Comparative Antitumor Effect of Preventive versus Therapeutic Vaccines Employing B16 Melanoma Cells Genetically Modified to Express GM-CSF and B7.2 in a Murine Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
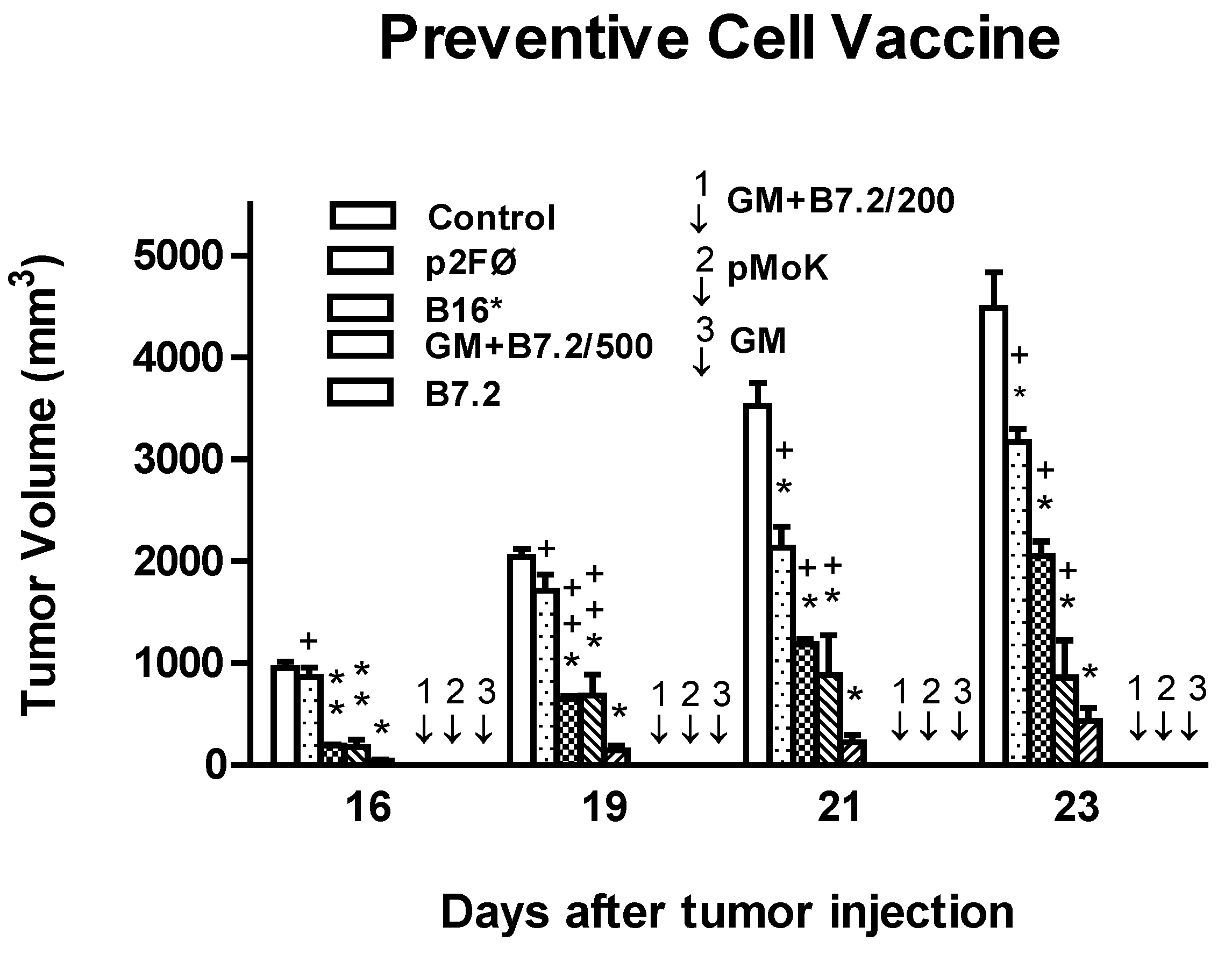
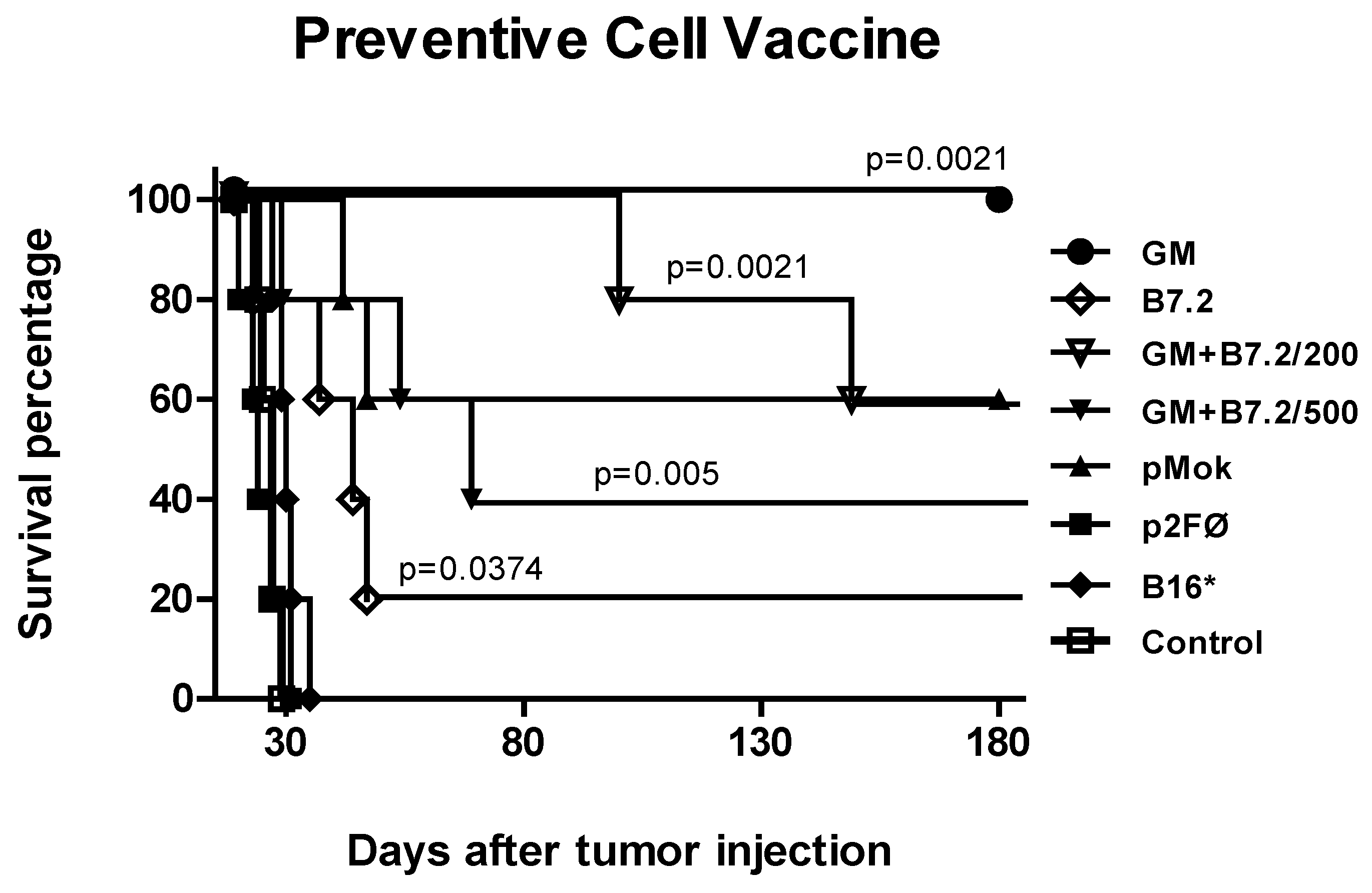
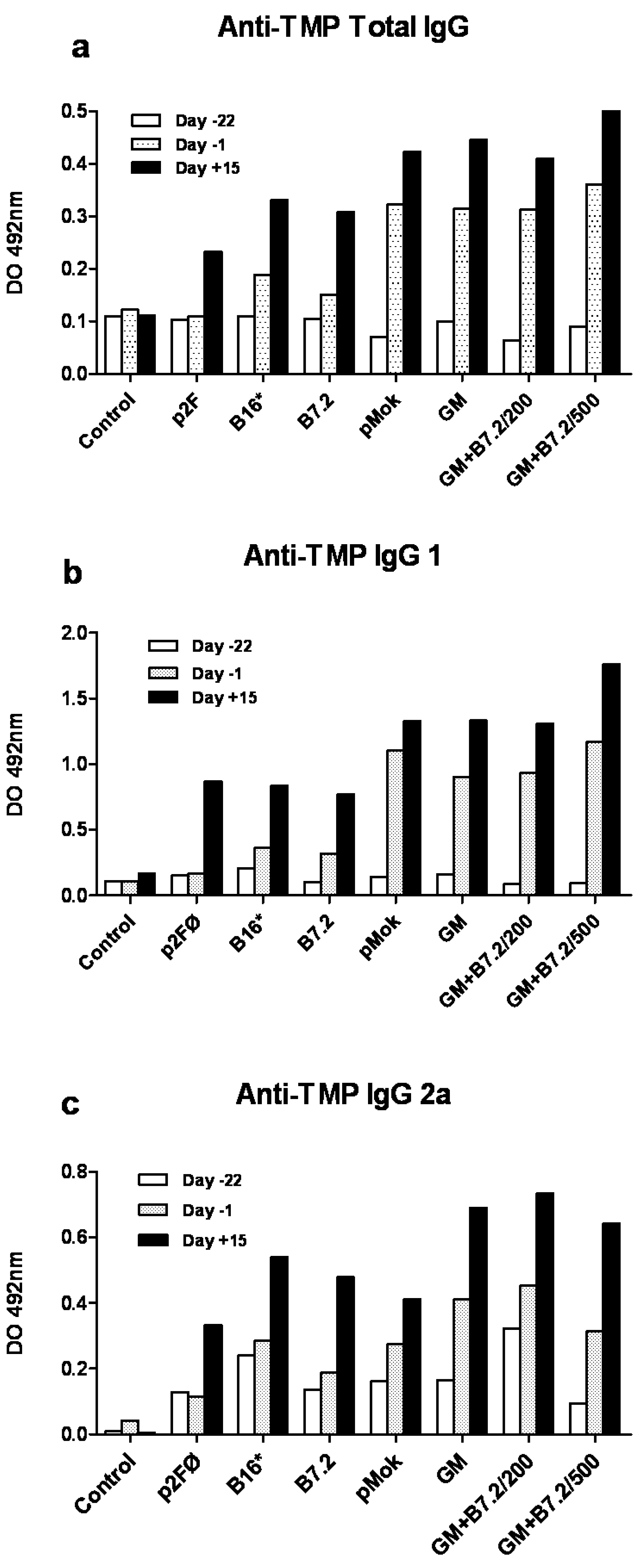
2.1. Preventive Vaccination


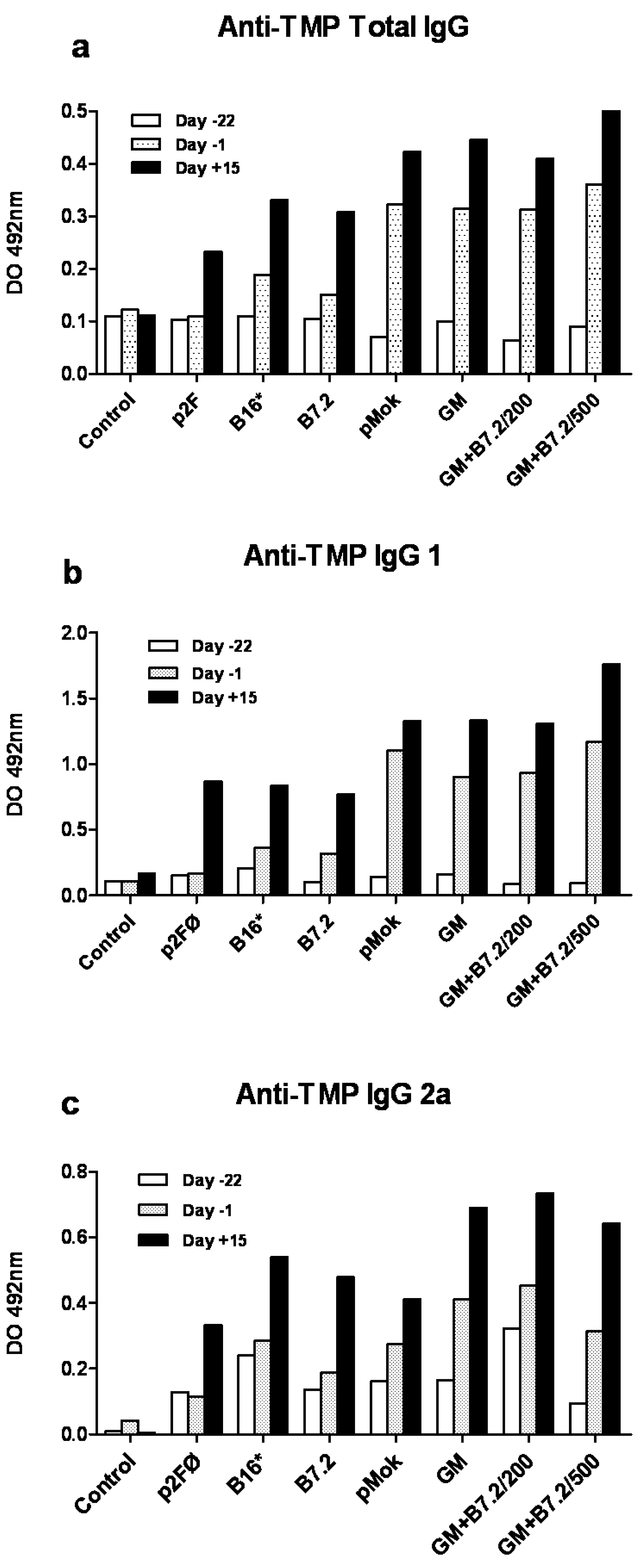
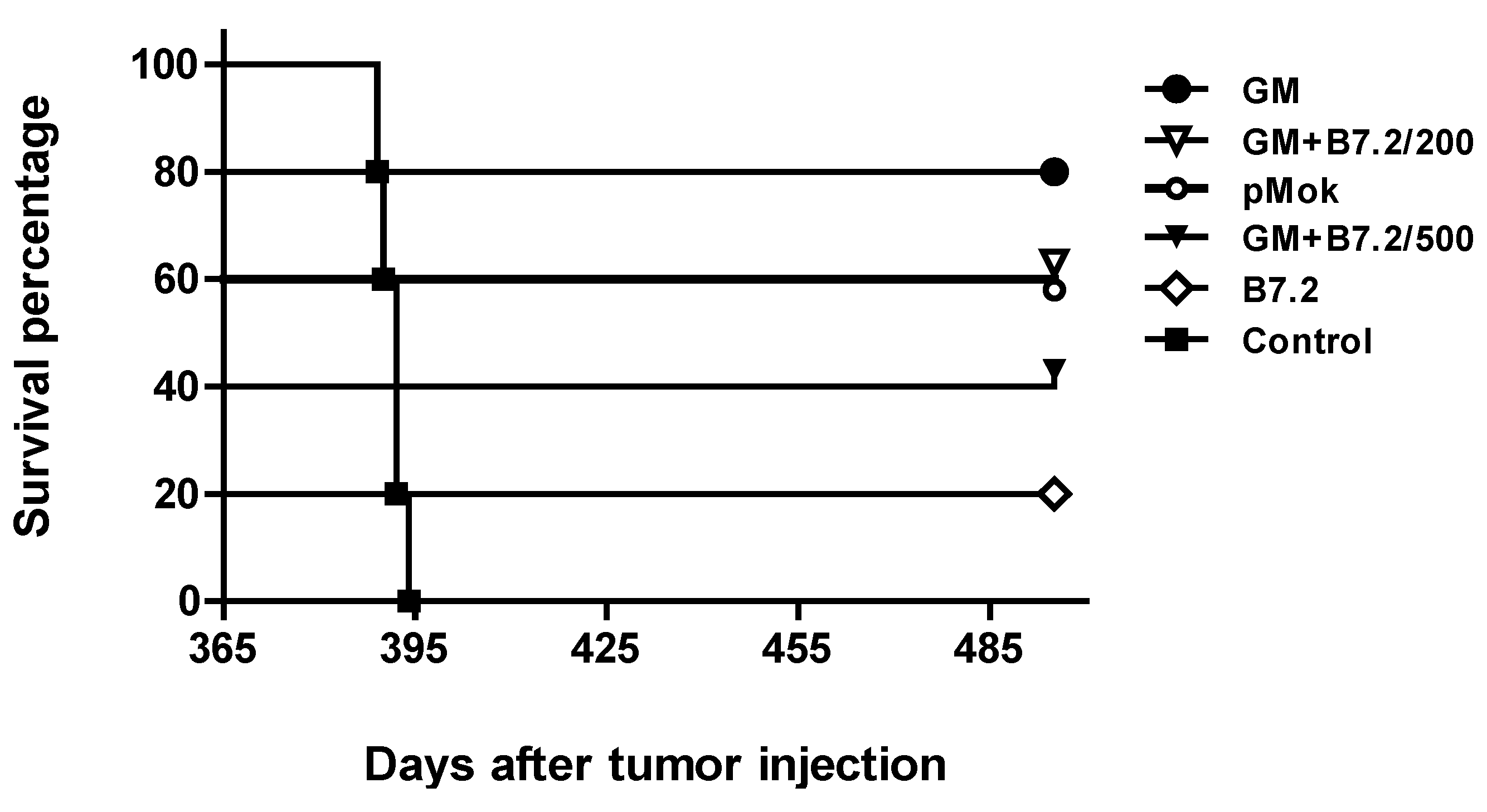

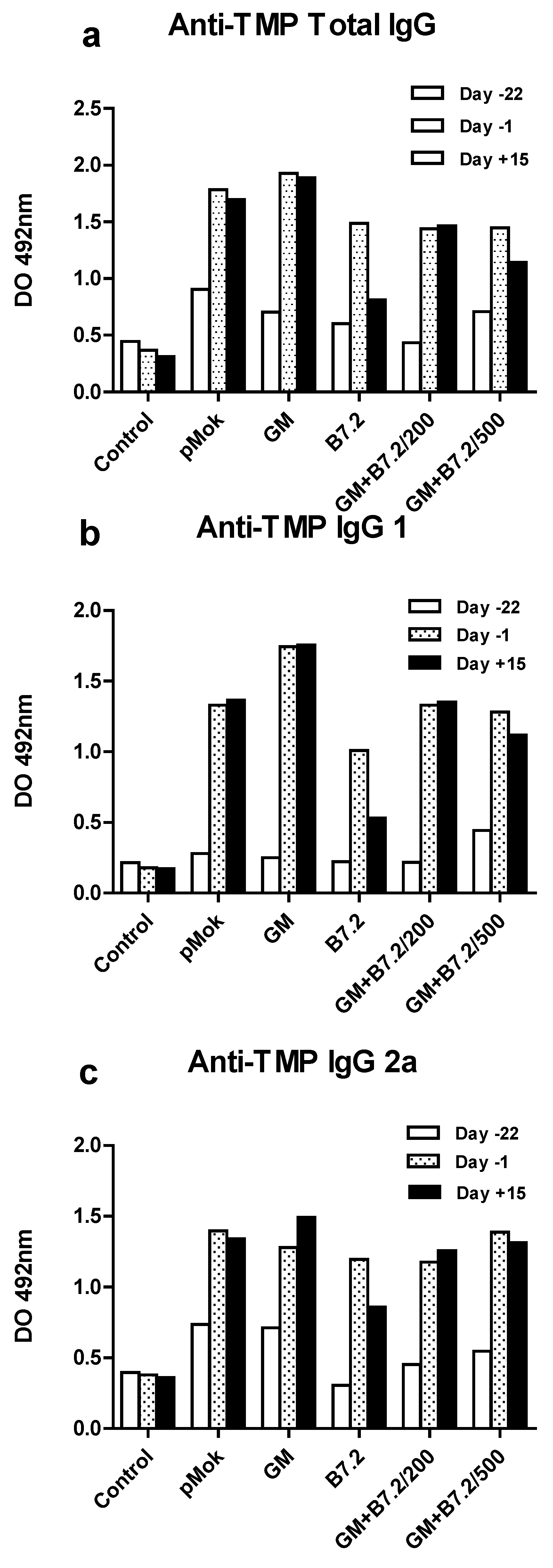
2.2. Therapeutic Vaccination
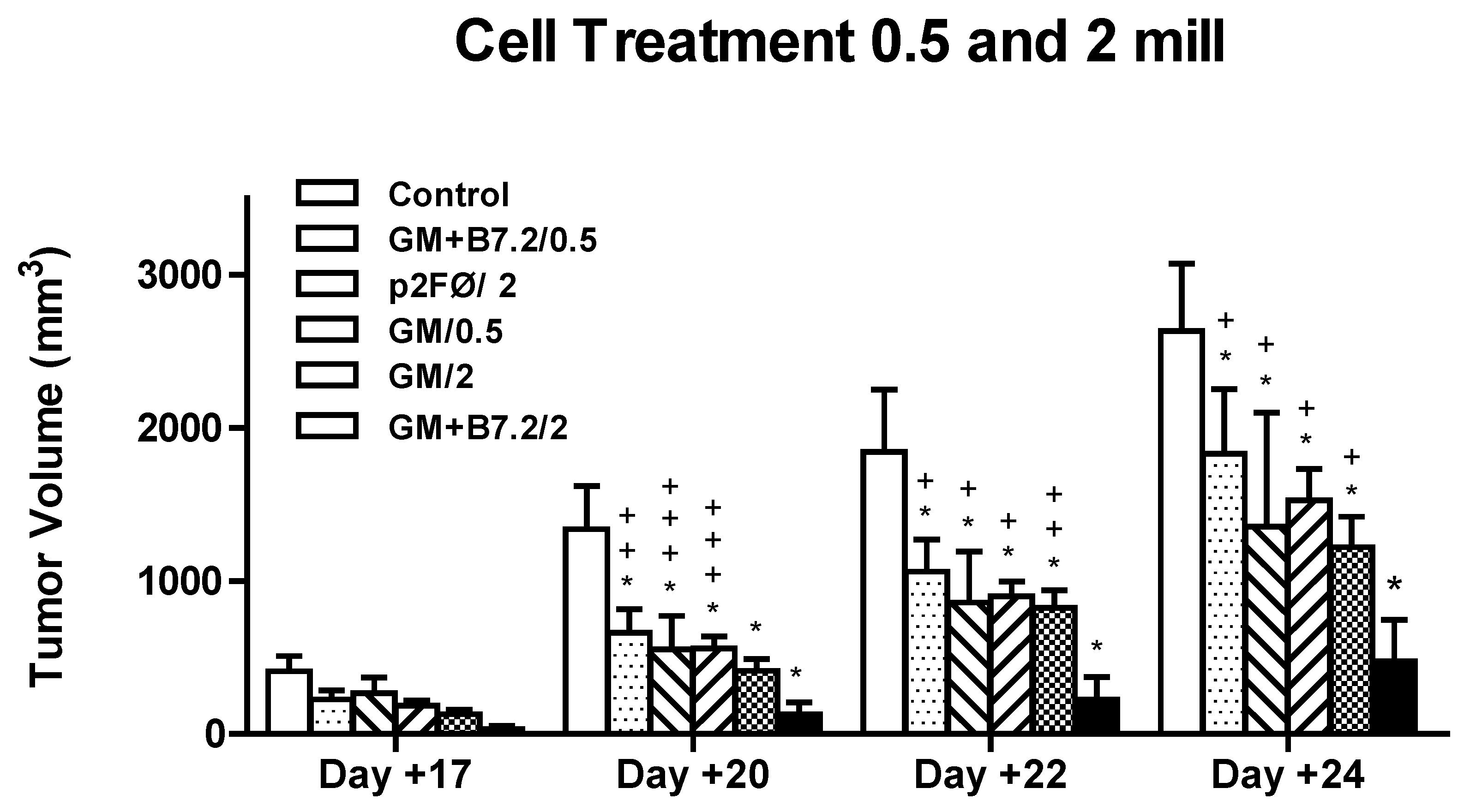
2.2.1. Therapeutic Antitumor Efficacy of Cellular Therapy at Low Doses
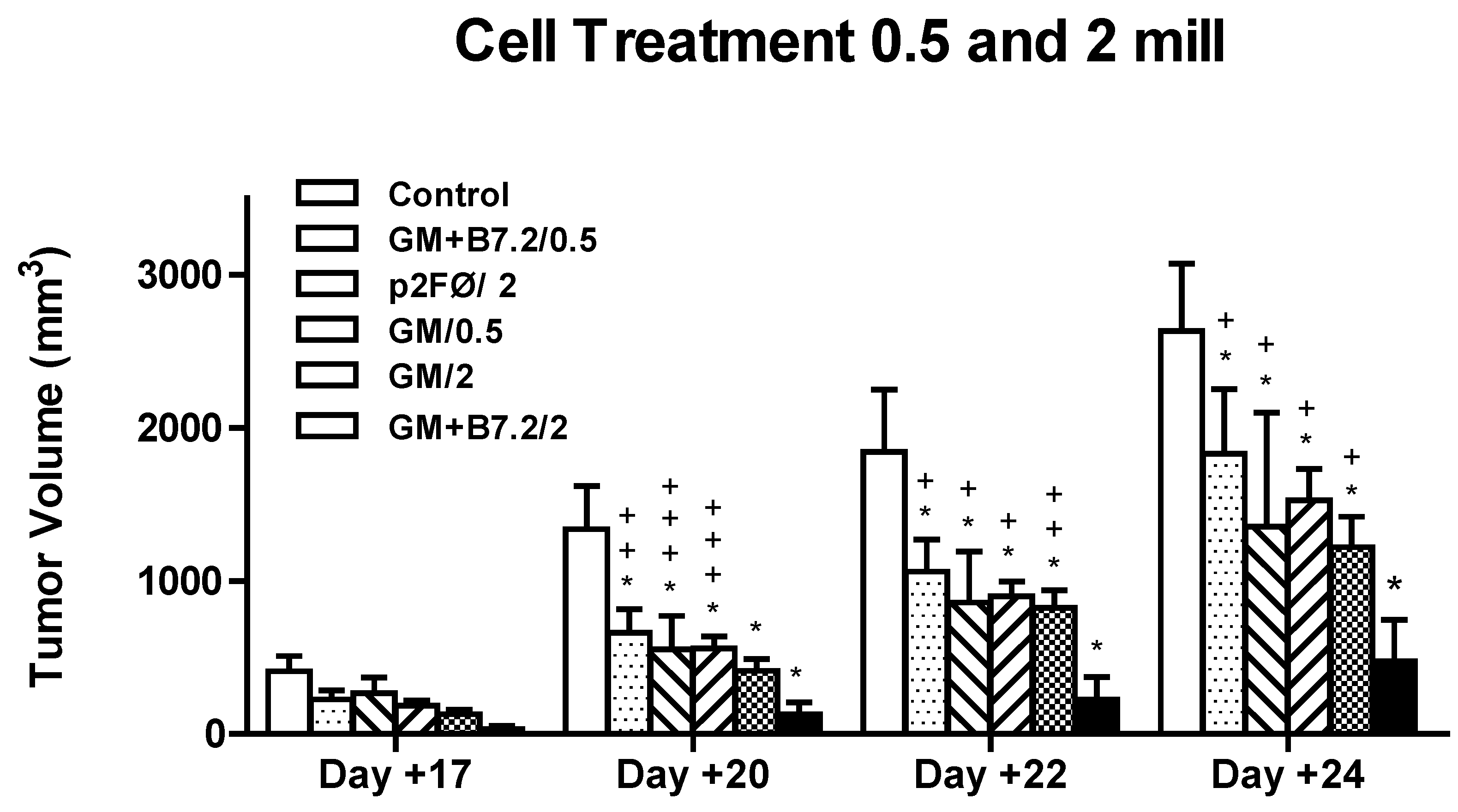
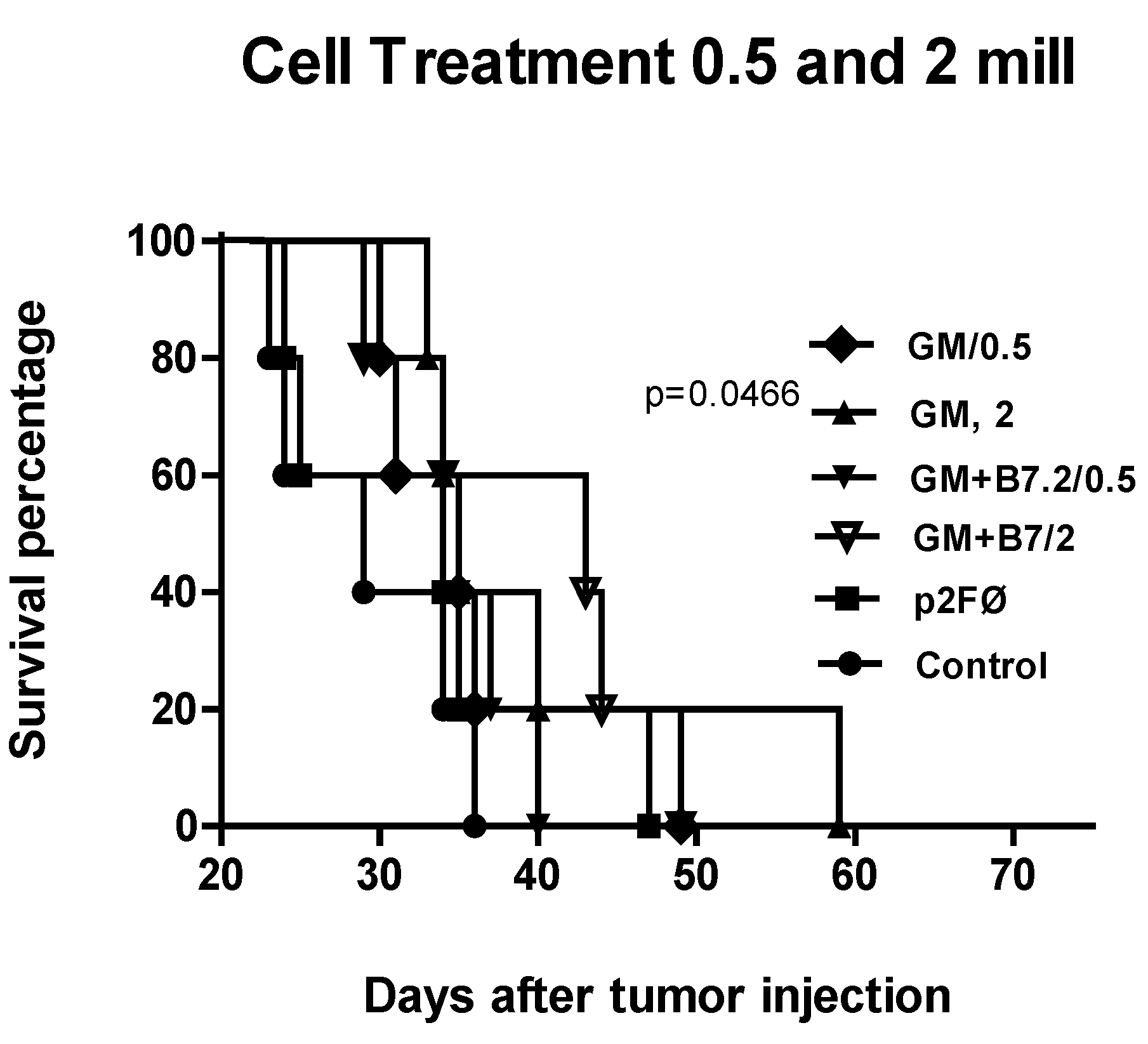

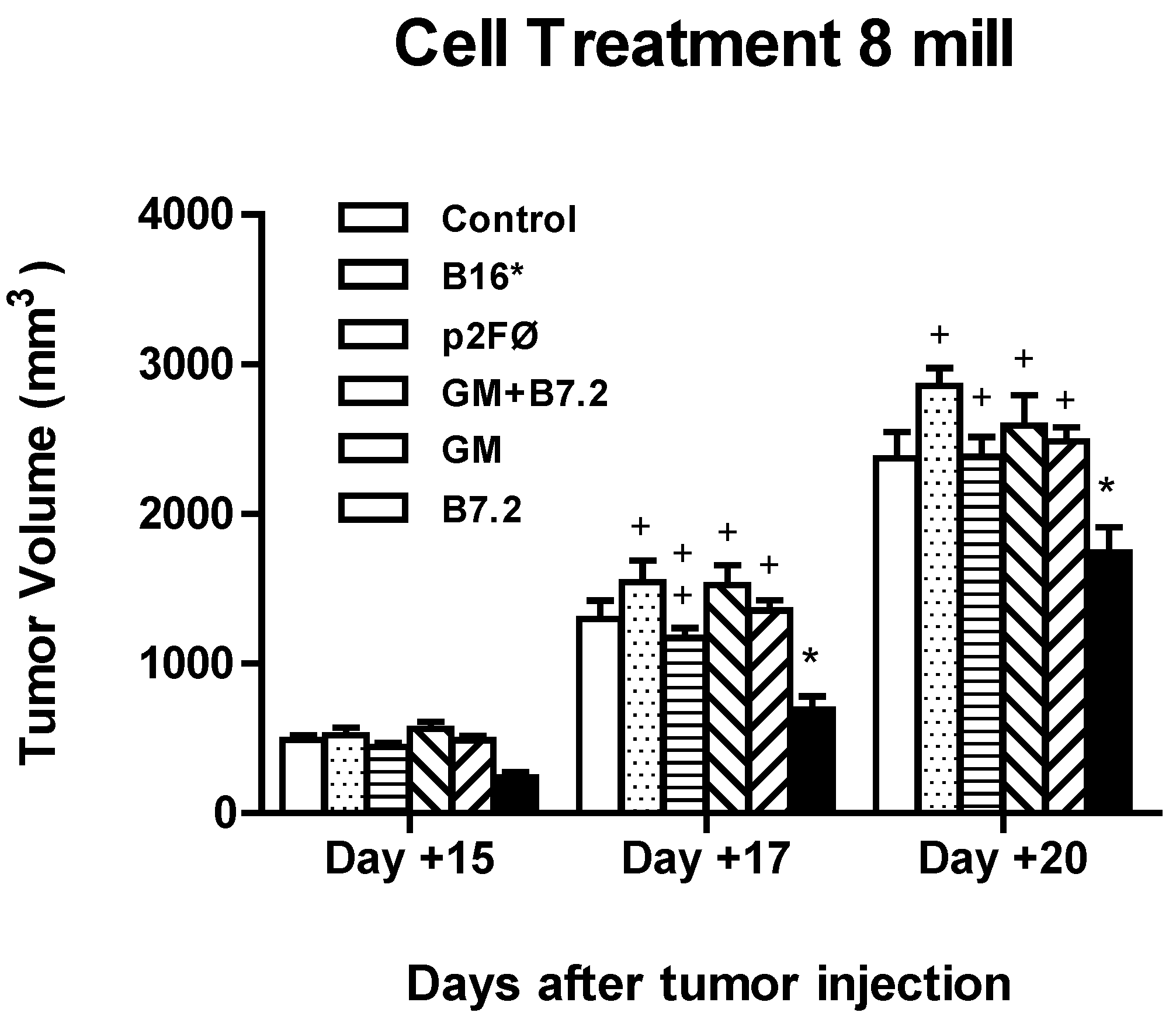
2.2.2. Therapeutic Antitumor Efficacy at High Cell Dose
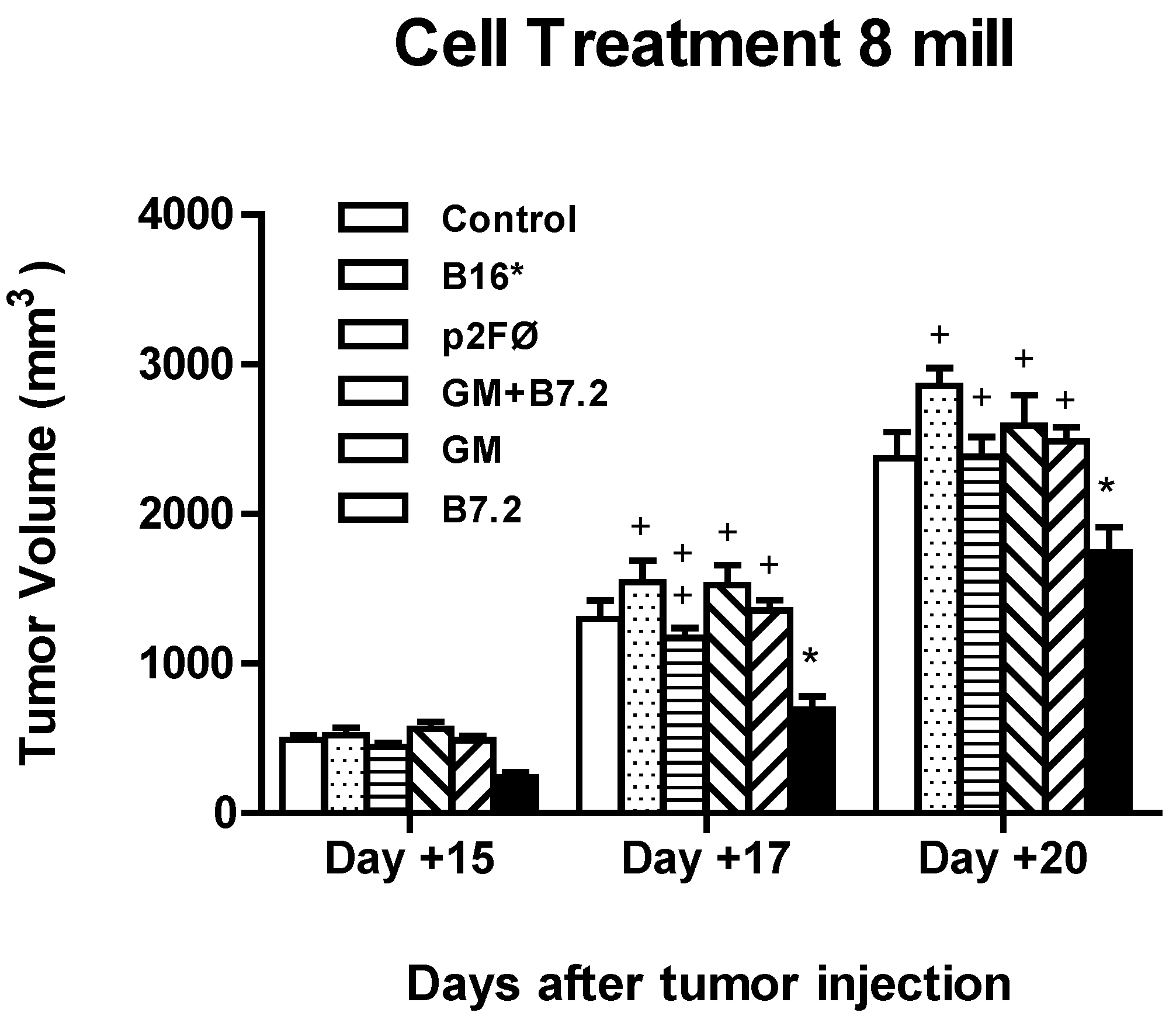
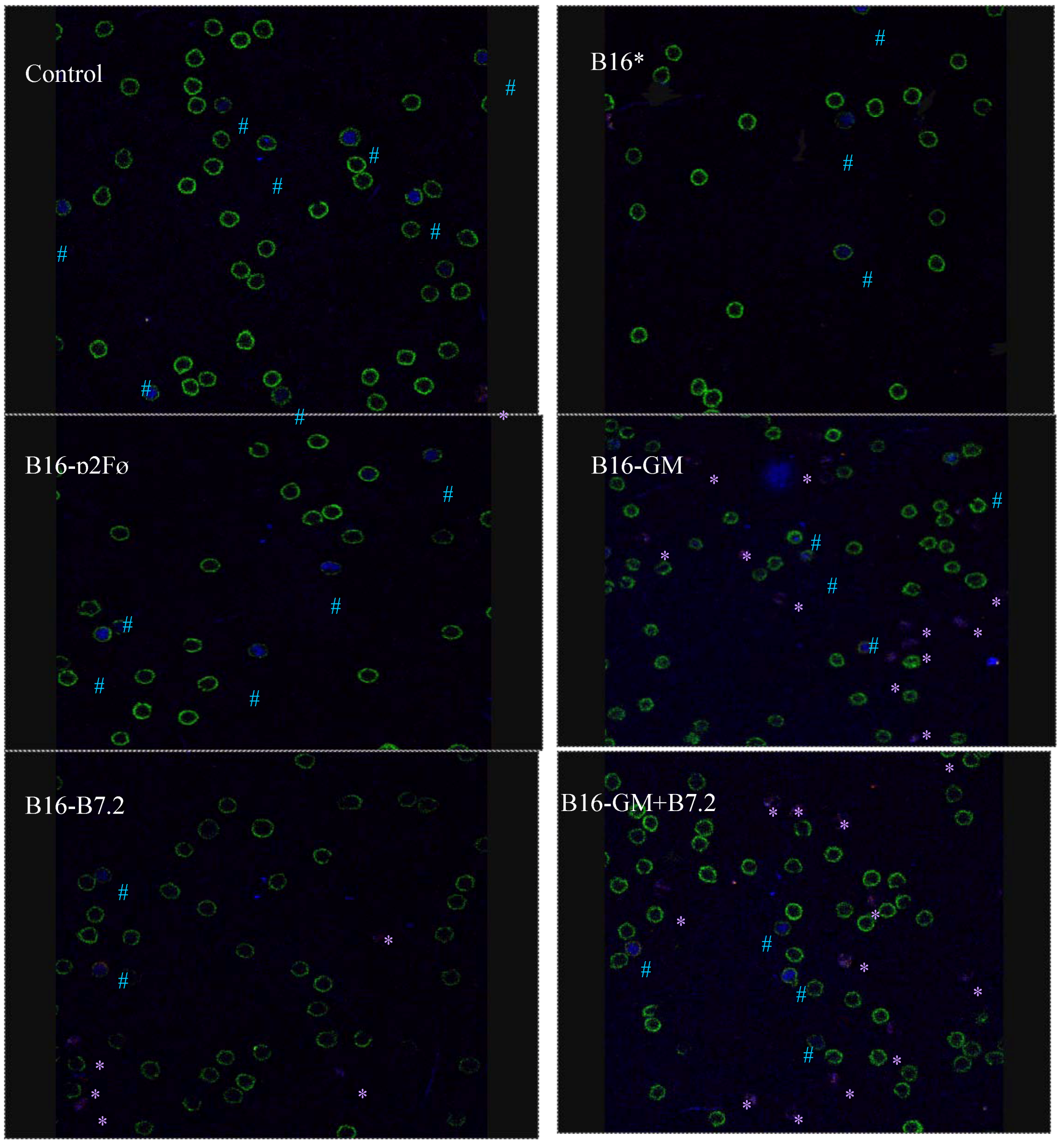
3. Experimental Section
3.1. Plasmids
3.2. Cells and Transfection Procedure
3.3. ELISA of m-GMCSF
3.4. Preventive Vaccination Procedure
3.5. Therapeutic Vaccination Procedure
3.6. Tumor Growth Measurement and Survival
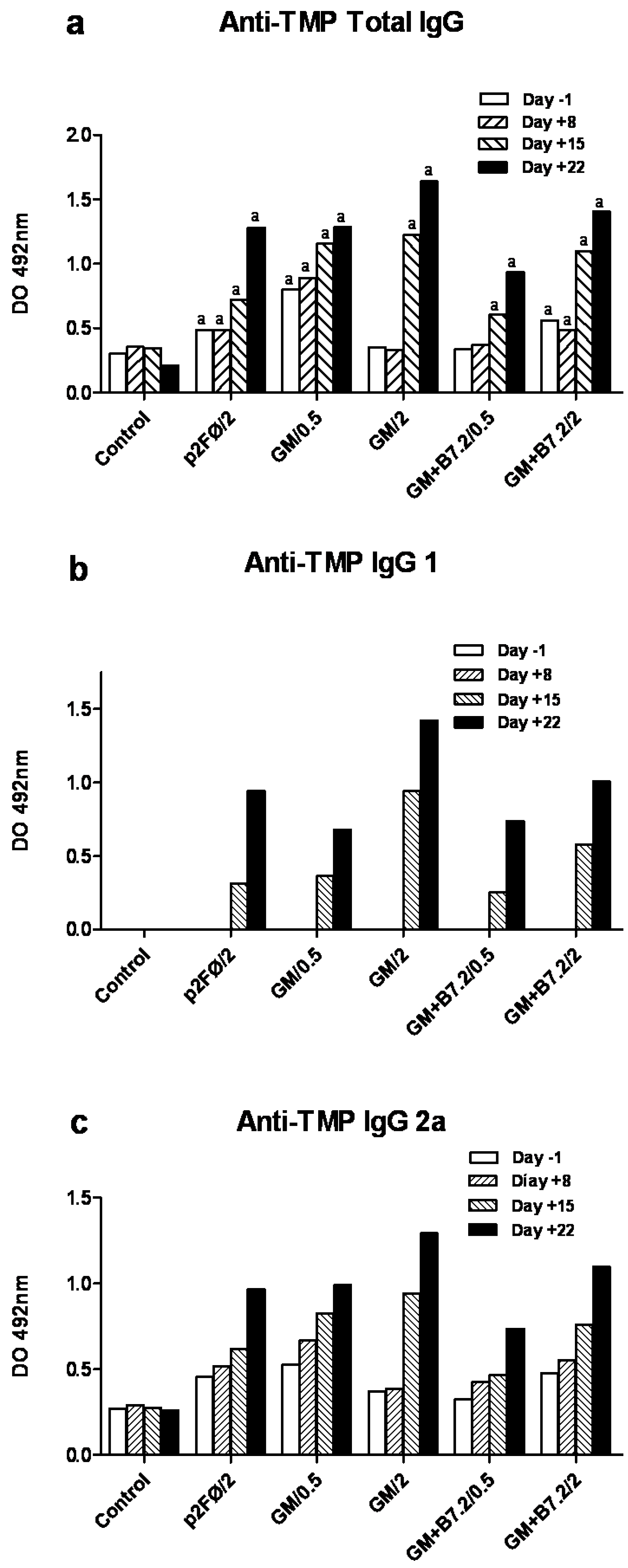
3.7. Specific Anti-TMP IgG ELISA
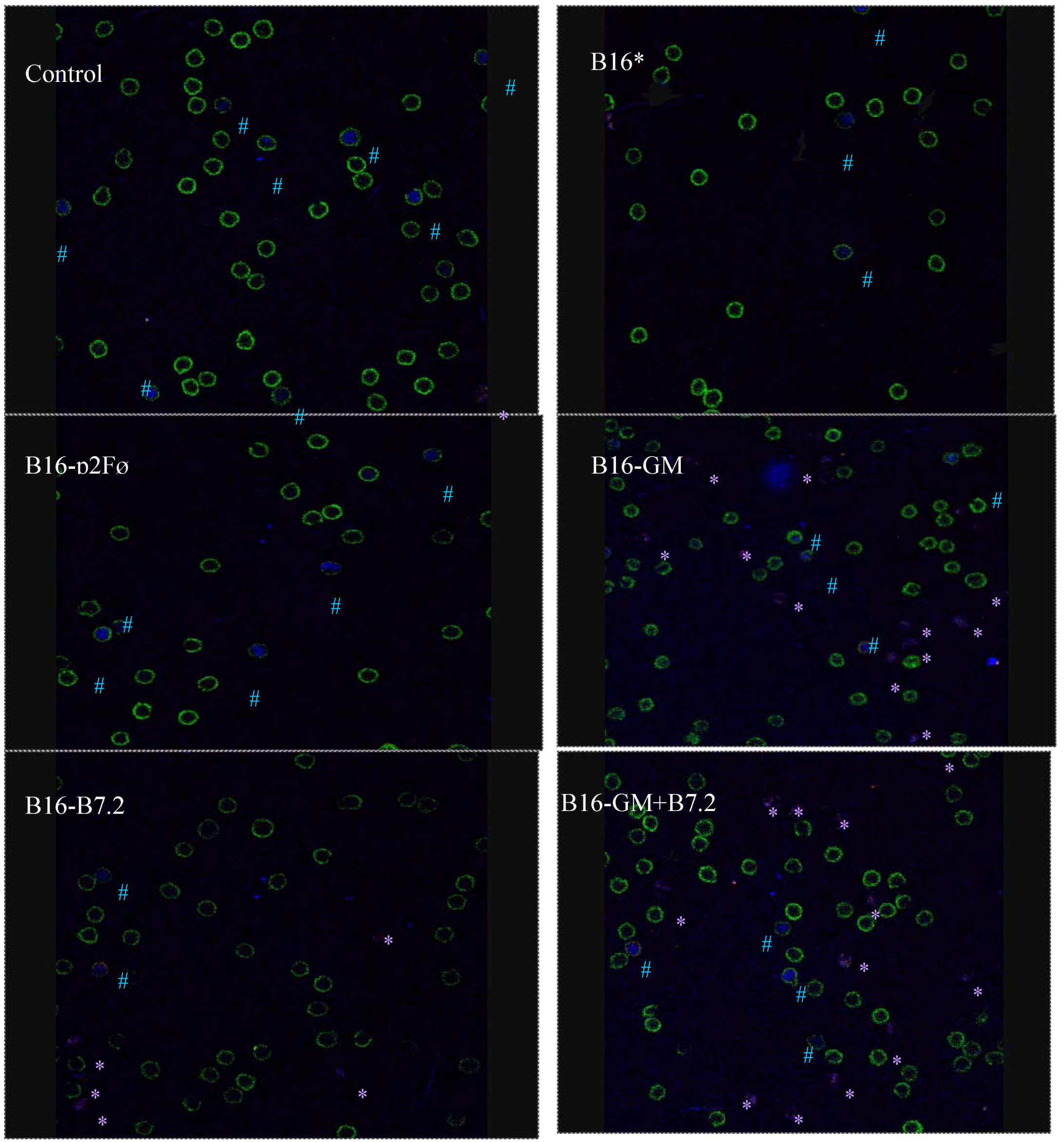
3.8. Characterization of Regulatory T Cells by Confocal Microscopy
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Kim, S.; Carew, J.F.; Kooby, D.A.; Shields, J.; Entwisle, C.; Patel, S.; Shah, J.P.; Fong, Y. Combination gene therapy using multiple immunomodulatory genes transferred by a defective infectious single-cycle herpes virus in squamous cell cancer. Cancer Gene Ther. 2000, 7, 1279–1285. [Google Scholar]
- Veelken, H.; Mackensen, A.; Lahn, M.; Kohler, G.; Becker, D.; Franke, B.; Brennscheidt, U.; Kulmburg, P.; Rosenthal, F.M.; Keller, H.; et al. A phase-I clinical study of autologous tumor cells plus interleukin-2-gene-transfected allogeneic fibroblasts as a vaccine in patients with cancer. Int. J. Cancer 1997, 70, 269–277. [Google Scholar] [CrossRef]
- Palmer, K.; Moore, J.; Everard, M.; Harris, J.D.; Rodgers, S.; Rees, R.C.; Murray, A.K.; Mascari, R.; Kirkwood, J.; Riches, P.G.; et al. Gene therapy with autologous, interleukin 2-secreting tumor cells in patients with malignant melanoma. Hum. Gene Ther. 1999, 10, 1261–1268. [Google Scholar] [CrossRef]
