach. J. Cancer Res. Clin. Oncol. 1995, 121, 621–627. [Google Scholar]
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor promotion via injury- and death-induced inflammation. Immunity 2011, 35, 467–477. [Google Scholar] [CrossRef]
- Slaga, T.J. Antiinflammatory steroids: potent inhibitors of tumor promotion. In Carcinogenesis,Volume 5: Modifiers of Chemical Carcinogenesis; Slaga, T.J., Ed.; Raven Press: New York, NY, USA, 1980; Volume 5, pp. 111–126. [Google Scholar]
- Fürstenberger, G.; Csuk-Glänzer, B.I.; Marks, F.; Keppler, D. Phorbol ester-induced leukotriene biosynthesis and tumor promotion in mouse epidermis. Carcinogenesis 1994, 15, 2823–2827. [Google Scholar] [CrossRef]
- Fujiki, H.; Suganuma, M. Unique features of the okadaic acid activity class of tumor promoters. J. Cancer Res. Clin. Oncol. 1999, 125, 150–155. [Google Scholar] [CrossRef]
- Suganuma, M.; Okabe, S.; Marino, M.W.; Sakai, A.; Sueoka, E.; Fujiki, H. Essential role of tumor necrosis factor α (TNF-α) in tumor promotion as revealed by TNF-α-deficient mice. Cancer Res. 1999, 59, 4516–4518. [Google Scholar]
- Fujiki, H.; Suganuma, M.; Okabe, S.; Sueoka, E.; Suga, K.; Imai, K.; Nakachi, K. A new concept of tumor promotion by tumor necrosis factor-α, and cancer preventive agents (-)-epigallocatechin gallate and green tea - a review. Cancer Detect. Prev. 2000, 24, 91–99. [Google Scholar]
- Corsini, E.; Galli, C.L. Cytokines and irritant contact dermatitis. Toxicol. Lett. 1998, 102-103, 277–282. [Google Scholar] [CrossRef]
- Lewis, R.W.; McCall, J.C.; Botham, P.A.; Kimber, I. Investigation of TNF-α release as a measure of skin irritancy. Toxicol. in Vitro 1993, 7, 393–395. [Google Scholar] [CrossRef]
- Fujiki, H.; Suganuma, M.; Hakii, H.; Bartolini, G.; Moore, R.E.; Takayama, S.; Sugimura, T. A two-stage mouse skin carcinogenesis study of lyngbyatoxin A. J. Cancer Res. Clin. Oncol. 1984, 108, 174–176. [Google Scholar] [CrossRef]
- Fujiki, H.; Suganuma, M.; Nakayasu, M.; Hakii, H.; Horiuchi, T.; Takayama, S.; Sugimura, T. Palytoxin is a non-12-O-tetradecanoylphorbol-13-acetate type tumor promoter in two-stage mouse skin carcinogenesis. Carcinogenesis 1986, 7, 707–710. [Google Scholar] [CrossRef]
- Gong, C.-X.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K. Dysregulation of protein phosphorylation/dephosphorylation in Alzheimer's disease: a therapeutic target. J. Biomed. Biotechnol. 2006. Article ID 31825. [Google Scholar]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer's Disease. Lancet 2006, 368, 387–403. [Google Scholar]
- Gómez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 2004, 41, 17–24. [Google Scholar]
- Lovestone, S.; Reynolds, C.H. The phosphorylation of tau: a critical stage in neurodevelopment and neurodegenerative processes. Neuroscience 1997, 78, 309–324. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Terro, F. Tau protein phosphatases in Alzheimer's disease: The leading role of PP2A. Ageing Res. Rev. 2013, 12, 39–49. [Google Scholar] [CrossRef]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Sun, L.; Liu, S.; Zhou, X.; Wang, X.; Liu, R.; Wang, Q.; Wang, J. Inhibition of protein phosphatase 2A-and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience 2003, 118, 1175–1182. [Google Scholar] [CrossRef]
