Conotoxins Targeting Neuronal Voltage-Gated Sodium Channel Subtypes: Potential Analgesics?



Abstract
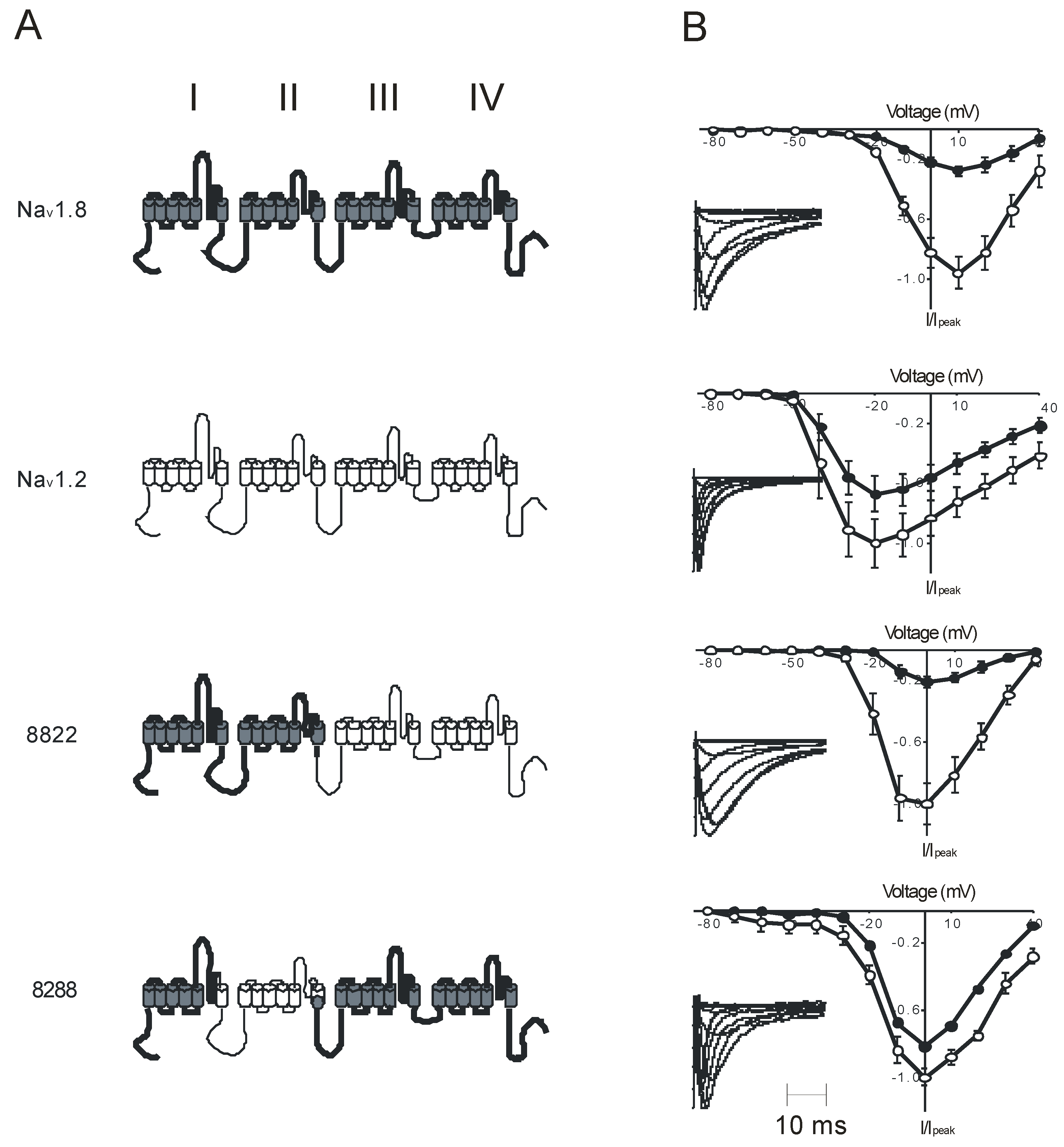
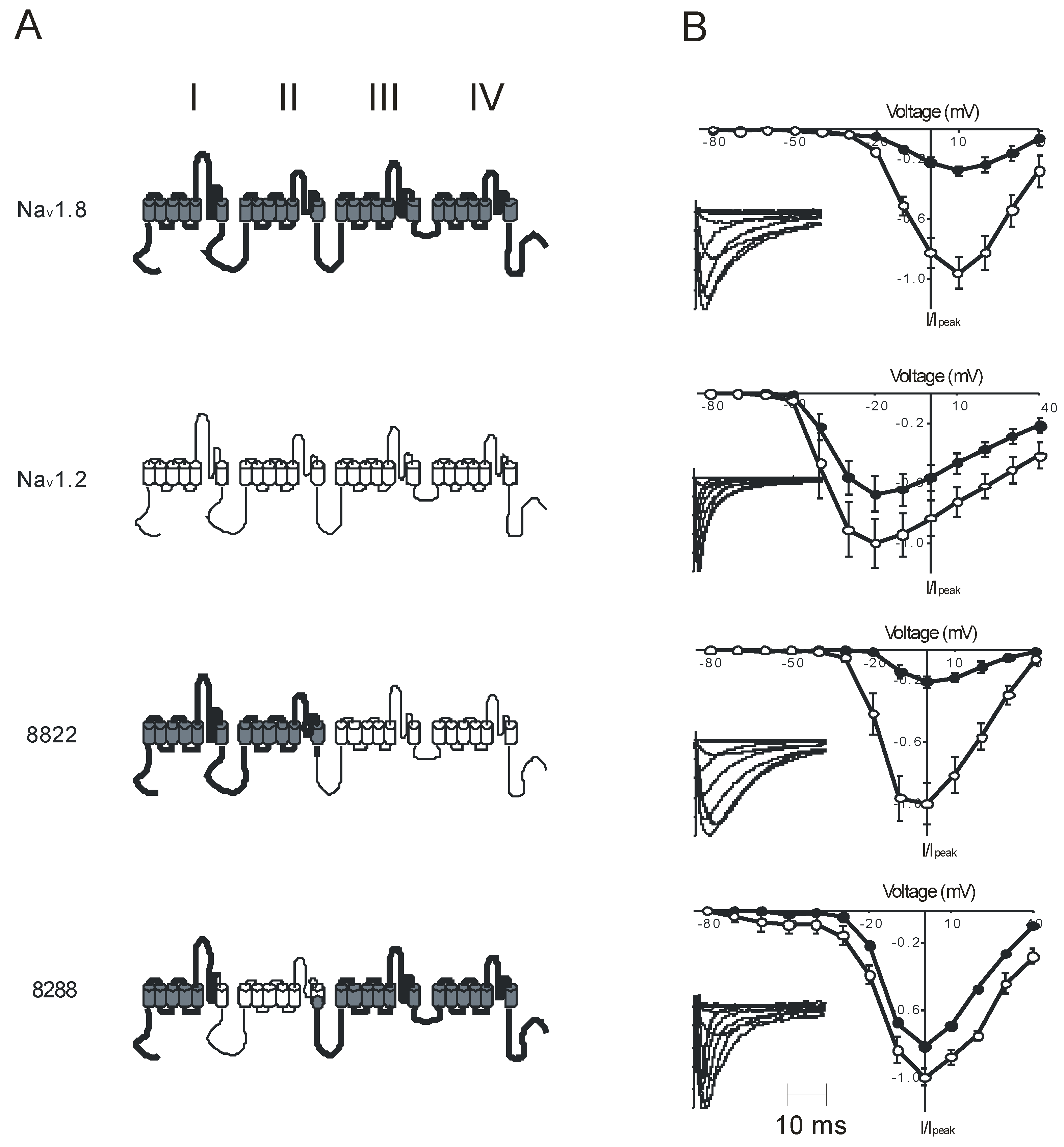
:1. Introduction
2. VGSC Subtypes Involved in Pain
2.1. Nav1.3
2.2. Nav1.7
2.3. Nav1.8
2.4. Nav1.9
3. Conotoxins
3.1. μ-Conotoxins

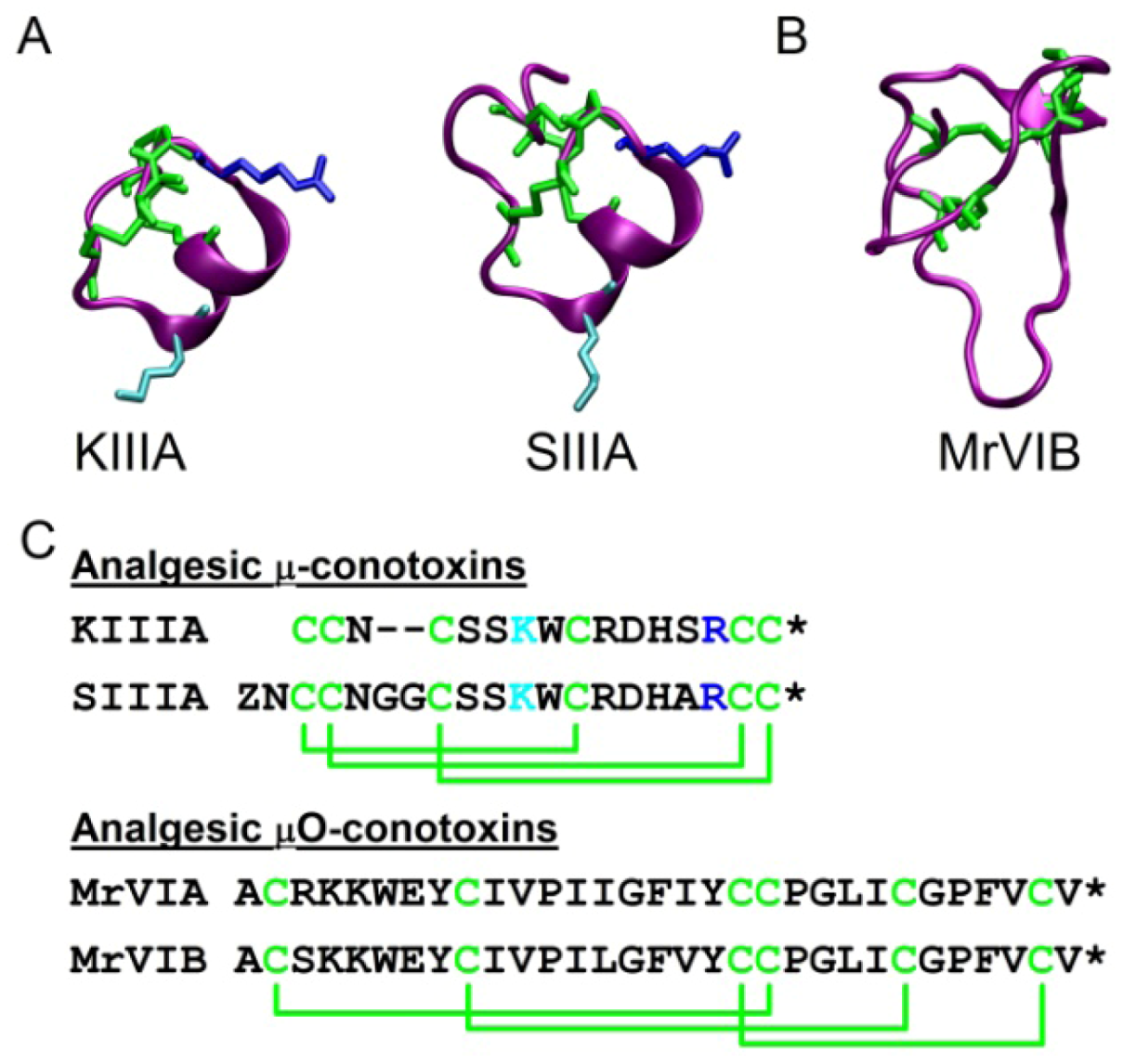{kind=link}
{kind=link}
| μ-Conotoxins | Conus species | Number of residues | VGSC subtypes (IC50) | References |
|---|---|---|---|---|
| KIIIA | C. kinoshitai | 16 | Nav1.3 (8 μM), | [100,101] |
| Nav1.7 (100–290 nM) | ||||
| SIIIA*/B | C. striatus | 20/20 | Nav1.3 (11 μM (SIIIA)), | [103,104,105] |
| Nav1.7 (65 μM (SIIIA)), | ||||
| Nav1.8 (insensitive to 10 μM) | ||||
| PIIIA | C. purpurascens | 22 | Nav1.3 (3.2 μM), | [96,106,107] |
| Nav1.7 (3.1–6.2 μM) | ||||