- Wittig, B.; Marten, A.; Dorbic, T.; Weineck, S.; Min, H.; Niemitz, S.; Trojaneck, B.; Flieger, D.; Kruopis, S.; Albers, A.; et al. Therapeutic vaccination against metastatic carcinoma by expression-modulated and immunomodified autologous tumor cells: A first clinical phase I/II trial. Hum. Gene Ther. 2001, 12, 267–278. [Google Scholar] [CrossRef]
- Moller, P.; Sun, Y.; Dorbic, T.; Alijagic, S.; Makki, A.; Jurgovsky, K.; Schroff, M.; Henz, B.M.; Wittig, B.; Schadendorf, D. Vaccination with IL-7 gene-modified autologous melanoma cells can enhance the anti-melanoma lytic activity in peripheral blood of patients with a good clinical performance status: A clinical phase I study. Br. J. Cancer 1998, 77, 1907–1916. [Google Scholar] [CrossRef]
- Maio, M.; Fonsatti, E.; Lamaj, E.; Altomonte, M.; Cattarossi, I.; Santantonio, C.; Melani, C.; Belli, F.; Arienti, F.; Colombo, M.P.; et al. Vaccination of stage IV patients with allogeneic IL-4- or IL-2-gene-transduced melanoma cells generates functional antibodies against vaccinating and autologous melanoma cells. Cancer Immunol. Immunother. 2002, 51, 9–14. [Google Scholar] [CrossRef]
- Arienti, F.; Belli, F.; Napolitano, F.; Sule-Suso, J.; Mazzocchi, A.; Gallino, G.F.; Cattelan, A.; Santantonio, C.; Rivoltini, L.; Melani, C.; et al. Vaccination of melanoma patients with interleukin 4 gene-transduced allogeneic melanoma cells. Hum. Gene Ther. 1999, 10, 2907–2916. [Google Scholar] [CrossRef]
- Bowman, L.C.; Grossmann, M.; Rill, D.; Brown, M.; Zhong, W.Y.; Alexander, B.; Leimig, T.; Coustan-Smith, E.; Campana, D.; Jenkins, J.; et al. Interleukin-2 gene-modified allogeneic tumor cells for treatment of relapsed neuroblastoma. Hum. Gene Ther. 1998, 9, 1303–1311. [Google Scholar] [CrossRef]
- Belli, F.; Arienti, F.; Sule-Suso, J.; Clemente, C.; Mascheroni, L.; Cattelan, A.; Santantonio, C.; Gallino, G.F.; Melani, C.; Rao, S.; et al. Active immunization of metastatic melanoma patients with interleukin-2-transduced allogeneic melanoma cells: Evaluation of efficacy and tolerability. Cancer Immunol. Immunother. 1997, 44, 197–203. [Google Scholar] [CrossRef]
- Olivares, J.; Kumar, P.; Yu, Y.; Maples, P.B.; Senzer, N.; Bedell, C.; Barve, M.; Tong, A.; Pappen, B.O.; Kuhn, J.; et al. Phase I trial of TGF-β2 antisense GM-CSF gene-modified autologous tumor cell (TAG) vaccine. Clin. Cancer Res. 2011, 1, 183–192. [Google Scholar]
- Agarwalla, P.; Barnard, Z.; Fecci, P.; Dranoff, G.; Curry, W.T., Jr. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J. Immunother. 2012, 35, 385–389. [Google Scholar] [CrossRef]