- Maidana, M.; Carlis, V.; Galhardi, F.G.; Yunes, J.S.; Geracitano, L.A.; Monserrat, J.M.; Barros, D.M. Effects of microcystins over short- and long-term memory and oxidative stress generation in hippocampus of rats. Chem.-Biol. Interact. 2006, 159, 223–234. [Google Scholar] [CrossRef]
- Li, G.; Yan, W.; Cai, F.; Li, C.; Chen, N.; Wang, J. Spatial learning and memory impairment and pathological change in rats induced by acute exposure to microcystin-LR. Environ. Toxicol. 2012. [Google Scholar] [CrossRef]
- Stein-Behrens, B.; Mattson, M.; Chang, I.; Yeh, M.; Sapolsky, R. Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J. Neurosci. 1994, 14, 5373–5380. [Google Scholar]
- Wang, Q.; Yu, S.; Simonyi, A.; Sun, G.Y.; Sun, A.Y. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol. Neurobiol. 2005, 31, 3–16. [Google Scholar] [CrossRef]
- Antequera, D.; Bolos, M.; Spuch, C.; Pascual, C.; Ferrer, I.; Fernandez-Bachiller, M.I.; Rodríguez-Franco, M.I.; Carro, E. Effects of a tacrine-8-hydroxyquinoline hybrid (IQM-622) on Aβ accumulation and cell death: involvement in hippocampal neuronal loss in Alzheimer's disease. Neurobiol. Dis. 2012, 46, 682–691. [Google Scholar] [CrossRef]
- Sun, L.; Wang, X.C.; Liu, S.; Wang, Q.; Wang, J.Z.; Bennecib, M.; Gong, C.-X.; Sengupta, A.; Grundke-Iqbal, I.; Iqbal, K. Bilateral injection of isoproterenol into hippocampus induces Alzheimer-like hyperphosphorylation of tau and spatial memory deficit in rat. FEBS Lett. 2005, 579, 251–258. [Google Scholar]
- Zhang, Y.-J.; Xu, Y.-F.; Liu, Y.-H.; Yin, J.; Li, H.-L.; Wang, Q.; Wang, J.-Z. Peroxynitrite induces Alzheimer-like tau modifications and accumulation in rat brain and its underlying mechanisms. FASEB J. 2006, 20, 1431–1442. [Google Scholar] [CrossRef]
- Janke, C.; Gärtner, U.; Holzer, M.; Arendt, T. Reversible in vivo phosphorylation of tau induced by okadaic acid and by unspecific brain lesion in rat. J. Brain Res. 1998, 39, 143–153. [Google Scholar]
- Wen, Y.; Yang, S.; Liu, R.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer's disease-like tauopathy in female rats. J. Biol. Chem. 2004, 279, 22684–22692. [Google Scholar]
- Wen, Y.; Yang, S.; Liu, R.; Simpkins, J.W. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004, 1022, 30–38. [Google Scholar]
- King, C.E.; Adlard, P.A.; Dickson, T.C.; Vickers, J.C. Neuronal response to physical injury and its relationship to the pathology of Alzheimer’s disease. Clin. Exp. Pharmacol. Physiol. 2000, 27, 548–552. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Munday, R. Is Protein Phosphatase Inhibition Responsible for the Toxic Effects of Okadaic Acid in Animals? Toxins 2013, 5, 267-285. https://doi.org/10.3390/toxins5020267
Munday R. Is Protein Phosphatase Inhibition Responsible for the Toxic Effects of Okadaic Acid in Animals? Toxins. 2013; 5(2):267-285. https://doi.org/10.3390/toxins5020267
Chicago/Turabian StyleMunday, Rex. 2013. "Is Protein Phosphatase Inhibition Responsible for the Toxic Effects of Okadaic Acid in Animals?" Toxins 5, no. 2: 267-285. https://doi.org/10.3390/toxins5020267