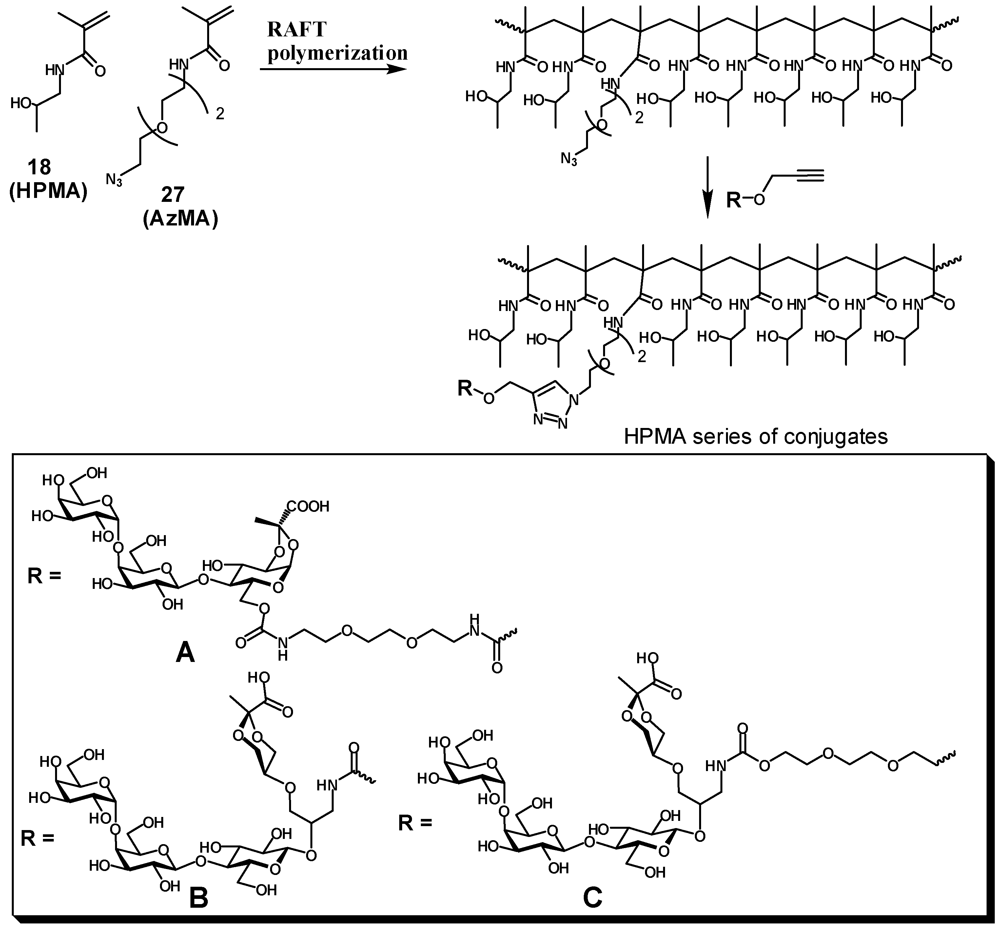
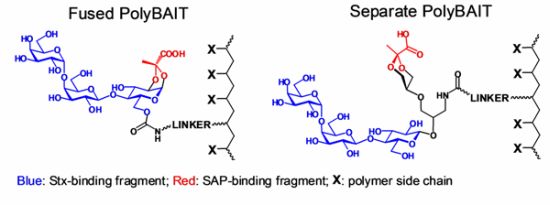
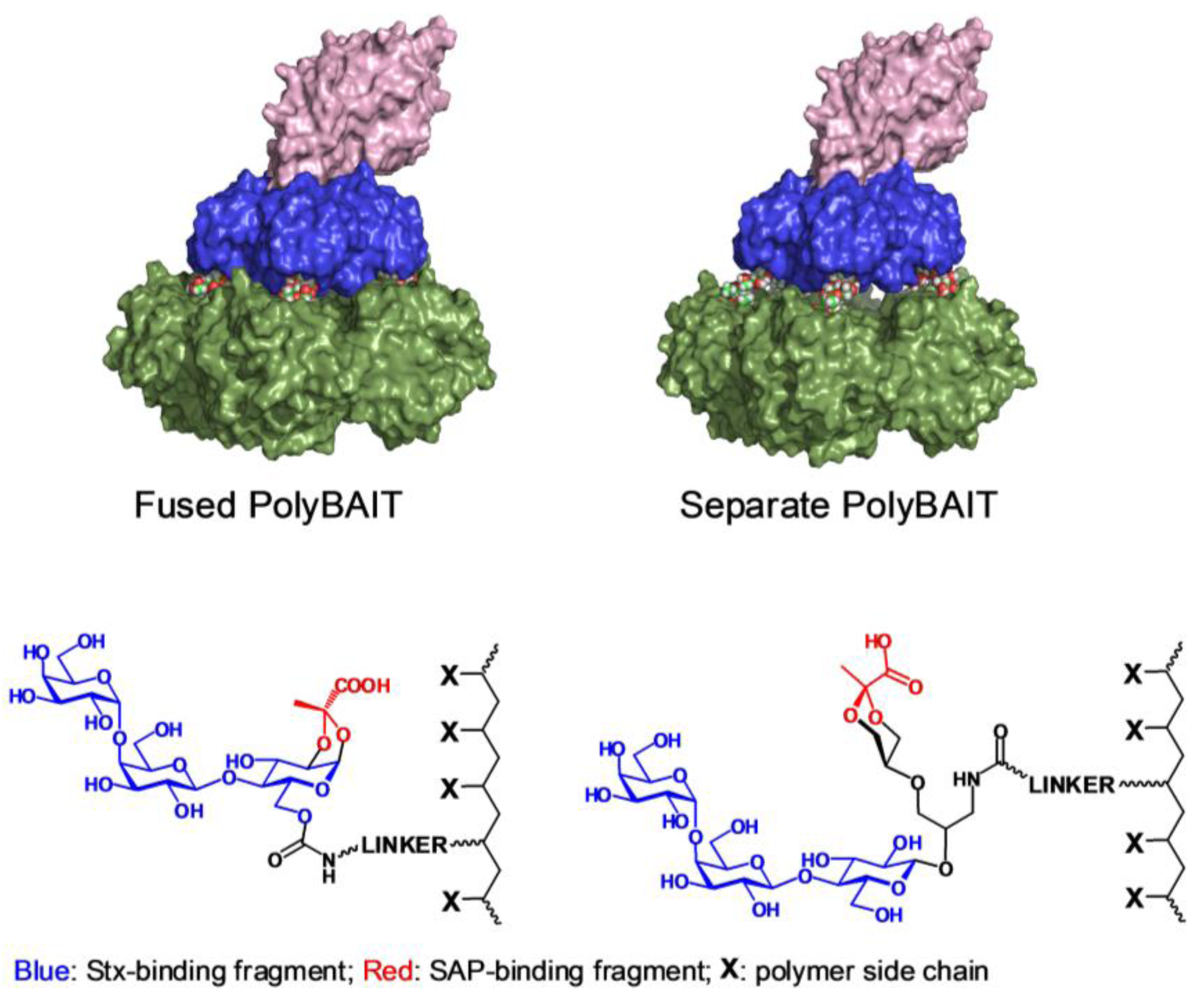
2.1. Synthesis of Heterobifunctional Inhibitors
Commercially available reagents were used as supplied without further purification. Evaporation and concentration in vacuo was conducted under water-aspirator pressure. All solution-phase reactions were carried out under nitrogen atmosphere. Reactions were monitored by analytical thin-layer chromatography (TLC) with pre-coated silica gel 60 F254 glass plate (Merck). Plates were visualized under UV light or stained by treatment with either cerium ammonium molybdate solution or 5% sulfuric acid in ethanol followed by heating at 180 °C. Purification of products was conducted by column chromatography using silica gel SiliaFlashF60 (40–63 μm, 60 Å) from SiliCycle® Inc. IR data were recorded on a Nic–Plan IR Microscope (solid film); only signals corresponding to functional groups indicative to the structure are reported. NMR spectra were recorded at 500 or 600 MHz, at 27 °C in CDCl3 or D2O. Chemical shifts are referenced to residual solvent (CDCl3) at 7.24 p.p.m. for 1H and 77.0 p.p.m. for 13C and relative to 0.1% external acetone at 2.225 p.p.m. for 1H for solutions in D2O. Electrospray ionization mass spectra were recorded on a Micromass Zabspec TOF-mass spectrometer.