- Jaffee, E.M.; Abrams, R.; Cameron, J.; Donehower, R.; Duerr, M.; Gossett, J.; Greten, T.F.; Grochow, L.; Hruban, R.; Kern, S.; Lillemoe, K.D.; O’Reilly, S.; et al. A phase I clinical trial of lethally irradiated allogeneic pancreatic tumor cells transfected with the GM-CSF gene for the treatment of pancreatic adenocarcinoma. Hum. Gene Ther. 1998, 9, 1951–1971. [Google Scholar] [CrossRef]
- Ojima, T.; Iwahashi, M.; Nakamura, M.; Matsuda, K.; Naka, T.; Nakamori, M.; Ueda, K.; Ishida, K.; Yamaue, H. The boosting effect of co-transduction with cytokine genes on cancer vaccine therapy using genetically modified dendritic cells expressing tumor-associated antigen. Int. J. Oncol. 2006, 289, 47–53. [Google Scholar]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete granulocyte—Macrophage colony-stimulating factor (GM-CSF) stimulates potent, specific, and long-lasting antitumor immunity in multiple murine tumor model systems, including malignant melanoma. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar]
- Parmiani, G.; Castelli, C.; Pilla, L.; Santinami, M.; Colombo, M.P.; Rivoltini, L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann. Oncol. 2007, 18, 226–232. [Google Scholar]
- Curiel-Lewandrowski, C.; Mahnke, K.; Labeur, M.; Roters, B.; Schmidt, W.; Granstein, R.D.; Luger, T.A.; Schwarz, T.; Grabbe, S. Transfection of immature murine bone marrow-derived dendritic cells with the granulocyte-macrophage colony-stimulating factor gene potently enhances their in vivo antigen-presenting capacity. J. Immunol. 1999, 163, 174–183. [Google Scholar]
- Dunussi-Joannopoulos, K.; Dranoff, G.; Weinstein, H.J.; Ferrara, J.L.; Bierer, B.E.; Croop, J.M. Gene immunotherapy in murine acute myeloid leukemia: Granulocyte-macrophage colony-stimulating factor tumor cell vaccines elicit more potent antitumor immunity compared with B7 family and other cytokine vaccines. Blood 1998, 91, 222–230. [Google Scholar]
- Soiffer, R.; Hodi, F.S.; Haluska, F.; Jung, K.; Gillessen, S.; Singer, S.; Tanabe, K.; Duda, R.; Mentzer, S.; Jaklitsch, M.; et al. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J. Clin. Oncol. 2003, 21, 3343–3350. [Google Scholar]
- Nemunaitis, J.; Sterman, D.; Jablons, D.; Smith, J.W.; Fox, B.; Maples, P.; Hamilton, S.; Borellini, F.; Lin, A.; Morali, S.; et al. Granulocyte-macrophage colony-stimulating factor gene-modified autologous tumor vaccines in non-small-cell lung cancer. J. Natl. Cancer Inst. 2004, 96, 326–331. [Google Scholar] [CrossRef]