| GIIIA/B/C | C. geographus | 22/22/22 | N.D. | |
| CnIIIA/B/C | C. consors | 22/25/22 | Nav1.3 (11 μM (CnIIIA)), | [98,107,108] |
| Nav1.7 (489 nM (CnIIIC)) | ||||
| Nav1.8 (insensitive to 1 μM (CnIIIC)) | ||||
| CIIIA | C. catus | 22 | N.D. | |
| SmIIIA | C. stercusmuscarum | 22 | Nav1.3 (40 nM), | [97] |
| Nav1.7 (1.3 μM) | ||||
| MIIIA | C. magus | 22 | Nav1.3 (7.7 nM), | [107] |
| Nav1.7 (97 μM) | ||||
| SxIIIA/B | C. striolatus | 22/23 | N.D. | |
| BuIIIA/B/C | C. bullatus | 23/24/26 | Nav1.3 (350 nM (BuIIIA); 200 nM (BuIIIB)) | [107] |
| TIIIA | C. tulipa | 22 | Nav1.3 (50 nM), | [107] |
| Nav1.7 (insensitive to 3 μM) | ||||
| Nav1.8 (insensitive to 3 μM) |
3.2. μ-Conotoxin Engineering
3.3. μO-Conotoxins
| μO-Conotoxins | Conus species | Number of residues | VGSC subtypes(IC50) | References |