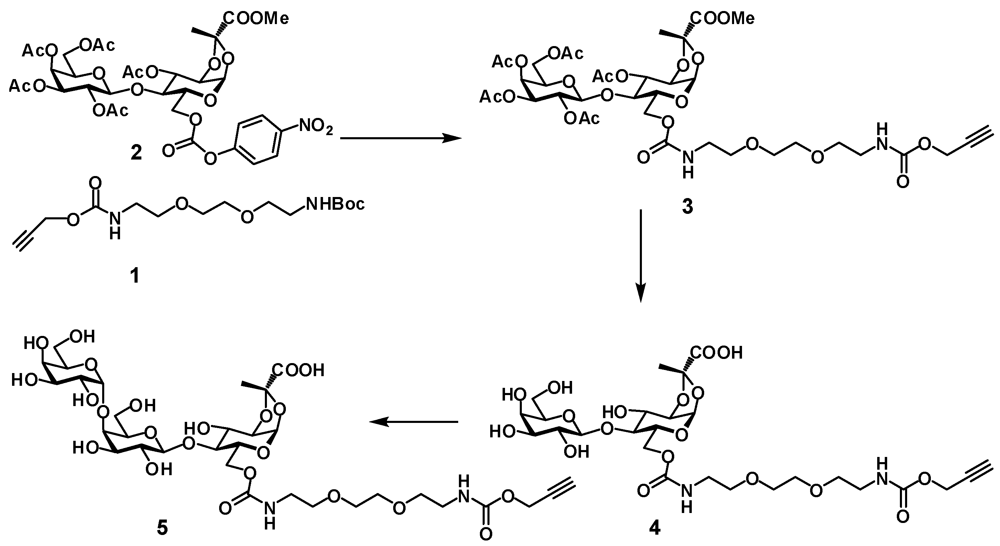
Prop-2-ynyl 2-(2-(2-t-butyloxycarbonylaminoethoxy)ethoxy)ethylcarbamate (1). To a solution of
tert-butyl 2-[2-(2-aminoethoxy)ethoxy]ethylcarbamate [
12] (698 mg, 2.81 mmol) in dry DCM (3 mL) at −78 °C triethylamine (0.85 g, 8.4 mmol) was added followed by propargyl chloroformate (522 mg, 4.4 mmol). The mixture was allowed to warm to room temperature and then stirred for 1.5 h. The mixture was diluted with DCM, washed with brine, concentrated and chromatographed on silica gel using hexane-acetone (7:3) to yield product
1 as a clear syrup (689 mg, 74%);
1H-NMR (CDCl
3) δ: 5.63 (bs, 1 H, NH), 5.26 (bs, 1 H, NH), 4.93 (d, 2 H,
J 2.3 Hz, CH
2), 3.88–3.86 (m, 4 H, OCH
2), 3.83–3.76 (m, 4 H, OCH
2), 3.66–3.60 (m, 2 H, NCH
2), 3.57–3.52 (m, 2 H, NCH
2), 2.70 (t, 1 H, CH), 1.70 (s, 9 H
tBu); ESI HRMS:
m/
z: calcd for C
15H
26N
2O
6Na: 353.1683; found: 353.1681 [M + Na]
+.
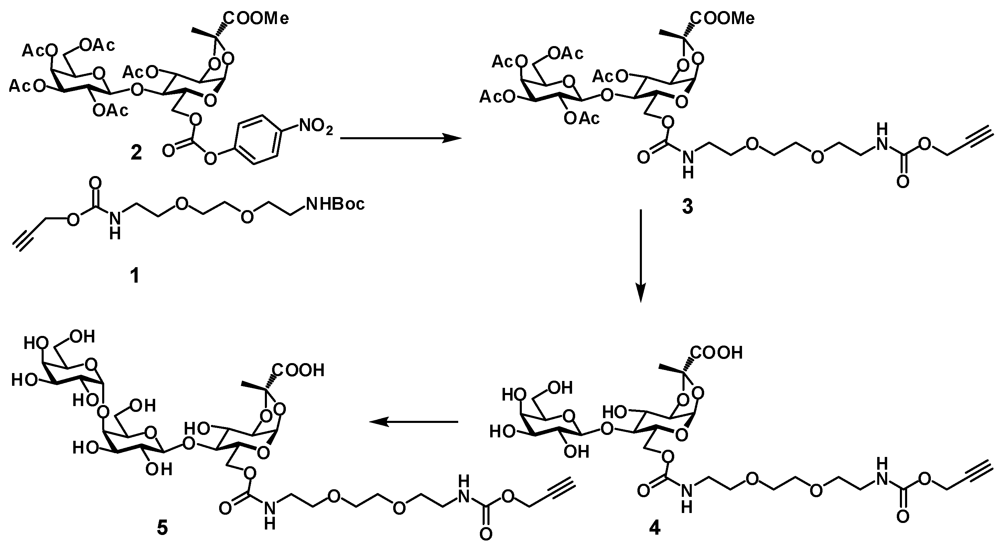
3-O-Acetyl-6-O-(10-oxo-3,6,11-trioxa-9-azatetradec-13-ynyl)carbamoyl-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-1,2-O-[(S)-1-(methoxycarbonyl)ethylidene]-α-D-glucopyranoside (3). Alkyne
1 (545 mg, 1.65 mmol) was dissolved in TFA (1.6 mL) and left at room temperature for 1 h. The mixture was concentrated, treated with Et
3N and concentrated again then 3-
O-acetyl-6-
O-(4-nitrophenyloxycarbonyl)-4-
O-(2,3,4,6-tetra-
O-acetyl-β-D-galactopyranosyl)-1,2-
O-[(
S)-1-(methoxycarbonyl) ethylidene]-α-D-glucopyranoside
2 [
11] (886 mg, 1.105 mmol) was added. The mixture was dissolved in dry DCM (10 mL) and triethylamine (310 μL, 2.21 mmol) was added. The reaction mixture was stirred at room temperature for 2 h. The mixture was concentrated and chromatographed on silica gel using hexane-acetone (2:1 to 1:1) to provide the title compound
3 as a white foam (0.93 g, 94%);
1H-NMR (CDCl
3) δ: 5.76 (d, 1 H,
J1,2 5.13 Hz, H-1) 1.53–1.52 (m, 1 H, H-3), 5.46–5.40 (m, 1 H, NH), 5.38 (dd, 1 H, H
J3',4' 3.5 Hz,
J4',5' 0.7 Hz, H-4'), 5.18 (dd, 1 H,
J1',2' 7.0 Hz,
J2',3' 10.4 Hz, H-2'), 5.01 (dd, 1 H, H-3'), 4.68 (d, 2 H,
J 2.2 Hz, CH
2), 4.62 (d, 1 H, H-1'),4.36–4.33 (m, 1 H, H-2), 4.24 (dd, 1 H,
J5,6a 2.2 Hz,
J6a,6b 11.7 Hz, H-6a), 4.18–4.10 (m, 3 H, H-6b, H-6'a, H-6'b), 3.96–3.93 (m, 1 H, H-5'), 3.92–3.88 (m, 1 H, H-5), 3.76 (s, 3 H, OCH
3), 3.64–3.54 (m, 8 H, OCH
2), 3.42–3.36 (m, 4 H, NCH
2), 2.48 (t, 1 H, CH), 2.18, 2.10, 2.03 1.98 (5 s, 15 H, 5 OAc), 1.76 (s, 3 H, CH
3); ESI HRMS:
m/z: calcd for C
37H
52N
2O
23Na: 915.2853; found 915.2846 [M + Na]
+.