- Salgia, R.; Lynch, T.; Skarin, A.; Lucca, J.; Lynch, C.; Jung, K.; Hodi, F.S.; Jaklitsch, M.; Mentzer, S.; Swanson, S.; et al. Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor augments antitumor immunity in some patients with metastatic non-small-cell lung carcinoma. J. Clin. Oncol. 2003, 21, 624–630. [Google Scholar]
- Soiffer, R.; Lynch, T.; Mihm, M.; Jung, K.; Rhuda, C.; Schmollinger, J.C.; Hodi, F.S.; Liebster, L.; Lam, P.; Mentzer, S.; et al. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc. Natl. Acad. Sci.USA 1998, 95, 13141–13146. [Google Scholar]
- Small, E.J.; Sacks, N.; Nemunaitis, J.; Urba, W.J.; Dula, E.; Centeno, A.S.; Nelson, W.G.; Ando, D.; Howard, C.; Borellini, F.; et al. Granulocyte macrophage colony-stimulating factor—Secreting allogeneic cellular immunotherapy for hormone-refractory prostate cancer. Clin. Cancer Res. 2007, 13, 3883–3891. [Google Scholar] [CrossRef]
- Jaffee, E.M.; Hruban, R.H.; Biedrzycki, B.; Laheru, D.; Schepers, K.; Sauter, P.R.; Goemann, M.; Coleman, J.; Grochow, L.; Donehower, R.C.; et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor—Secreting tumor vaccine for pancreatic cancer: A phase I trial of safety and immune activation. J. Clin. Oncol. 2001, 19, 145–156. [Google Scholar]
- Borrello, I.; Pardoll, D. GM-CSF-based cellular vaccines: A review of the clinical experience. Cytokine Growth Factor Rev. 2002, 13, 185–193. [Google Scholar] [CrossRef]
- Moret-Tatay, I.; Diaz, J.; Marco, F.M.; Crespo, A.; Aliño, S.F. Complete tumor prevention by engineered tumor cell vaccines employing nonviral vectors. Cancer Gene Ther. 2003, 10, 887–897. [Google Scholar] [CrossRef]
- Moret-Tatay, I.; Sanmartín, I.; Marco, F.M.; Díaz, J.; Aliño, S.F. Nonviral therapeutic cell vaccine mediates potent antitumor effects. Vaccine 2006, 24, 3937–3945. [Google Scholar]
- Herrero, M.J.; Botella, R.; Dasí, F.; Algás, R.; Sánchez, M.; Aliño, S.F. Antigens and cytokine genes in antitumor vaccines: The importance of the temporal delivery sequence in antitumor signals. Ann. N. Y. Acad. Sci. 2006, 1091, 412–424. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, X.; Li, J.; Hu, Z.; Zhou, D.; Gao, J.; Tan, W. A novel therapeutic vaccine of mouse GM-CSF surface modified MB49 cells against metastatic bladder cancer. J. Urol. 2012, 187, 1071–1079. [Google Scholar]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef]