|---|---|---|---|---|
| MrVIA | C. marmoreus | 31 | Nav1.7 (345 nM) | [123,134] |
| MrVIB* | C. marmoreus | 31 | Nav1.3 (1 μM), | [62,123,134] |
| Nav1.7 (345 nM), | ||||
| Nav1.8 (1–326 nM) | ||||
| MfVIA | C. magnificus | 32 | Nav1.3 (2175 nM), | [133] |
| Nav1.7 (2317–5491 nM), | ||||
| Nav1.8 (529 nM) | ||||
| LtVIIA | C. litteratus | 29 | N.D. | [135,136] |
| LtVIC | C. litteratus | 28 | N.D. | [135,136] |
4. β-Subunits Modulate the Effects of Conotoxins
5. Multiple Sites of μ- and μO-Conotoxin Action
6. Conotoxins—Analgesics of the Future?
Conflict of Interest
Acknowledgements
References
- Hille, B.; Armstrong, C.M.; MacKinnon, R. Ion channels: from idea to reality. Nat. Med. 1999, 5, 1105–1109. [Google Scholar] [CrossRef]
- Payandeh, J.; Gamal El-Din, T.M.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar]
- Payandeh, J.; Gamal El-Din, T.M.; Scheuer, T.; Zheng, N.; Catterall, W.A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012, 486, 135–139. [Google Scholar]
- Zhang, X.; Ren, W.; DeCaen, P.; Yan, C.; Tao, X.; Tang, L.; Wang, J.; Hasegawa, K.; Kumasaka, T.; He, J.; Wang, J.; Clapham, D.E.; Yan, N. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 2012, 486, 130–134. [Google Scholar]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure–function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar]
- Al-Sabi, A.; McArthur, J.; Ostroumov, V.; French, R.J. Marine toxins that target voltage-gated sodium channels. Mar. Drugs 2006, 4, 157–192. [Google Scholar] [CrossRef]
- Lee, C.H.; Ruben, P.C. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels (Austin) 2008, 2, 407–412. [Google Scholar]
- Rogart, R.B.; Cribbs, L.L.; Muglia, L.K.; Kephart, D.D.; Kaiser, M.W. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc. Natl. Acad. Sci. USA 1989, 86, 8170–8174. [Google Scholar] [CrossRef]
- Satin, J.; Kyle, J.W.; Chen, M.; Bell, P.; Cribbs, L.L.; Fozzard, H.A.; Rogart, R.B. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science 1992, 256, 1202–1205. [Google Scholar]
- Isom, L.L. Sodium channel beta subunits: anything but auxiliary. Neuroscientist 2011, 7, 42–54. [Google Scholar] [CrossRef]
- Meadows, L.; Malhotra, J.D.; Stetzer, A.; Isom, L.L.; Ragsdale, D.S. The intracellular segment of the sodium channel β1 subunit is required for its efficient association with the channel α subunit. J. Neurochem. 2001, 76, 1871–1878. [Google Scholar] [CrossRef]
- Zimmer, T.; Benndorf, K. The human heart and rat brain IIA Na+ channels interact with different molecular regions of the β1 subunit. J. Gen. Physiol. 2002, 120, 887–895. [Google Scholar] [CrossRef]
- Carter, G.T.; Sullivan, M.D. Antidepressants in pain management. Curr. Opin. Investig. Drugs 2002, 3, 454–458. [Google Scholar]
- Reisner, L. Antidepressants for chronic neuropathic pain. Curr. Pain Headache Rep. 2003, 7, 24–33. [Google Scholar] [CrossRef]
- Bouhassira, D.; Lantéri-Minet, M.; Attal, N.; Laurent, B.; Touboul, C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008, 136, 380–387. [Google Scholar] [CrossRef]
- Jensen, M.P.; Chodroff, M.J.; Dworkin, R.H. The impact of neuropathic pain on health-related quality of life. Neurology 2007, 68, 1178–1182. [Google Scholar] [CrossRef]
- Dworkin, R.H.; Malone, D.C.; Panarites, C.J.; Armstrong, E.P.; Pham, S.V. Impact of post-therapeutic neuralgia and painful diabetic peripheral neuropathy on health care costs. J. Pain. 2010, 11, 360–368. [Google Scholar] [CrossRef]
- Akopian, A.N.; Souslova, V.; England, S.; Okuse, K.; Ogata, N.; Ure, J.; Smith, A.; Kerr, B.J.; McMahon, S.B.; Boyce, S.; Hill, R.; Stanfa, L.C.; Dickenson, A.H.; Wood, J.N. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 1999, 2, 541–548. [Google Scholar] [CrossRef]
- Black, J.A.; Cummins, T.R.; Plumpton, C.; Chen, Y.H.; Hormuzdiar, W.; Clare, J.J.; Waxman, S.G. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neuron. J. Neurophysiol. 1999, 82, 2776–2785. [Google Scholar]
- Coward, K.; Plumpton, C.; Facer, P.; Birch, R.; Carlstedt, T.; Tate, S.; Bountra, C.; Anand, P. Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain 2000, 85, 41–50. [Google Scholar] [CrossRef]
- Cummins, T.R.; Dib-Hajj, S.D.; Black, J.A.; Waxman, S.G. Sodium channels and the molecular pathophysiology of pain. Prog. Brain Res. 2000, 129, 3–19. [Google Scholar] [CrossRef]
- Cummins, T.R.; Waxman, S.G. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J. Neurosci. 1997, 17, 3503–3514. [Google Scholar]
- Dib-Hajj, S.; Black, J.A.; Felts, P.; Waxman, S.G. Down-regulation of transcripts for Na+ channel α-SNS in spinal sensory neurons following axotomy. Proc. Natl. Acad. Sci. USA 1996, 93, 14950–14954. [Google Scholar]
- Dib-Hajj, S.D.; Black, J.A.; Cummins, T.R.; Kenney, A.M.; Kocsis, J.D.; Waxman, S.G. Rescue of α-SNS sodium channel expression in small dorsal root ganglion neurons after axotomy by nerve growth factor in vivo. J. Neurophysiol. 1998, 79, 2668–2676. [Google Scholar]
- Dib-Hajj, S.D.; Fjell, J.; Cummins, T.R.; Zheng, Z.; Fried, K.; LaMotte, R.; Black, J.A.; Waxman, S.G. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 1999, 83, 591–600. [Google Scholar] [CrossRef]
- Fjell, J.; Cummins, T.R.; Davis, B.M.; Albers, K.M.; Fried, K.; Waxman, S.G.; Black, J.A. Sodium channel expression in NGF-overexpressing transgenic mice. J. Neurosci. Res. 1999, 57, 39–47. [Google Scholar] [CrossRef]
- Gold, M. Tetrodotoxin-resistant Na+ currents and inflammatory hyperalgesia. Proc. Natl. Acad. Sci. USA 1999, 96, 7645–7649. [Google Scholar] [CrossRef]
- Leffler, A.; Cummins, T.R.; Dib-Hajj, S.D.; Hormuzdiar, W.N.; Black, J.A.; Waxman, S.G. GDNF and NGF reverse changes in repriming of TTX-sensitive Na+ currents following axotomy of dorsal root ganglion neurons. J. Neurophysiol. 2002, 88, 650–658. [Google Scholar]