1,2-O-[(S)-1-(carboxy)ethylidene]-4-O-(β-D-galactopyranosyl)-6-O-(10-oxo-3,6,11-trioxa-9-azatetradec-13-ynyl)carbamoyl-α-D-glucopyranoside (4). Disaccharide 3 (480 mg, 0.537 mmol) was dissolved in dry methanol (4 mL) and 1 M NaOMe (0.54 mL) was added. The solution was stirred at room temperature for 16 h. The mixture was concentrated and dissolved in water. After 0.5 h TLC indicated that hydrolysis of the methyl ester was complete. The mixture was neutralized with 5 M acidic acid and lyophilized to provide the product 4 as white foam (387 mg, 94%). 1H-NMR (D2O) δ: 5.62 (d, 1 H, J1,2 5.0 Hz, H-1), 4.67 (d, 1 H, J 2.1 Hz, CH2), 4.44 (d, 1 H, J1',2' 7.8 Hz, H-1'), 4.41–4.37 (m, 2 H, J2,3 4.1 Hz, J6a,6b 11.9 Hz, H-3, H-6a), 4.24 (dd, 1 H, J5,6b 5.2 Hz, H-6b), 4.18 (dd, 1 H, H-2), 4.06–4.01 (m, 1 H, H-5), 3.91 (d, 1 H, J3',4' 3.4 Hz, H-4'), 3.82–3.60 (m, 13 H, H-4, H-3', H-5', H-6'a, H-6'b, 4 × OCH2), 3.55 (dd, 1 H, H-2'), 3.36–3.32 (m, 4 H, 2 × NCH2), 2.9 (t, 1 H, CH), 1.64 (s, 3 H, CH3). ESI HRMS: m/z: calcd for C26H39N2O18: 667.2203; found 667.2205 [M − H]−.
1,2-O-[(S)-1-(carboxy)ethylidene]-4-O-[4-O-(α-D-galactopyranosyl)-β-D-galactopyranosyl]-6-O-(10-oxo-3,6,11-trioxa-9-azatetradec-13-ynyl)carbamoyl-α-D-glucopyranoside (5). Compound
4 (379 mg, 0.537 mmol) was dissolved in water (5.34 mL) and the pH was adjusted to 7.5 by addition of solid NaHCO
3. Then 1 M HEPES buffer (pH: 8, 3.2 mL) was added followed by 0.4 M DTT solution (500 μL) and alkaline phosphatase (1 U/μL, 50 μL, Sigma). UDP-glucose (578 mg, 0.849 mmol) was dissolved in the reaction mixture and the fusion enzyme GalT/UDP-4'-epimerase [
21] (950 μL) was added. The reaction mixture was incubated at 37 °C. After 24 h the reaction mixture was treated with Dowex H
+ resin, filtered and freeze-dried. NMR of the crude product confirmed the reaction was complete. The mixture was purified on silica gel using DCM-methanol (2:1 with 2% acetic acid) to afford after 2 passages pure trisaccharide
5 (270 mg, 61%). Impure fractions were further purified on HPLC (C
18) using water (with 0.1% TFA)–acetonitrile to yield an additional amount of trisaccharide (51 mg, 11%);
1H-NMR (D
2O) δ: 5.64 (d, 1 H,
J1,2 5.0 Hz, H-1), 4.94 (d, 1 H,
J1'',2'' 4.0 Hz, H-1''), 4.67 (d, 2 H,
J 2.2 Hz, CH
2), 4.51 (d, 1 H,
J1',2' 7.8 Hz, H-1'), 4.41–4.33 (m, 3 H, H-3, H-6a, H-5''), 4.28–4.21 (m, 2 H, H-2, H-6b), 4.04–4.00 (m, 3 H, H-5, H-4', H-4''), 3.94–3.90 (m, 2 H, H-6'a, H-3''), 3.85–3.75 (m, 4 H, H-4, H-5', H-6'b, H-2''), 3.72–3.66 (m, 7 H, H-3', H-6''a, H-6''b, 2 × OCH
2), 3.63–3.56 (m, 5 H, H-2', 2 × OCH
2 ), 3.34–3.32 (m, 4 H, 2 × NCH
2), 2.90 (t, 1 H, CH), 1.69 (s, 3 H, CH
3). ESI HRMS:
m/z: calcd for C
32H
50N
2O
23: 829.2732; found: 829.2734 [M − H]
−.