- Chong, H.; Todryk, S.; Hutchinson, G.; Hart, I.R.; Vile, R.G. Tumor cell expression of B7 costimulatory molecules and interleukin-12 or granulocyte-macrophage colony-stimulating factor induces a local antitumor response and may generate systemic protective immunity. Gene Ther. 1998, 5, 223–232. [Google Scholar]
- Kim, K.Y.; Kang, M.A.; Nam, M.J. Enhancement of natural killer cell-mediated cytotoxicity by coexpression of GM-CSF/B70 in hepatoma. Cancer Lett. 2001, 166, 33–40. [Google Scholar] [CrossRef]
- Mukherjee, S.; Nelson, D.; Loh, S.; van Bruggen, I.; Palmer, L.J.; Leong, C.; Garlepp, M.J.; Robinson, B.W. The immune anti-tumor effects of GM-CSF and B7-1 gene transfection are enhanced by surgical debulking of tumor. Cancer Gene Ther. 2001, 8, 580–588. [Google Scholar] [CrossRef]
- Parney, I.F.; Farr-Jones, M.A.; Kane, K.; Chang, L.J.; Petruk, K.C. Human autologous in vitro models of glioma immunogene therapy using B7-2, GM-CSF and IL-12. Can. J. Neurol. Sci. 2002, 29, 267–275. [Google Scholar]
- Parney, I.F.; Chang, L.J.; Farr-Jones, M.A.; Hao, C.; Smylie, M.; Petruk, K.C. Technical hurdles in a pilot clinical trial of combined B7-2 and GM-CSF immunogene therapy for glioblastomas and melanomas. J. Neurooncol. 2006, 78, 71–80. [Google Scholar] [CrossRef]
- Zajac, P.; Oertli, D.; Marti, W.; Adamina, M.; Bolli, M.; Guller, U.; Noppen, C.; Padovan, E.; Schultz-Thater, E.; Heberer, M.; et al. Phase I/II Clinical trial of a nonreplicative vaccinia virus expressing multiple HLA-A0201-restricted tumor-associated epitopes and costimulatory molecules in metastatic melanoma patients. Hum. Gene Ther. 2003, 14, 1497–1510. [Google Scholar] [CrossRef]
- Pizzoferrato, E. B7-2 expression above a threshold elicits anti-tumor immunity as effective as interleukin-12 and prolongs survival in murine B-cell lymphoma. Int. J. Cancer 2004, 110, 61–69. [Google Scholar] [CrossRef]
- Shi, F.S.; Weber, S.; Gan, J.; Rakhmilevich, A.L.; Mahvi, D.M. Granulocyte-macrophage colony-stimulating factor (GM-CSF) secreted by cDNA-transfected tumor cells induces a more potent antitumor response than exogenous GM-CSF. Cancer Gene Ther. 1999, 6, 81–88. [Google Scholar]
- Herrero, M.J.; Botella, R.; Algás, R.; Marco, F.; Lledó, S.; Aliño, S.F. Nonviral Cancer Vaccines: From Free Antigens to Engineered Cells. In New Gene Therapy and Cancer Research; Gustafsson, W.B., Ed.; Nova Publishers: New York, NY, USA, 2008. [Google Scholar]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef]