- Yoshimura, N.; Seki, S.; Novakovic, S.D.; Tzoumaka, E.; Erickson, V.L.; Erickson, K.A.; Chancellor, M.B.; de Groat, W.C. The involvement of the tetrodotoxin-resistant sodium channel Nav1.8 (PN3/SNS) in a rat model of visceral pain. J. Neurosci. 2001, 21, 8690–8696. [Google Scholar]
- Waxman, S.G.; Dib-Hajj, S.; Cummins, T.R.; Black, J.A. Sodium channels and pain. Proc. Natl. Acad. Sci. USA 1999, 96, 7635–7639. [Google Scholar] [CrossRef]
- Djouhri, L.; Fang, X.; Koutsikou, S.; Lawson, S.N. Partial nerve injury induces electrophysiological changes in conducting (uninjured) nociceptive and nonnociceptive DRG neurons: Possible relationships to aspects of peripheral neuropathic pain and paresthesias. Pain 2012, 153, 1824–1836. [Google Scholar] [CrossRef]
- Waxman, S.G.; Kocsis, J.; Black, J.A. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is re-expressed following axotomy. J. Neurophysiol. 1994, 72, 466–470. [Google Scholar]
- Hains, B.C.; Saab, C.Y.; Klein, J.P.; Craner, M.J.; Waxman, S.G. Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J. Neurosci. 2004, 24, 4832–4839. [Google Scholar] [CrossRef]
- Black, J.A.; Lui, S.; Tanaka, M.; Cummins, T.R.; Waxman, S.G. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 2004, 108, 237–247. [Google Scholar] [CrossRef]
- Lampert, A.; Hains, B.C.; Waxman, S.G. Upregulation of persistent and ramp sodium current in dorsal horn neurons after spinal cord injury. Exp. Brain Res. 2006, 174, 660–666. [Google Scholar] [CrossRef]
- He, X.H.; Zang, Y.; Chen, X.; Pang, R.P.; Xu, J.T.; Zhou, X.; Wei, X.H.; Li, Y.Y.; Xin, W.J.; Qin, Z.H.; Liu, X.G. TNF-α contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain 2010, 151, 266–279. [Google Scholar] [CrossRef]
- Siqueira, S.; Alves, B.; Malpartida, H.M.; Teixeira, M.J.; Siqueira, J.T. Abnormal expression of voltage-gated sodium channels Nav1.7, Nav1.3 and Nav1.8 in trigeminal neuralgia. Neuroscience 2009, 164, 573–577. [Google Scholar]
- Yoshida, S. Tetrodotoxin-resistant sodium channels. Cell. Mol. Neurobiol. 1994, 14, 227–244. [Google Scholar] [CrossRef]
- Momin, A.; Wood, J.N. Sensory neuron voltage-gated sodium channels as analgesic drug targets. Curr. Opin. Neurobiol. 2008, 18, 383–388. [Google Scholar] [CrossRef]
- Nassar, M.A.; Baker, M.D.; Levato, A.; Ingram, R.; Mallucci, G.; McMahon, S.B.; Wood, J.N. Nerve injury induces robust allodynia and ectopic discharges in Nav1.3 null mutant mice. Mol. Pain 2006, 2, 33. [Google Scholar] [CrossRef]
- Klugbauer, N.; Lacinova, L.; Flockerzi, V.; Hofmann, F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995, 14, 1084–1090. [Google Scholar]
- Felts, P.A.; Yokoyama, S.; Dib-Hajj, S.; Black, J.A.; Waxman, S.G. Sodium channel α-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res. Mol. Brain Res. 1997, 45, 71–82. [Google Scholar]
- Sangameswaran, L.; Fish, L.M.; Koch, B.D.; Rabert, D.K.; Delgado, S.G.; Ilnicka, M.; Jakeman, L.B.; Novakovic, S.; Wong, K.; Sze, P.; Tzoumaka, E.; Stewart, G.R.; Herman, R.C.; Chan, H.; Eglen, R.M.; Hunter, J.C. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J. Biol. Chem. 1997, 272, 14805–14809. [Google Scholar]
- Toledo-Aral, J.J.; Moss, B.L.; He, Z.J.; Koszowski, A.G.; Whisenand, T.; Levinson, S.R.; Wolf, J.J.; Silos-Santiago, I.; Halegoua, S.; Mandel, G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. USA 1997, 94, 1527–1532. [Google Scholar]
- Theile, J.W.; Cummins, T.R. Recent developments regarding voltage-gated sodium channel blockers for the treatment of inherited and acquired neuropathic pain syndromes. Front. Pharmacol. 2011, 2, 54. [Google Scholar]
- Vijayaragavan, K.; O'Leary, M.; Chahine, M. Gating properties of Nav1.7 and Nav1.8 peripheral nerve sodium channels. J. Neurosci. 2001, 21, 7909–7918. [Google Scholar]
- Waxman, S.G.; Dib-Hajj, S. Erythermalgia: molecular basis for an inherited pain syndrome. Trends Mol. Med. 2005, 11, 555–562. [Google Scholar] [CrossRef]
- Fertleman, C.R.; Baker, M.D.; Parker, K.A.; Moffatt, S.; Elmslie, F.V.; Abrahamsen, B.; Ostman, J.; Klugbauer, N.; Wood, J.N.; Gardiner, R.M.; Rees, M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 2006, 52, 767–774. [Google Scholar] [CrossRef]
- Goldberg, Y.P.; MacFarlane, J.; MacDonald, M.L.; Thompson, J.; Dube, M.P.; Mattice, M.; Fraser, R.; Young, C.; Hossain, S.; Pape, T.; Payne, B.; Radomski, C.; Donaldson, G.; Ives, E.; Cox, J.; Younghusband, H.B.; Green, R.; Duff, A.; Boltshauser, E.; Grinspan, G.A.; Dimon, J.H.; Sibley, B.G.; Andria, G.; Toscano, E.; Kerdraon, J.; Bowsher, D.; Pimstone, S.N.; Samuels, M.E.; Sherrington, R.; Hayden, M.R. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 2007, 71, 311–319. [Google Scholar] [CrossRef]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; Al-Gazali, L.; Hamamy, H.; Valente, E.M.; Gorman, S.; Williams, R.; McHale, D.P.; Wood, J.N.; Gribble, F.M.; Woods, C.G. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar]
- Lampert, A.; O'Reilly, A.O.; Reeh, P.; Leffler, A. Sodium channelopathies and pain. Pflugers Arch. 2010, 460, 249–263. [Google Scholar] [CrossRef]
- McGowan, E.; Hoyt, S.; Li, X.; Lyons, K.A.; Abbadie, C. A peripherally acting Nav1.7 sodium channel blocker reverses hyperalgesia and allodynia on rat models of inflammatory and neuropathic pain. Anesth. Analg. 2009, 109, 951–958. [Google Scholar] [CrossRef]
- Ahn, H.S.; Black, J.; Zhao, P.; Tyrrell, L.; Waxman, S.G.; Dib-Hajj, S.D. Nav1.7 is the predominant sodium channel in rodent olfactory sensory neurons. Mol. Pain 2011, 7, 32. [Google Scholar] [CrossRef]