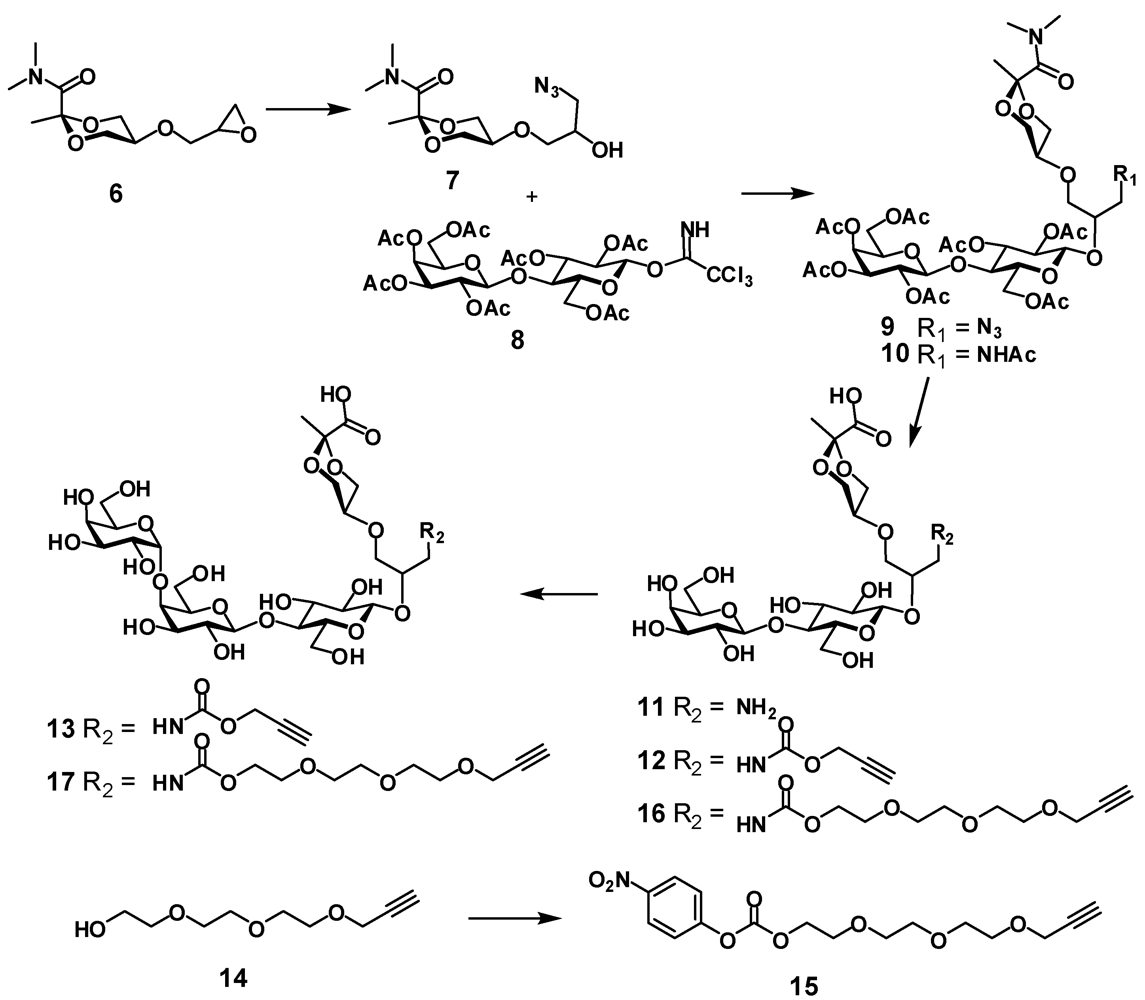
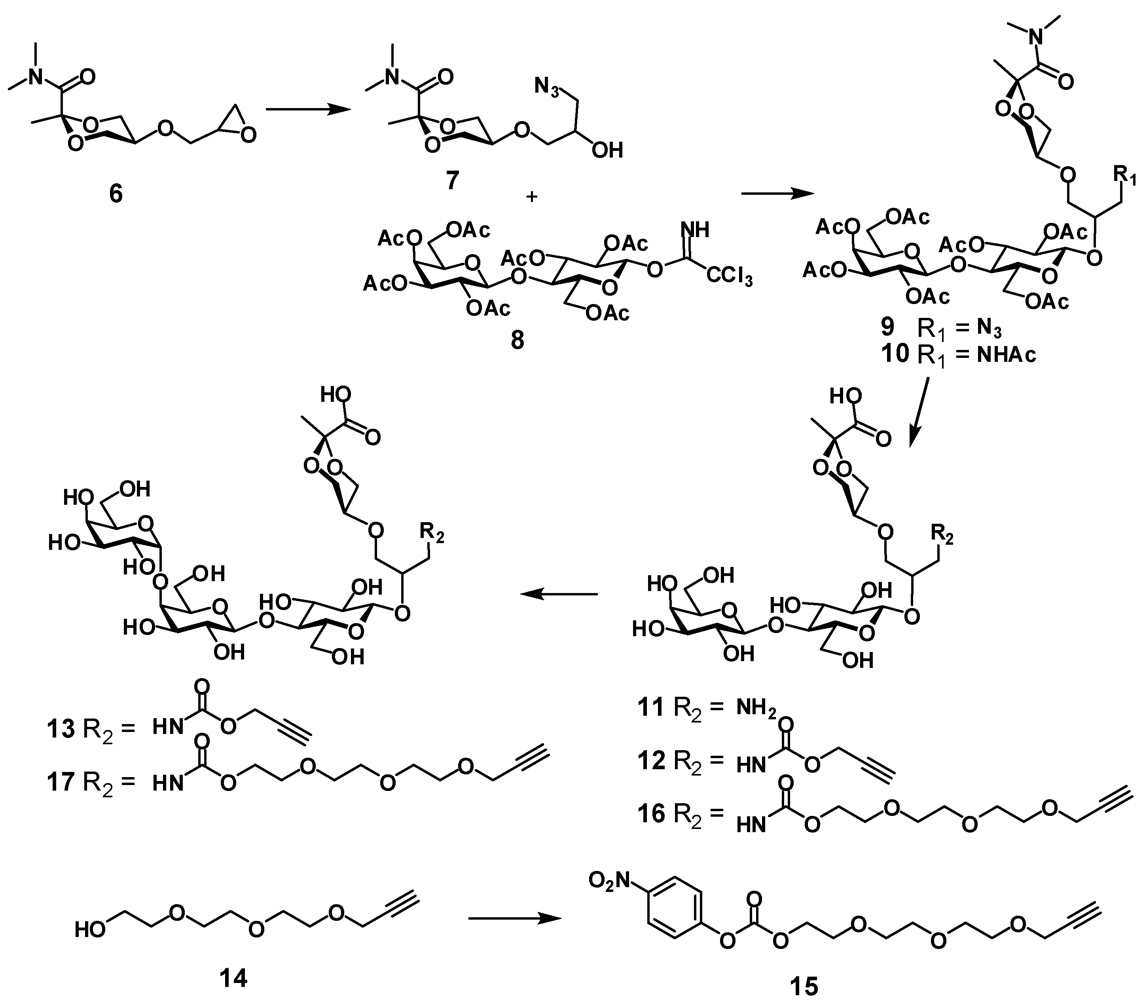
(R,S)-5-(3-azido-2-hydroxypropoxy)-N,N,2-trimethyl-1,3-dioxane-2-carboxamide (7). Epoxide
6 [
13] (4.595 g, 18.734 mmol) was dissolved in dry DMF (20 mL) then sodium azide (3.66 g, 56.2 mmol) and ammonium chloride (3.01 g, 56.2 mmol) were added and the mixture was stirred for 5 h at 50 C then overnight at room temperature. Water (100 mL) was added and the mixture was extracted with ethyl acetate (3 × 50 mL). Combined organic layers were washed with saturated NH
4Cl and brine, then dried with MgSO
4, filtered and concentrated. The residue was purified on silica gel using hexane-acetone (3:1) to provide the title compound
7 as a clear syrup (2.9 g, 54%).
1H-NMR (CDCl
3) δ: 4.10–4.06 (m, 2 H, H-4
e, H-6
e), 3.92–3.82 (m, 1 H, OCH), 3.70–3.48 (m, 5 H, H-4
a, H-5, H-6
a, OCH
2), 3.40–3.28 (m, 2 H, NCH
2), 3.24 and 3.02 (2s, 6 H, NMe
2), 1.52 (s, 3 H, CH
3); ESI HRMS:
m/z: calcd for C
11H
20N
4O
5Na: 311.1326; found: 311.1329 [M + Na]
+.