- Rodríguez-Lecompte, J.C.; Kruth, S.; Gyorffy, S.; Wan, Y.H.; Gauldie, J. Cell-based cancer gene therapy: Breaking tolerance or inducing autoimmunity. Anim. Health Res. Rev. 2004, 5, 227–234. [Google Scholar] [CrossRef]
- Herrero, M.J.; Botella, R.; Algás, R.; Marco, F.; Aliño, S.F. Bead-selected antitumor genetic cell vaccines. Clin. Med. Oncol. 2008, 2, 257–265. [Google Scholar]
- Terando, A.M.; Faries, M.B.; Morton, D.L. Vaccine therapy for melanoma: current status and future directions. Vaccine 2007, 25, B4–B16. [Google Scholar] [CrossRef]
- Zou, W. Regulatory T cells, tumor immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef]
- De Visser, K.; Eichten, A.; Coussens, L. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Serafini, P.; Borrello, I.; Bronte, V. Myeloid suppressor cells in cancer: Recruitment, phenotype, properties, and mechanisms of immune suppression. Semin. Cancer Biol. 2006, 16, 53–65. [Google Scholar] [CrossRef]
- Bossiotis, V.A.; Freeman, G.J.; Gribben, J.G.; Badler, L.M. The role of B7-1/B7-2:CD28/CTLA-4 pathways in the prevention of anergy, induction of productive immunity and down-regulation of the immune response. Immunol. Rev. 1996, 15, 5–26. [Google Scholar]
- Sperling, A.; Bluestone, J.A. The complexities of T-cell co-stimulation: CD28 and beyond. Immunol. Rev. 1996, 153, 155–182. [Google Scholar] [CrossRef]
- Marengere, L.E.M.; Waterhouse, H.W.; Duncan, G.S.; Mittrucker, H.W.; Feng, G.S.; Mak, T.W. Regulation of T cell receptor signalling by tyrosine phosphatase SYP association with CTLA-4. Science 1996, 272, 1170–1173. [Google Scholar]
- Onizuka, S.; Tawara, I.; Shimizu, J.; Sakaguchi, S.; Fujita, T.; Nakayama, E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer Res. 1999, 59, 3128–3133. [Google Scholar]
- Steitz, J.; Brück, J.; Lenz, J.; Knop, J.; Tüting, T. Depletion of CD4+CD25+ T cells and treatment with tyrosinase-related protein 2-transduced dendritic cells enhance the IFN α-induced CD8+ Tcell dependent immune defense of B16 melanoma. Cancer Res. 2001, 61, 8643–8646. [Google Scholar]
- Nagai, H.; Horikawa, T.; Hara, I.; Fukunaga, A.; Oniki, S.; Oka, M.; Nishigori, C.; Ichihashi, M. In vivo elimination of CD25+ regulatory T cells leads to tumor rejection of B16F10 melanoma, when combined with IL-12 gene transfer. Exp. Dermatol. 2004, 13, 613–620. [Google Scholar] [CrossRef]