- Weiss, J.; Pyrski, M.; Jacobi, E.; Bufe, B.; Willnecker, V.; Schick, B.; Zizzari, P.; Gossage, S.J.; Greer, C.A.; Leinders-Zufall, T.; Woods, C.G.; Wood, J.N.; Zufall, F. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature 2011, 472, 186–190. [Google Scholar]
- Rupasinghe, D.B.; Knapp, O.; Blomster, L.V.; Schmid, A.B.; Adams, D.J.; King, G.F.; Ruitenberg, M.J. Localization of Nav1.7 in the normal and injured rodent olfactory system indicates a critical role in olfaction, pheromone sensing and immune function. Channels (Austin) 2012, 6, 103–110. [Google Scholar]
- Gold, M.S.; Weinreich, D.; Kim, C.S.; Wang, R.; Treanor, J.; Porreca, F.; Lai, J. Redistribution of Nav1.8 in uninjured axons enables neuropathic pain. J. Neurosci. 2003, 23, 158–166. [Google Scholar]
- Zimmermann, K.; Leffler, A.; Babes, A.; Cendan, C.M.; Carr, R.W.; Kobayashi, J.; Nau, C.; Wood, J.N.; Reeh, P.W. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature 2007, 447, 855–858. [Google Scholar]
- Coward, K.; Jowett, A.; Plumpton, C.; Powell, A.; Birch, R.; Tate, S.; Bountra, C.; Anand, P. Sodium channel β1 and β2 subunits parallel SNS/PN3 α-subunit changes in injured human sensory neurons. Neuroreport 2001, 12, 483–488. [Google Scholar] [CrossRef]
- Meadows, L.S.; Chen, Y.H.; Powell, A.J.; Clare, J.J.; Ragsdale, D.S. Functional modulation of human brain Nav1.3 sodium channels, expressed in mammalian cells, by auxiliary β1, β2 and β3 subunits. Neuroscience 2002, 114, 745–753. [Google Scholar]
- Nassar, M.A.; Levato, A.; Stirling, L.C.; Wood, J.N. Neuropathic pain develops normally in mice lacking both Nav1.7 and Nav1.8. Pain 2005, 1, 24. [Google Scholar]
- Gilchrist, J.; Bosmans, F. Animal toxins can alter the function of Nav1.8 and Nav1.9. Toxins. 2012, 4, 620–632. [Google Scholar] [CrossRef]
- Ekberg, J.; Jayamanne, A.; Vaughan, C.W.; Aslan, S.; Thomas, L.; Mould, J.; Drinkwater, R.; Baker, M.D.; Abrahamsen, B.; Wood, J.N.; Adams, D.J.; Christie, M.J.; Lewis, R.J. μO-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. Proc. Natl. Acad. Sci. USA 2006, 103, 17030–17035. [Google Scholar]
- Kerr, B.J.; Souslova, V.; McMahon, S.B.; Wood, J.N. A role for the TTX-resistant sodium channel Nav1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport 2001, 12, 3077–3080. [Google Scholar]
- Decosterd, I.; Ji, R.R.; Abdi, S.; Tate, S.; Woolf, C.J. The pattern of expression of the voltage-gated sodium channels Nav1.8 and Nav1.9 does not change in uninjured primary sensory neurons in experimental neuropathic pain models. Pain 2002, 96, 269–277. [Google Scholar] [CrossRef]
- Leo, S.; D'Hooge, R.; Meert, T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav. Brain. Res. 2010, 208, 149–157. [Google Scholar] [CrossRef]
- Cummins, T.R.; Dib-Hajj, S.D.; Black, J.A.; Akopian, A.N.; Wood, J.N.; Waxman, S.G. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J. Neurosci. 1999, 19, RC43. [Google Scholar]
- Herzog, R.I.; Cummins, T.R.; Waxman, S.G. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J. Neurophysiol. 2001, 86, 1351–1364. [Google Scholar]
- Baker, M.D.; Chandra, S.; Ding, Y.; Waxman, S.G.; Wood, J.N. GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurones. J. Physiol. 2003, 548, 373–382. [Google Scholar] [CrossRef]
- Priest, B.T.; Murphy, B.A.; Lindia, J.A.; Diaz, C.; Abbadie, C.; Ritter, A.M.; Liberator, P.; Iyer, L.M.; Kash, S.F.; Kohler, M.G.; Kaczorowski, G.J.; MacIntyre, D.E.; Martin, W.J. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel Nav1.9 to sensory transmission and nociceptive behavior. Proc. Natl. Acad. Sci. USA 2005, 102, 9382–9387. [Google Scholar]
- Amaya, F.; Wang, H.; Costigan, M.; Allchorne, A.J.; Hatcher, J.P.; Egerton, J.; Stean, T.; Morisset, V.; Grose, D.; Gunthorpe, M.J.; Chessell, I.P.; Tate, S.; Green, P.J.; Woolf, C.J. The voltage-gated sodium channel Nav1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 2006, 26, 12852–12860. [Google Scholar]
- Yu, Y.Q.; Zhao, F.; Guan, S.M.; Chen, J. Antisense-mediated knockdown of Nav1.8, but not Nav1.9, generates inhibitory effects on complete Freund's adjuvant-induced inflammatory pain in rat. PLoS One 2011, 6, e19865. [Google Scholar]
- Oliver, B.M. Conus venom peptides: reflections from the biology of clades and species. Annu. Rev. Ecol. Syst. 2002, 33, 25–47. [Google Scholar] [CrossRef]
- Terlau, H.; Oliver, B.M. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- Rice, R.D.; Halstead, B.W. Report of fatal cone shell sting by Conus geographus Linnaeus. Toxicon 1968, 5, 223–224. [Google Scholar] [CrossRef]
- Davis, J.; Jones, A.; Lewis, R.J. Remarkable inter- and intra-species complexity of conotoxins revealed by LC/MS. Peptides 2009, 30, 1222–1227. [Google Scholar] [CrossRef]
- Hui, K.; Lipkind, G.; Fozzard, H.A.; French, R.J. Electrostatic and steric contributions to block of the skeletal muscle sodium channel by μ-conotoxin. J. Gen. Physiol. 2002, 119, 45–54. [Google Scholar] [CrossRef]
- Leipold, E.; DeBie, H.; Zorn, S.; Borges, A.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. μO-conotoxins inhibit Nav channels by interfering with their voltage sensors in domain-2. Channels (Austin) 2007, 1, 253–262. [Google Scholar]
- Gilly, W.F.; Richmond, T.A.; Duda, T.F., Jr.; Elliger, C.; Lebaric, Z.; Schulz, J.; Bingham, J.P.; Sweedler, J.V. A diverse family of novel peptide toxins from an unusual cone snail, Conus californicus. J. Exp. Biol. 2011, 214, 147–161. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Q.; Pi, C.; Zhao, Y.; Zhou, M.; Wang, L.; Chen, S.; Xu, A. Isolation and characterization of a T-superfamily conotoxin from Conus litteratus with targeting tetrodotoxin-sensitive sodium channels. Peptides 2007, 28, 2313–2319. [Google Scholar] [CrossRef]