(R,S)-5-{3-azido-2-[2,3,6-tri-O-acetyl-4-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-β-D-glucopyranosyloxy]propoxy}-N,N,2-trimethyl-1,3-dioxane-2-carboxamide (9). Azide
7 (1.05 g, 3.64 mmol) and lactosyl trichloroacetimidate
8 [
14] (3.37 g, 4.32 mmol) were dried together under vacuum overnight then dissolved in dry DCM (30 mL) and powdered molecular sieves 4 Å were added and the mixture was stirred under argon for 0.5 h and cooled to 0 °C. TMSOTf (50 μL, 61 mg, 0.27 mmol) was added slowly then the ice bath was removed and stirring continued at room temperature. After 40 min the mixture was neutralized with a drop of Et
3N, filtered through celite, concentrated and chromatographed on silica gel using hexane-acetone (3:1 to 3:2) to provide the title product
9 as white foam (2.4 g, 69 %);
1H-NMR (CDCl
3) δ: 5.36–5.34 (m, 1 H,
J3',4' 3.4 Hz, H-4'), 5.20−5.16 (m, 1 H, H-3), 5.13–5.08 (m, 1 H,
J1',2' 7.8 Hz, H-2'), 4.98–4.94 (m, 1 H, H-3'), 4.90-4.84 (m, 1 H,
J1,2 7.9 Hz, H-2), 4.63 (d, 1 H, H-1), 4.60-4.56 (m, 1 H, H-6a), 4.52–4.49 (m, 1 H, H-1'), 4.15–4.02 (m, 5 H, H-4e
pyr, H-6e
pyr, H-6b, H-6a', H-6b'), 3.90–3.85 (m, 1 H, H-5'), 3.83–3.68 (m, 2 H, H-4, CH), 3.65–3.44 (m, 6 H, H-5, H-4a
pyr, H-5
pyr, H-6a
pyr, OCH
2), 3.41–3.35 and 3.30–3.22 (2 m, 2 H, NCH
2), 3.22 and 3.02 (2 s, 6 H, NMe
2), 2.16–1.97 (ms, 21 H, 7 × OAc), 1.50 (s, 3 H, CH
3). ESI HRMS:
m/z: calcd for C
47H
54N
4O
22Na: 929.3; found: 929.3 [M + Na]
+.
(R,S)-5-{3-acetamido-2-[2,3,6-tri-O-acetyl-4-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-β-D-glucopyranosyloxy]propoxy}-N,N,2-trimethyl-1,3-dioxane-2-carboxamide (10). To a solution of azide 9 (945 mg, 1.04 mmol) in dry THF (19 mL) triphenylphosphine (423 mg, 1.6 mmol) was added and the mixture was stirred at 65 °C for 6 h then water (2.5 mL) was added and the mixture was stirred at 65 °C for 6.5 h, then concentrated, co-evaporated with toluene and dried under vacuum. The residue was dissolved in pyridine (20 mL) and acetic anhydride (10 mL) was added then the mixture was left at room temperature overnight. The mixture was concentrated, co-evaporated with toluene and the residue was chromatographed on silica gel using hexane-acetone (1:1) to provide N-acetate 10 as a white foam (635 mg; 65 %); 1H-NMR (CDCl3) δ: 6.07 (t, 0.45 H, J 5.5 Hz, NH), 5.89 (t, 0.55 H, J 5.5 Hz, NH), 5.35 (dd, 1 H, J3',4' 3.4 Hz, J4',5' 0.3 Hz, H-4'), 5.18 (dd, 1 H, J2,3 = J3.4 = 9.4 Hz, H-3), 5.13–5.10 (m, 1 H, J1',2' 7.9 Hz, J2',3' 10.4 Hz, H-2'), 4.98 (dd, 1 H, H-3'), 4.88–4.83 (m, 1 H, H-2), 4.63 (d, 0.55 H, H-1), 4.60–4.56 (m, 1 H, H-6a), 4.55 (d, 0.45 H, H-1), 4.50 (d, 1 H, H-1'), 4.16–4.02 (m, 5 H, H-6b, H-6'a, H-6'b, H-4epyr, H-6epyr), 3.90–3.86 (m, 1 H, H-5'), 3.79–3.70 (m, 2 H, H-4, OCH), 3.66–3.41 (m, 7 H, H-5, H-4apyr, H-5pyr, H-6apyr, NCH, OCH2), 3.22 (s, 3 H, NCH3), 3.21–3.12 (m, 1 H, NCH), 2.17–2.04 (ms, 18 H, 6 x OAc), 1.96 (s, 6 H, NAc, OAc), 1.50 (2 s, 3 H, CH3); ESI HRMS: m/z: calcd for C39H58N2O23Na: 945.3323, found: 945.3315 [M + Na]+.
(R,S)-5-{3-(prop-2-yn)oxycarbonylamino-2-[4-(β-D-galactopyranosyl)-β-D-glucopyranosyloxy]propoxy}-2-methyl-1,3-dioxane-2-carboxylic acid (12). Acetate 10 (186 mg, 0.2 mmol) was dissolved in methanol (4 mL) and transferred to a plastic Falcon tube, then 4 M NaOH (0.5 mL, 2 mmol) was added. The mixture was stirred at 80 °C. After 3 days the reaction was not complete as checked by NMR. More 4 M NaOH (0.1 mL, 0.4 mmol) was added and stirring was continued overnight at 85 °C. The mixture containing hydrolysis product 11 was treated with dry ice until the solution reached pH 8–9. Then propargyl chloroformate (40 μL, 0.4 mmol) was added and the mixture was stirred at room temperature. After 2 h the mixture was purified on HPLC (C-18) using water containing 0.1% TFA—acetonitrile gradient. Two fractions were isolated, concentrated and freeze-dried. First fraction was the product of incomplete hydrolysis of 10 (R,S)-5-{3-acetamido-2-[4-(β-D-galactopyranosyl)-β-D-glucopyranosyloxy]propoxy}-2-methyl-1,3-dioxane-2-carboxylic acid (11a) (54 mg, 45%), 1H-NMR (D2O) δ: 4.56 (d, 1 H, J1,2 7.9 Hz, H-1), 4.44 (d, 1 H, J1',2' 7.8 Hz, H-1'), 4.16 (dd, Jvic 4.5 Hz, Jgem 11.4 Hz, H-4epyr, H-6epyr), 4.02–3.99 (m, 1 H, CH), 3.95 (dd, 1 H, J5,6a 1.7 Hz, J6a,6b 12.1 Hz, H-6a), 3.06 (d, 1 H, J3',4' 3.3 Hz, H-4'), 3.82 (m, 14 H, H-3, H-4, H-5, H-6b, H-2', H-3', H-5', H-6'a, H-6'b, H-4apyr, H-5pyr, H-6apyr, OCH2), 3.40–3.29 (m, 3 H, H-2, NCH2), 2.00 (s, 3 H, OAc), 1.00 (s, 3 H, CH3); ESI HRMS: m/z: calcd for C23H39NO17: 624.211; found: 624.2104 [M+ Na]+. This intermediate can be recycled.
The second fraction was the target compound 12 (59 mg, 46%); 1H-NMR (D2O) δ: 4.65 (bs, 2 H, CH2 propargyl), 4.56–4.50 (m, 1 H, J1.2 7.9 Hz, H-1), 4.42 (d, 1 H, J1',2' 7.8 Hz, H-1'), 4.24–4.16 (m, 2 H, H-4epyr, H-6epyr), 4.00–3.88 (m, 3 H, J3',4' 3.3 Hz, H-6a, H-4', CH), 3.86–3.50 (m, 14 H, J2',3 9.9 Hz, H-3, H-4, H-5, H-6b, H-2', H-3', H-5', H-6'a, H-6'b, H-4apyr, H-5pyr, H-6apyr, OCH2), 3.30–3.24 (m, 3 H, H-2, NCH2), 2.90 (bs, 1 H, CHpropargyl), 1.52 (s, 3 H, CH3); ESI HRMS: m/z: calcd for C25H38NO18: 640.2094; found: 640.2091 [M − H]−.