- Rech, A.J.; Vonderheide, R.H. Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann. N. Y. Acad. Sci. 2009, 1174, 99–106. [Google Scholar] [CrossRef]
- Rech, A.J.; Mick, R.; Martin, S.; Recio, A.; Aqui, N.A.; Powell, D.J., Jr.; Colligon, T.A.; Trosko, J.A.; Leinbach, L.I.; Pletcher, C.H.; et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with inmunotherapy in cancer patients. Sci. Transl. Med. 2012, 4, 134ra62. [Google Scholar] [CrossRef]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD4+CD25+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar]
- Ascierto, P.A.; Marincola, F.M.; Ribas, A. Anti-CTLA4 monoclonal antibodies: The past and the future in clinical application. J. Transl. Med. 2011, 9, 196. [Google Scholar] [CrossRef]
- Attia, P.; Maker, A.V.; Haworth, L.R.; Rogers-Freezer, L.; Rosenberg, S.A. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389IL2-ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J. Immunother. 2005, 28, 582–592. [Google Scholar] [CrossRef]
- Dannull, J.; Su, Z.; Rizzieri, D.; Yang, B.K.; Coleman, D.; Yancey, D.; Zhang, A.; Dahm, P.; Chao, N.; Gilboa, E.; et al. Enhancement of vaccine-mediated antitumor immnunity in cancer patients after depletion of regulatory T cells. J. Clin. Invest. 2005, 115, 3623–3633. [Google Scholar] [CrossRef]
- Sharma, R.K.; Elpek, K.G.; Yolcu, E.S.; Schabowsky, R.H.; Zhao, H.; Bandura-Morgan, L.; Shirwan, H. Costimulation as a platform for the development of vaccines: A peptide-based vaccine containing a novel form of 4-1BB ligand eradicates established tumors. Cancer Res. 2009, 69, 4319–4326. [Google Scholar]
- Sharma, R.K.; Schabowsky, R.H.; Srivastava, A.K.; Elpek, K.G.; Madireddi, S.; Zhao, H.; Zhong, Z.; Miller, R.W.; Macleod, K.J.; Yolcu, E.S.; et al. 4-1BB ligand as an effective multifunctional immunomodulator and antigen delivery vehicle for the development of therapeutic cancer vaccines. Cancer Res. 2010, 70, 3945–3954. [Google Scholar]
- Madireddi, S.; Schabowsky, R.H.; Srivastava, A.K.; Sharma, R.K.; Yolcu, E.S.; Shirwan, H. SA-4-1BBL costimulation inhibits conversion of conventional CD4+ T cells into CD4+FoxP3+ T regulatory cells by production of IFN-γ. PLoS One 2012, 7, e42459. [Google Scholar]