- Leipold, E.; Hansel, A.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. Molecular interaction of δ-conotoxins with voltage-gated sodium channels. FEBS Lett. 2005, 579, 3881–3884. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Fainzilber, M.; van der Schors, R.; Lodder, J.C.; Li, K.W.; Geraerts, W.P.; Kits, K.S. New sodium channel-blocking conotoxins also affect calcium currents in Lymnaea neurons. Biochemistry 1995, 34, 5364–5371. [Google Scholar] [CrossRef]
- Hasson, A.; Shon, K.J.; Olivera, B.M.; Spira, M.E. Alterations of voltage-activated sodium current by a novel conotoxin from the venom of Conus gloriamaris. J. Neurophysiol. 1995, 73, 1295–1301. [Google Scholar]
- Shichor, I.; Fainzilber, M.; Pelhate, M.; Malecot, C.O.; Zlotkin, E.; Gordon, D. Interactions of δ-conotoxins with alkaloid neurotoxins reveal differences between the silent and effective binding sites on voltage-sensitive sodium channels. J. Neurochem. 1996, 167, 2451–2460. [Google Scholar]
- Barbier, J.; Lamthanh, H.; Le Gall, F.; Favreau, P.; Benoit, E.; Chen, H.; Gilles, N.; Ilan, N.; Heinemann, S.H.; Gordon, D.; Ménez, A.; Molgó, J. A δ-conotoxin from Conus ermineus venom inhibits inactivation in vertebrate neuronal Na+ channels but not in skeletal and cardiac muscles. J. Biol. Chem. 2004, 279, 4680–4685. [Google Scholar]
- Sudarslal, S.; Majumdar, S.; Ramasamy, P.; Dhawan, R.; Pal, P.P.; Ramaswami, M.; Lala, A.K.; Sikdar, S.K.; Sarma, S.P.; Krishnan, K.S.; Balaram, P. Sodium channel modulating activity in a δ-conotoxin from an Indian marine snail. FEBS Lett. 2003, 553, 209–212. [Google Scholar] [CrossRef]
- Sarma, S.P.; Kumar, G.S.; Sudarslal, S.; Iengar, P.; Ramasamy, P.; Sikdar, S.K.; Krishnan, K.S.; Balaram, P. Solution structure of δ-Am2766: a highly hydrophobic δ-conotoxin from Conus amadis that inhibits inactivation of neuronal voltage-gated sodium channels. Chem. Biodivers. 2005, 2, 535–556. [Google Scholar] [CrossRef]
- Wang, L.; Lui, J.; Pi, C.; Zeng, X.; Zhou, M.; Jiang, X.; Chen, S.; Ren, Z.; Xu, A. Identification of a novel M-superfamily conotoxin with the ability to enhance tetrodotoxin sensitive sodium currents. Arch. Toxicol. 2009, 83, 925–932. [Google Scholar] [CrossRef]
- Fiedler, B.; Zhang, M.M.; Buczek, O.; Azam, L.; Bulaj, G.; Norton, R.S.; Olivera, B.M.; Yoshikami, D. Specificity, affinity and efficacy of ι-conotoxin RXIA, an agonist of voltage-gated sodium channels Nav1.2, 1.6 and 1.7. Biochem. Pharmacol. 2008, 75, 2334–2344. [Google Scholar]
- Li, R.A.; Ennis, I.; Xue, T.; Nguyen, H.M.; Tomaselli, G.F.; Goldin, A.L.; Marbán, E. Molecular basis of isoform-specific μ-conotoxin block of cardiac, skeletal muscle, and brain Na+ channels. J. Biol. Chem. 2003, 278, 8717–8724. [Google Scholar]
- Spence, I.; Gillessen, D.; Gregson, R.P.; Quinn, R.J. Characterization of the neurotoxic constituents of Conus geographus (L) venom. Life Sci. 1977, 21, 1759–1769. [Google Scholar] [CrossRef]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M.; Zeikus, R.D.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 1985, 260, 9280–9288. [Google Scholar]
- Becker, S.; Prusak-Sochaczewski, E.; Zamponi, G.; Beck-Sickinger, A.G.; Gordon, R.D.; French, R.J. Action of derivatives of μ-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry 1992, 31, 8229–8238. [Google Scholar]
- Wakamatsu, K.; Kohda, D.; Hatanaka, H.; Lancelin, J.M.; Ishida, Y.; Oya, M.; Nakamura, H.; Inagaki, F.; Sato, K. Structure–activity relationships of μ-conotoxin GIIIA: structure determination of active and inactive sodium channel blocker peptides by NMR and simulated annealing calculations. Biochemistry 1992, 31, 12577–12584. [Google Scholar]
- Hill, J.M.; Alewood, P.F.; Craik, D.J. Three-dimensional solution structure of μ-conotoxin GIIIB, a specific blocker of skeletal muscle sodium channels. Biochemistry 1996, 35, 8824–8835. [Google Scholar] [CrossRef]
- Nielsen, K.J.; Watson, M.; Adams, D.J.; Hammarström, A.K.; Gage, P.W.; Hill, J.M.; Craik, D.J.; Thomas, L.; Adams, D.; Alewood, P.F.; Lewis, R.J. Solution structure of μ-conotoxin PIIIA, a preferential inhibitor of persistent tetrodotoxin-sensitive sodium channels. J. Biol. Chem. 2002, 277, 27247–27255. [Google Scholar]
- Keizer, D.W.; West, P.J.; Lee, E.F.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structural basis for tetrodotoxin-resistant sodium channel binding by μ-conotoxin SmIIIA. J. Biol. Chem. 2003, 278, 46805–46813. [Google Scholar]
- Favreau, P.; Benoit, E.; Hocking, H.G.; Carlier, L.; D'hoedt, D.; Leipold, E.; Markgraf, R.; Schlumberger, S.; Córdova, M.A.; Gaertner, H.; Paolini-Bertrand, M.; Hartley, O.; Tytgat, J.; Heinemann, S.H.; Bertrand, D.; Boelens, R.; Stöcklin, R.; Molgó, J. Pharmacological characterization of a novel μ-conopeptide, CnIIIC, indicates potent and preferential inhibition of sodium channel subtypes (Nav 1.2/1.4) and reveals unusual activity on neuronal nicotinic acetylcholine receptors. Br. J. Pharmacol. 2012, 166, 1654–1668. [Google Scholar]
- Shon, K.J.; Olivera, B.M.; Watkins, M.; Jacobsen, R.B.; Gray, W.R.; Floresca, C.Z.; Cruz, L.J.; Hillyard, D.R.; Brink, A.; Terlau, H.; Yoshikami, D. μ-Conotoxin PIIIA, a new peptide for discriminating among tetrodotoxin-sensitive Na channel subtypes. J. Neurosci. 1998, 18, 4473–4481. [Google Scholar]
- McArthur, J.R.; Simgh, G.; McMaster, D.; Winkfein, R.; Tieleman, D.P.; French, R.J. Interactions of key charged residues contributing to selective block of neuronal sodium channels by μ-conotoxin KIIIA. Mol. Pharmacol. 2011, 80, 573–584. [Google Scholar] [CrossRef]