(R,S)-5-{3-(prop-2-yn)oxycarbonylamino-2-[α-D-galactopyranosyl-(1-4)-β-D-galactopyranosyl-(1-4)-β-D-glucopyranosyloxy]propoxy}-2-methyl-1,3-dioxane-2-carboxylic acid (13). Lactose derivative
12 (110 mg, 0.17 mmol) was dissolved in water (1.5 mL) and neutralized with dry NaHCO
3. Then other components were added in the following order: 1 M HEPES buffer (pH: 8; 0.93 mL), DTT solution (0.4 M, 145 μL), alkaline phosphatase (1 U/μL, 14.5 μL, Sigma), UDP-glucose (157 mg, 0.257 mmol) and crude fusion enzyme GalT/UDP-4'-epimerase [
21] (0.3 mL). The mixture was incubated for 40 h at 37 °C. The mixture was treated with Dowex H
+ resin, filtered and purified on HPLC (C-18) using water with 0.1% TFA and acetonitrile gradient. The fraction containing product was concentrated and lyophilized to afford the product
13 as a white foam (116 mg, 85 %);
1H-NMR (D
2O) δ: 4.92 (d, 1 H,
J1'',2'' 4.0 Hz, H-1''), 4.66 (d, 2 H,
J 1.8 Hz, CH
2 propargyl), 4.56–4.52 ( m, 1 H,
J1,2 7.6 Hz, H-1), 4.50 (d, 1 H,
J1',2' 7.8 Hz, H-1'), 4.34–4.31 (m, 1 H,
J5'',6''a =
J5'',6''b= 6.4 Hz, H-5''), 4.22 (m, 2 H, H-4e
pyr, H-6e
pyr), 4.03–4.00 (m, 2 H,
J3',4' 3.2 Hz,
J3'',4'' 3.3 Hz, H-4', H-4''), 3.99–3.94 (m, 2 H, H-5, CH), 3.93–3.54 (m, 18 H, H-3, H-4, H-6a, H-6b, H-2', H-3', H-5', H-6'a, H-6'b, H-2'',H-3'', H-6''a, H-6''b, H-4a
pyr, H-5
pyr, H-6a
pyr, OCH
2), 3.38–3.27 (m, 3 H, H-2, NCH
2), 2.89 (bs, 1 H, CH
propargyl), 1.52 (s, 3 H, CH
3); ESI HRMS:
m/z: calcd for C
31H
49NO
23Na: 826.2588; found: 826.2588 [M + Na]
+.
4-Nitrophenyl 2-(2-(2-(prop-2-ynyloxy)ethoxy)ethoxy)ethyl carbonate (15). To a solution of 2-(2-(2-(prop-2-ynyloxy)ethoxy)ethoxy)ethanol
14 [
15] (1.953 g, 10.37 mmol) and 4-nitrophenyl chloroformate (2.51 g, 12.5 mmol) in dry DCM pyridine (2 mL) was slowly added at room temperature. After 30 min the reaction was quenched by addition of methanol, diluted with DCM and washed with brine. The organic layer was collected, concentrated and the residue was chromatographed on silica gel using hexane acetone (3:1) to provide
15 as a clear syrup (3.345 g; 91%);
1H-NMR (CDCl
3) δ: 8.30–8.26 and 7.42–7.37 (2 m, 4 H, C
6H
4), 4.46–4.42 (m, 2 H, CH
2OCO), 4.20 (d, 2 H,
J 3.4 Hz, CH
2 propargyl), 3.84–3.80 (m, 2H, OCH
2), 3.73–3.67 (m, 8 H, 4 × CH
2), 2.43 (t, 1 H, CH
propargyl); ESI HRMS:
m/z: calcd for C
16H
19NO
8: 376.1003; found: 376.1004 [M+ Na]
+.
(R,S)-5-{2-[β-D-galactopyranosyl-(1-4)-β-D-glucopyranosyloxy]-5-oxo-6,9,12,15-tetraoxa-4-azaoctadec-17-ynyloxy}-2-methyl-1,3-dioxane-2-carboxylic acid (16). To a solution of 10 (545 mg, 0.59 mmol) in methanol (12 mL) in a plastic tube NaOH (4 M, 2.25 mL, 9 mmol) was added and the mixture was stirred at 80 °C for 3 days. The mixture was diluted with water and treated with dry ice until the solution pH was ~11 at which point a solution of 4-nitrophenylcarbonate 15 (0.6 g, 1.7 mmol) in methanol (2 mL) was added. The mixture was stirred overnight at room temperature. The reaction was not complete. Et3N (410 μL, 3 mmol) and more 15 (0.3 g, 0.85 mmol) were added and the mixture was stirred overnight. The reaction was not complete. The mixture was concentrated and purified by HPLC (C-18) using water with 0.1% of TFA and acetonitrile gradient to provide compound 16 (285 mg; 62%) as a white foam; 1H-NMR (D2O) δ: 4.55–4.54 (m, 1 H, J1,2 7.9 Hz, H-1), 4.43 (d, 1 H, J1',2' 7.8 Hz, H-1'), 4.23 (d, 2 H, J 2.4 Hz, CH2propargyl), 4.22–4.15 (m, 4 H, H-4epyr, H-6epyr, CH2OCO), 3.98–3.92 (m, 2 H, H-6a, CH), 3.91 (d, 1 H, J3',4' 3.4 Hz, H-4'), 3.81–3.50 (m, 24 H, J2',3' 9.9 Hz, H-3, H-4, H-5, H-6b, H-2', H-3', H-5', H-6'a, H-6'b, H-4apyr, H-5pyr, H-6apyr, 6 × OCH2), 3.37–3.26 (m, 3 H, H-2', NCH2), 2.89 (t, 1 H, CH propargyl), 1.50 (s, 3 H ,CH3); ESI HRMS: m/z: calcd for C31H51NO21Na: 796.2846; found: 796.2841 [M+ Na]+.
(R,S)-5-{2-[α-D-galactopyranosyl-(1-4)-β-D-galactopyranosyl-(1-4)-β-D-glucopyranosyloxy]-5-oxo-6,9,12,15-tetraoxa-4-azaoctadec-17-ynyloxy}-2-methyl-1,3-dioxane-2-carboxylic acid (17). Lactose derivative
16 (273 mg, 0.35 mmol) was dissolved in water (3 mL) and neutralized with dry NaHCO
3. Then other components were added in the following order: 1 M HEPES buffer (pH: 8; 1.86 mL), DTT solution (0.4 M, 300 μL), alkaline phosphatase (1 U/μL, 30 μL, Sigma), UDP-glucose (320 mg, 0.52 mmol) and crude fusion enzyme GalT/UDP-4'-epimerase [
21] (0.5 mL). The mixture was incubated for 24 h at 37 °C then treated with Dowex H
+ resin, filtered and purified on HPLC (C-18) using water with 0.1% TFA and acetonitrile gradient. The fraction containing product was concentrated and lyophilized to afford the product
17 as a white foam (272 mg; 82 %);
1H-NMR (D
2O) δ: 4.94 (m, 1 H,
J1'',2'' 4.0 Hz, H-1''), 4.67–4.65 (m, 1 H,
J1,2 7.8 Hz, H-1), 4.50 (d, 1 H,
J1',2' 7.7 Hz, H-1'), 4.36-4.32 (m, 1 H,
J 5'',6''a =
J5'',6''b = 6.4 Hz, H-5''), 4.24 (d, 2 H,
J 2.4 Hz, CH
2 propargyl), 4.22–4.16 (m, 4 H, H-4e
pyr, H-6e
pyr, CH
2OCO), 4.04–4.01 (m, 2 H, H-4', H-4''), 4.00-–3.52 (m, 30 H, H-3, H-4, H-5, H-6a, H-6b, H-2', H-3', H-5', H-6'a, H-6'b, H-2'', H-3'', H-6''a, H-6''b, H-4a
pyr, H-5
pyr, H-6a
pyr, OCH, 6 × OCH
2), 3.38–3.27 (m, 3 H, H-2, NCH
2), 2.58 (t, 1 H, CH
propargyl), 1.50 (s, CH
3); ESI HRMS:
m/z: calcd for C
37H
61NO
26Na: 958.3374; found 958.3366 [M + Na]
+.