- Murphy, K.A.; Lechner, M.G.; Popescu, F.E.; Bedi, J.; Decker, S.A.; Hu, P.; Erickson, J.R.; O’Sullivan, M.G.; Swier, L.; Salazar, A.M.; et al. An in vivo immunotherapy screen of costimulatory molecules identifies Fc-OX40L as a potent reagent for the treatment of established murine gliomas. Clin. Cancer Res. 2012, 18, 4657–4668. [Google Scholar]
- Horna, P.; Cuenca, A.; Cheng, F.; Brayer, J.; Wang, H.W.; Borrello, I.; Levitsky, H.; Sotomayor, E.M. In vivo disruption of tolerogenic cross-presentation mechanisms uncovers an effective T-cell activation by Bcell lymphomas leading to antitumor immunity. Blood 2006, 107, 2871–2878. [Google Scholar] [CrossRef]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef]
- Frey, A.B. Myeloid supresor cells regulate the adaptive immune response to cancer. J. Clin. Invest. 2006, 116, 2587–2590. [Google Scholar] [CrossRef]
- Filipazzi, P.; Valenti, R.; Huber, V.; Pilla, L.; Canese, P.; Iero, M.; Castelli, C.; Mariani, L.; Parmiani, G.; Rivoltini, L. Identification of a new subset of myeloid supresor cells in Peripherals blood of melanoma patients with modulation by a GM-CSF-based antitumor vaccine. J. Clin. Oncol. 2007, 25, 2546–2553. [Google Scholar] [CrossRef]
- Guillem, V.M.; Aliño, S.F. Transfection pathways of nonspecific and targeted PEI-polyplexes. Gene Ther. Mol. Biol. 2004, 8, 369–384. [Google Scholar]
- Bordier, C. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 1981, 25, 1604–1607. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miguel, A.; Herrero, M.J.; Sendra, L.; Botella, R.; Algás, R.; Sánchez, M.; Aliño, S.F. Comparative Antitumor Effect of Preventive versus Therapeutic Vaccines Employing B16 Melanoma Cells Genetically Modified to Express GM-CSF and B7.2 in a Murine Model. Toxins 2012, 4, 1058-1081. https://doi.org/10.3390/toxins4111058
Miguel A, Herrero MJ, Sendra L, Botella R, Algás R, Sánchez M, Aliño SF. Comparative Antitumor Effect of Preventive versus Therapeutic Vaccines Employing B16 Melanoma Cells Genetically Modified to Express GM-CSF and B7.2 in a Murine Model. Toxins. 2012; 4(11):1058-1081. https://doi.org/10.3390/toxins4111058
Chicago/Turabian StyleMiguel, Antonio, María José Herrero, Luis Sendra, Rafael Botella, Rosa Algás, Maria Sánchez, and Salvador F. Aliño. 2012. "Comparative Antitumor Effect of Preventive versus Therapeutic Vaccines Employing B16 Melanoma Cells Genetically Modified to Express GM-CSF and B7.2 in a Murine Model" Toxins 4, no. 11: 1058-1081. https://doi.org/10.3390/toxins4111058