- Zhang, M.M.; Green, B.R.; Catlin, P.; Fiedler, B.; Azam, L.; Chadwick, A.; Terlau, H.; McArthur, J.R.; French, R.J.; Gulyas, J.; Rivier, J.E.; Smith, B.J.; Norton, R.S.; Olivera, B.M.; Yoshikami, D.; Bulaj, G. Structure/function characterization of μ-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J. Biol. Chem. 2007, 282, 30699–30706. [Google Scholar]
- Zhang, M.M.; Han, T.S.; Olivera, B.M.; Bulaj, G.; Yoshikami, D. μ-Conotoxin KIIIA derivatives with divergent affinities versus efficacies in blocking voltage-gated sodium channels. Biochemistry 2010, 49, 4804–4812. [Google Scholar] [CrossRef]
- Green, B.R.; Catlin, P.; Zhang, M.M.; Fiedler, B.; Bayudan, W.; Morrison, A.; Norton, R.S.; Smith, B.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Conotoxins containing nonnatural backbone spacers: cladistic-based design, chemical synthesis, and improved analgesic activity. Chem. Biol. 2007, 14, 399–407. [Google Scholar] [CrossRef]
- Leipold, E.; Markgraf, R.; Miloslavina, A.; Kijas, M.; Schirmeyer, J.; Imhof, D.; Heinemann, S.H. Molecular determinants for the subtype specificity of μ-conotoxin SIIIA targeting neuronal voltage-gated sodium channels. Neuropharmacology 2011, 61, 105–111. [Google Scholar] [CrossRef]
- Yao, S.; Zhang, M.M.; Yoshikami, D.; Azam, L.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure, dynamics, and selectivity of the sodium channel blocker μ-conotoxin SIIIA. Biochemistry 2008, 47, 10940–10949. [Google Scholar] [CrossRef]
- McArthur, J.R.; Ostroumov, V.; Al-Sabi, A.; McMaster, D.; French, R.J. Multiple, distributed interactions of μ-conotoxin PIIIA associated with broad targeting among voltage-gated sodium channels. Biochemistry 2011, 50, 116–124. [Google Scholar]
- Wilson, M.J.; Yoshikami, D.; Azam, L.; Gajewiak, J.; Olivera, B.M.; Bulaj, G.; Zhang, M.M. μ-Conotoxins that differentially block sodium channels Nav1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 10302–10307. [Google Scholar]
- Markgraf, R.; Leipold, E.; Schirmeyer, J.; Paolini-Bertrand, M.; Hartley, O.; Heinemann, S.H. Mechanism and molecular basis for the sodium channel subtype specificity of μ-conopeptide CnIIIC. Br. J. Pharmacol. 2012, 167, 576–586. [Google Scholar] [CrossRef]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms - a rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef]
- McArthur, J.R.; Singh, G.; O'Mara, M.L.; McMaster, D.; Ostroumov, V.; Tieleman, D.P.; French, R.J. Orientation of μ-conotoxin PIIIA in a sodium channel vestibule, based on voltage dependence of its binding. Mol. Pharmacol. 2011, 80, 219–227. [Google Scholar] [CrossRef]
- Li, R.A.; Ennis, I.I.; Vélez, P.; Tomaselli, G.F.; Marbán, E. Novel structural determinants of μ-conotoxin (GIIIB) block in rat skeletal muscle (μ1) Na+ channels. J. Biol. Chem. 2000, 275, 27551–27558. [Google Scholar]
- Choudhary, G.; Aliste, M.; Tieleman, D.P.; French, R.J.; Dudley, S.C., Jr. Docking of μ-conotoxin GIIIA in the sodium channel outer vestibule. Channels (Austin) 2007, 1, 344–352. [Google Scholar]
- Chang, N.S.; French, R.J.; Lipkind, G.M.; Fozzard, H.A.; Dudley, S., Jr. Predominant interactions between μ-conotoxin Arg-13 and the skeletal muscle Na+ channel localized by mutant cycle analysis. Biochemistry 1998, 37, 4407–4419. [Google Scholar] [CrossRef]
- Tietze, A.A.; Tietze, D.; Ohlenschläger, O.; Leipold, E.; Ullrich, F.; Kühl, T.; Mischo, A.; Buntkowsky, G.; Görlach, M.; Heinemann, S.H.; Imhof, D. Structurally diverse μ-conotoxin PIIIA isomers block sodium channel Nav 1.4. Angew. Chem. Int. Ed. Engl. 2012, 51, 4058–4061. [Google Scholar]
- Walewska, A.; Zhang, M.M.; Skalicky, J.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Integrated oxidative folding of cysteine/selenocysteine containing peptides: improving chemical synthesis of conotoxins. Angew. Chem. Int. Ed. Engl. 2009, 48, 2221–2224. [Google Scholar] [CrossRef]
- Besse, D.S.; Fourmy, D.; Diercks, T.; Kessler, H.; Moroder, L. The redox potential of selenocystine in unconstrained cyclic peptides. Angew. Chem. Int. Ed. 1997, 36, 883–885. [Google Scholar] [CrossRef]
- Norton, R.S. μ-Conotoxins as leads in the development of new analgesics. Molecules 2010, 15, 2825–2844. [Google Scholar] [CrossRef]
- Khoo, K.K.; Wilson, M.J.; Smith, B.J.; Zhang, M.M.; Gulyas, J.; Yoshikami, D.; Rivier, J.E.; Bulaj, G.; Norton, R.S. Lactam-stabilized helical analogues of the analgesic μ-conotoxin KIIIA. J. Med. Chem. 2011, 54, 7558–7566. [Google Scholar]
- Stevens, M.; Peigneur, S.; Dyubankova, N.; Lescrinier, E.; Herdewijn, P.; Tytgat, J. Design of bioactive peptides from naturally occurring μ-conotoxin structures. J. Biol. Chem. 2012, 37, 31382–31392. [Google Scholar]
- Khoo, K.K.; Feng, Z.P.; Smith, B.J.; Zhang, M.M.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure of the analgesic μ-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry 2009, 48, 1210–1219. [Google Scholar] [CrossRef]
- Schroeder, C.I.; Adams, D.; Thomas, L.; Alewood, P.F.; Lewis, R.J. N- and C-terminal extensions of μ-conotoxins increase potency and selectivity for neuronal sodium channels. Biopolymers 2012, 98, 161–165. [Google Scholar] [CrossRef]
- Han, T.S.; Zhang, M.M.; Walewska, A.; Gruszczynski, P.; Robertson, C.R.; Cheatham, T.E.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Structurally minimized μ-conotoxin analogues as sodium channel blockers: implications for designing conopeptide-based therapeutics. ChemMedChem. 2009, 4, 406–614. [Google Scholar] [CrossRef]
- Stürzebecher, A.S.; Hu, J.; Smith, E.S.; Frahm, S.; Santos-Torres, J.; Kampfrath, B.; Auer, S.; Lewin, G.R.; Ibañez-Tallon, I. An in vivo tethered toxin approach for the cell-autonomous inactivation of voltage-gated sodium channel currents in nociceptors. J. Physiol. 2010, 588, 1695–1707. [Google Scholar]