N-[3-(tert-Butyloxycarbonylamino)propyl]methacrylamide (19). To a solution of 1,3-diaminopropane (1 mL) in DCM (4 mL) methacrylic acid NHS ester (0.36 g, 1.96 mmol) was added. The mixture was stirred at room temperature for 15 min. The mixture was concentrated and the residue dissolved in methanol. To the solution di(tert-butoxy)carbonyl oxide (2.5 g, 10 mmol) was added followed by triethylamine (3 mL, 25 mmol). After 1 h the mixture was concentrated and the residue was chromatographed on silica gel using hexane-ethyl acetate (3:2) to give the title product 19 (0.2 g, 42%); 1H-NMR (CDCl3) δ: 6.74 (broad s, 1 H, NH), 5.73 (s, 1 H, CH2), 5.31 (s, 1 H, CH2), 4.96 (broad s, 1 H, NH), 3.35 (dd, 2 H, J 6.2 Hz, NCH2), 3.18 (dd, 2 H, J 5.9 Hz, NCH2), 1.96 (s, 3 H, CH3), 1.67–1.58 (m, 2 H, CH2), 1.43 (s, 9 H, tBu). ESI HRMS: m/z: calcd for C12H22N2O3Na ([M + Na]+): 265.15226; found: 265.15212.
tert-Butyl 2-(2-((4-nitrophenoxy)carbonyloxy)ethoxy)ethylcarbamate (21). 2-(2-Aminoethoxy)-ethanol (2.17 g; 20.6 mmol) was dissolved in methanol (10 mL) and di(tert-butoxy)carbonyl oxide (6.75 g; 30.9 mmol) was added followed by triethylamine (3.13 g, 30.9 mmol). After 30 min the mixture was concentrated and dried overnight under a vacuum provided by an oil pump. The residue was dissolved in dry DCM (20 mL) and p-nitrophenyl chloroformate (5.18 g, 25.7 mmol) was added followed by pyridine (3.26 g, 41.2 mmol). After 30 min the reaction was quenched by addition of methanol, then the mixture was diluted with DCM, washed with brine, concentrated and the residue was chromatographed on silica gel using hexane-acetone (4:1) to provide the product as slightly yellow syrup (7.146 g, 94%); 1H-NMR (CDCl3) δ: 8.24 and 7.40 (2 d, 4 H, C6H4), 4.90 (broad s, 1 H, NH), 4.43 (ddd, 2 H, J 4.6 Hz, J 2.9 Hz, OCH2), 3.77 (ddd, 2 H, OCH2), 3.58 (t, 2 H, J 5.2 Hz, NCH2 ), 3.38–3.00 (m, 2 H, NCH2), 1.45 (s, 9 H, t-Bu). ESI HRMS: m/z: calcd for C16H22N2O8Na ([M + Na]+): 393.12684; found 393.12652.
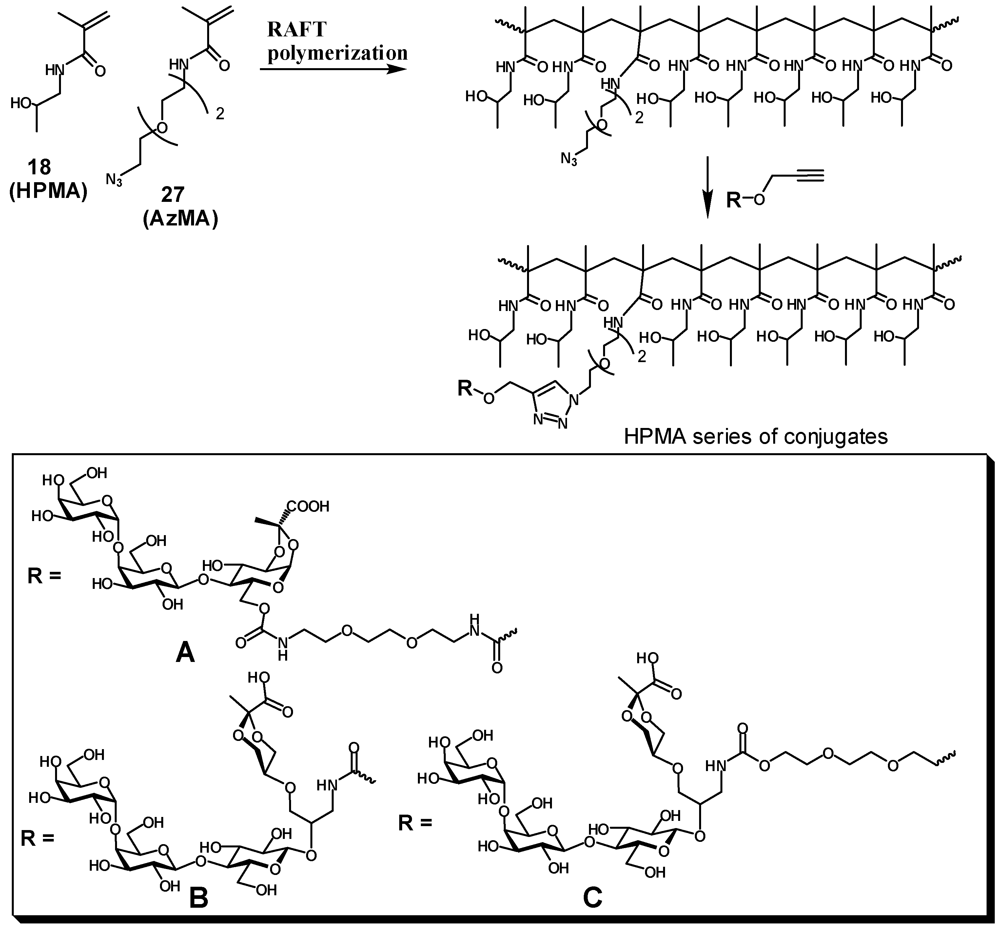
Copolymer of N-(2-hydroxypropyl)methacrylamide with methacrylamide 19 (20). A solution of N-(2-hydroxypropyl)methacrylamide (502.5 mg, 3.5 mmol) and 19 (42 mg, 0.174 mmol) in degassed water (7 mL) was ultrasonicated under vacuum. A solution of ammonium persulfate (7 mg in 70 μL of water) was added to the mixture followed by an aq. solution of cysteamine hydrochloride (98 μL; 1 mg/100 μL). The reaction mixture was vortexed and incubated at 50 °C overnight then concentrated and dissolved in treated with TFA (8 mL). After 2 h the mixture was concentrated, the residue was dissolved in water, dialyzed extensively against deionized water, then freeze-dried to provide a white powder of aminated HPMA polymer 20 (445 mg, 82%); 1H-NMR (D2O) δ: 4.00–3.90 (broad s, 1 H, CHOH), 3.30–3.00 (m, 2.2 H, CH2N, CH2NH2), 2.00–1.60 (m, 2 H, CH2), 1.60–0.80 (m, 6.3 H, CH2, CH3). Integration of 1H-NMR signals indicated ~5% incorporation of propylenediamine.