- Bulaj, G.; Zhang, M.M.; Green, B.R.; Fiedler, B.; Layer, R.T.; Wei, S.; Nielsen, J.S.; Low, S.J.; Klein, B.D.; Wagstaff, J.D.; Chicoine, L.; Harty, T.P.; Terlau, H.; Yoshikami, D.; Olivera, B.M. Synthetic μO-conotoxin MrVIB blocks TTX-resistant sodium channel Nav1.8 and has a long-lasting analgesic activity. Biochemistry 2006, 45, 7404–7414. [Google Scholar]
- Daly, N.L.; Ekberg, J.A.; Thomas, L.; Adams, D.J.; Lewis, R.J.; Craik, D.J. Structures of μO-conotoxins from Conus marmoreus. Inhibitors of tetrodotoxin (TTX)-sensitive and TTX-resistant sodium channels in mammalian sensory neurons. J. Biol. Chem. 2004, 279, 25774–25782. [Google Scholar]
- Terlau, H.; Stocker, M.; Shon, K.J.; McIntosh, J.M.; Olivera, B.M. μO-conotoxin MrVIA inhibits mammalian sodium channels, but not through site I. J. Neurophysiol. 1996, 76, 1423–1429. [Google Scholar]
- Wilson, M.J.; Zhang, M.M.; Azam, L.; Olivera, B.M.; Bulaj, G.; Yoshikami, D. Navβ subunits modulate the inhibition of Nav1.8 by the analgesic gating modifier μO-conotoxin MrVIB. J. Pharmacol. Exp. Ther. 2011, 338, 687–693. [Google Scholar]
- Zorn, S.; Leipold, E.; Hansel, A.; Bulaj, G.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. The μO-conotoxin MrVIA inhibits voltage-gated sodium channels by associating with domain-3. FEBS Lett. 2006, 580, 1360–1364. [Google Scholar] [CrossRef]
- Knapp, O.; Nevin, S.; Yasuda, T.; Lawrence, N.; Lewis, R.J.; Adams, D.J. Biophysical properties of Nav1.8/Nav1.2 chimeras and inhibition by μO-conotoxin MrVIB. Br. J. Pharmacol. 2012, 166, 2148–2160. [Google Scholar]
- Bosmans, F.; Puopolo, M.; Martin-Eauclaire, M.F.; Bean, B.P.; Swartz, K.J. Functional properties and toxin pharmacology of a dorsal root ganglion sodium channel viewed through its voltage sensors. J. Gen. Physiol. 2011, 138, 59–72. [Google Scholar] [CrossRef]
- Bosmans, F.; Martin-Eauclaire, M.F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef]
- Bosmans, F.; Swartz, K.J. Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 2010, 31, 175–182. [Google Scholar] [CrossRef]
- Vetter, I.; Dekan, Z.; Knapp, O.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Isolation, characterization and total regioselective synthesis of the novel μO-conotoxin MfVIA from Conus magnificus that targets voltage-gated sodium channels. Biochem. Pharmacol. 2012, 4, 540–548. [Google Scholar]
- McIntosh, J.M.; Hasson, A.; Spira, M.E.; Gray, W.R.; Li, W.; Marsh, M.; Hillyard, D.R.; Olivera, B.M. A new family of conotoxins that blocks voltage-gated sodium channels. J. Biol. Chem. 1995, 270, 16796–16802. [Google Scholar]
- Pi, C.; Lui, J.; Wang, L.; Jiang, X.; Liu, Y.; Peng, C.; Chen, S.; Xu, A. Soluble expression, purification and functional identification of a disulfide-rich conotoxin derived from Conus litteratus. J. Biotechnol. 2007, 128, 184–193. [Google Scholar] [CrossRef]
- Wang, L.; Pi, C.; Liu, J.; Chen, S.; Peng, C.; Sun, D.; Zhou, M.; Xiang, H.; Ren, Z.; Xu, A. Identification and characterization of a novel O-superfamily conotoxin from Conus litteratus. J. Pept. Sci. 2008, 14, 1077–1083. [Google Scholar] [CrossRef]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef]
- de Araujo, A.D.; Callaghan, B.; Nevin, S.T.; Daly, N.L.; Craik, D.J.; Moretta, M.; Hopping, G.; Christie, M.J.; Adams, D.J.; Alewood, P.F. Total synthesis of the analgesic conotoxin MrVIB through selenocysteine-assisted folding. Angew. Chem. Int. Ed. Engl. 2011, 50, 6527–6529. [Google Scholar]
- Ekberg, J.; Adams, D.J. Neuronal voltage-gated sodium channel subtypes: key roles in inflammatory and neuropathic pain. Int. J. Biochem. Cell. Biol. 2006, 38, 2005–2010. [Google Scholar]
- Miljanich, G.P. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef]
- Staats, P.S.; Yearwood, T.; Charapata, S.G.; Presley, R.W.; Wallace, M.S.; Byas-Smith, M.; Fisher, R.; Bryce, D.A.; Mangieri, E.A.; Luther, R.R.; Mayo, M.; McGuire, D.; Ellis, D. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. JAMA 2004, 291, 63–70. [Google Scholar]
- Stix, G. A toxin against pain. Sci. Am. 2005, 292, 70–75. [Google Scholar]
- Edgerton, G.B.; Blumenthal, K.M.; Hanck, D.A. Inhibition of the activation pathway of the T-type calcium channel Cav3.1 by ProTxII. Toxicon 2010, 56, 624–636. [Google Scholar] [CrossRef]
- Sokolov, S.; Kraus, R.L.; Scheuer, T.; Catterall, W.A. Inhibition of sodium channel gating by trapping the domain II voltage sensor with protoxin II. Mol. Pharmacol. 2008, 73, 1020–1028. [Google Scholar]
- Siemens, J.; Zhou, S.; Piskorowski, R.; Nikai, T.; Lumpkin, E.A.; Basbaum, A.I.; King, D.; Julius, D. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006, 444, 208–212. [Google Scholar]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 2010, 49, 6545–6548. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Knapp, O.; McArthur, J.R.; Adams, D.J. Conotoxins Targeting Neuronal Voltage-Gated Sodium Channel Subtypes: Potential Analgesics? Toxins 2012, 4, 1236-1260. https://doi.org/10.3390/toxins4111236
Knapp O, McArthur JR, Adams DJ. Conotoxins Targeting Neuronal Voltage-Gated Sodium Channel Subtypes: Potential Analgesics? Toxins. 2012; 4(11):1236-1260. https://doi.org/10.3390/toxins4111236
Chicago/Turabian StyleKnapp, Oliver, Jeffrey R. McArthur, and David J. Adams. 2012. "Conotoxins Targeting Neuronal Voltage-Gated Sodium Channel Subtypes: Potential Analgesics?" Toxins 4, no. 11: 1236-1260. https://doi.org/10.3390/toxins4111236
APA StyleKnapp, O., McArthur, J. R., & Adams, D. J. (2012). Conotoxins Targeting Neuronal Voltage-Gated Sodium Channel Subtypes: Potential Analgesics? Toxins, 4(11), 1236-1260. https://doi.org/10.3390/toxins4111236