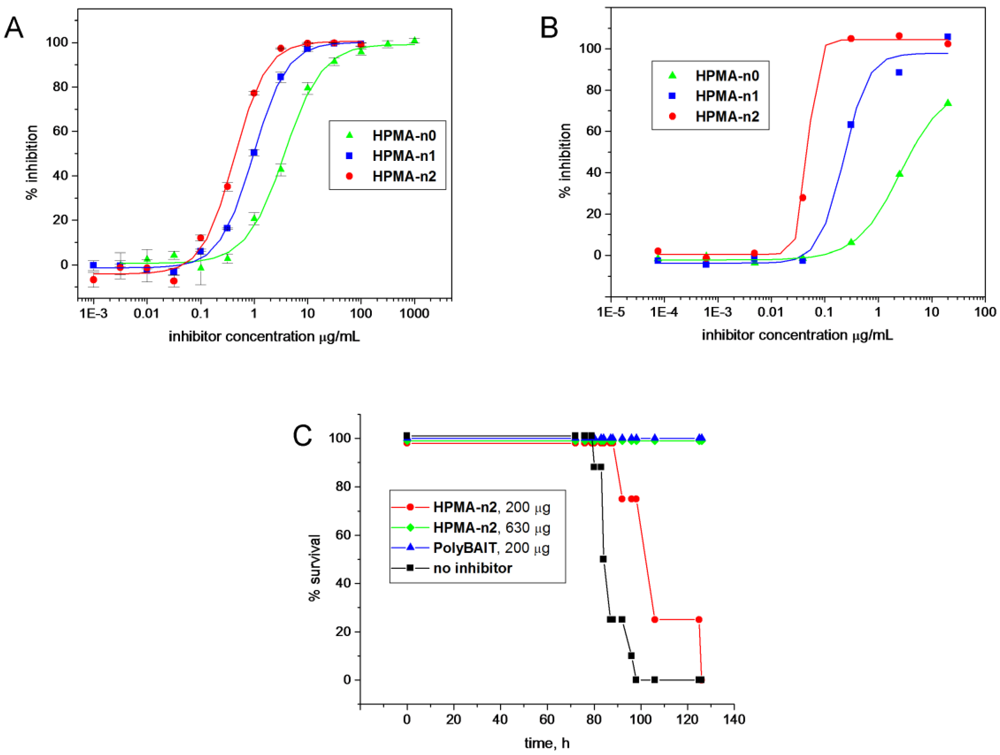
2.4. Enzymatic Galactosylation of HPMA-Lactose Polymers. Preparation of Trisaccharide Conjugates HPMA-n0, HPMA-n1, and HPMA-n2
Polymer
24 (41.5 mg) was dissolved in water (506 μL) then HEPES buffer (43 μL) was added, followed by aq. DTT solution (0.4 M, 10.6 μL) and alkaline phosphatase (1 U/μL, 1.06 μL). UDP-Glc (12.6 mg, 0.02 mmol) was added and followed by crude fusion enzyme GalT/UDP-4'-epimerase [
21] (32 μL) and the mixture was incubated at 37 °C overnight. Then it was treated with aq. 10% trichloroacetic acid (620 μL) and centrifuged. The solution was diluted with water and dialyzed against deionized water. The dialyzed solution was filtered through Milipore membrane (0.45 μm) and lyophilized to give the product as a white powder.
1H-NMR of the sample indicated the reaction was 70% complete. Enzymatic reaction was repeated to achieve complete conversion to give
HPMA-n0 as a white powder (32 mg);
1H-NMR (D
2O) δ: 5.68 (bs, 1H, H-1), 4.96 (d, 1 H,
J1”,2'' 3.5 Hz, H-1''), 4.52 (d, 1H,
J1,2 7.6 Hz, H-1), 4.46–4.34 (m, 3 H, H-3, H-5''), 4.28–4.18 (m, 2 H, H-2), 4.08–4.01 (m, 3 H, H-5, H-4', H-4''), 3.98–3.76 (m, 30 H, H-6a, H-6b, H-5', H-6a',H-6b', H-2'', H-3'', H-6a'', H-6b'', C
HOH), 3.76-3.68 (m, 2H, H-4, H-3'), 3.60 (dd, 1 H,
J2',3' 9.7 Hz, H-2'), 3.60–3.00 (m, 50 H, NCH
2), 2.1–0.6 (m, 198 H, CH
2, CH
3).
Polymer 25 was processed in a similar manner to give conjugate HPMA-n1 as white powder; 1H-NMR (D2O) δ: 5.64 (bs, 1 H, H-1), 4.96 (d, 1 H, J1'',2'' 3.3 Hz, H-1''), 4.52 (d, 1 H, J1,'2' 7.9 Hz, H-1'), 4.44 (m, 3 H, H-3, H-6a, H-5''), 4.30–4.16 (m, 4 H, H-2, H-6b, CH2OCO), 4.04 (bs, 3 H, H-5, H-4', H-4''), 4.00–3.62 (m, 32 H, H-4, H-3',H-5', H-6a', H-6b', H-2'', H-3'', H-6a'',H-6b'', CH,CH2), 3.60 (dd, 1-H, J2',3' 8.7 Hz, H-2'), 3.40–3.00 (m, 43 H, NCH2), 2.10–0.80 (m, 157 H, CH2, CH3).
Polymer 26 was processed in a similar manner to give conjugate HPMA-n2 as white powder; 1H-NMR (D2O) δ: 5.66 (bs, 1H, H-1), 4.95 (d, 1 H, H-1''), 4.52 (d, 1 H, H-1'), 4.22–4.36 (m, 3 H, H-3, H-6a, H-5''), 4.28–4.16 (m, 4 H, H-2, H-6b, CH2OCO), 4.04 (bs, 3 H, H-5, H-4', H-4''), 3.98–3.56 (m, 38 H, H-4, H-2', H-3', H-5', H-6a', H-6b', H-2'', H-3'', H-6a'', H-6b'', CH, CH2), 3.40–3.00 (m, 49 H, NCH2), 2.10–0.80 (m, 175 H, CH2, CH3).
N-(2-(2-(2-azidoethoxy)ethoxy)ethyl)methacrylamide (27, AzMA). To a solution of 2-(2-(2-azidoethoxy)ethoxy)ethanamine [
16] (3.17 g, 18.2 mmol) in DCM (15 mL) and methacrylic anhydride (4.2 g, 1.5 eq) was added followed by Et
3N (2 g, ~3 eq) and the mixture was stirred for 30 min. Chromatography on the silica gel in hexane—ethyl acetate (up to 70%) gave
27 (2.8 g, 63%).
1H-NMR (CDCl
3) δ: 6.26 (s, 1 H, NH), 5.68–5.67 (m, 1 H, CH
2=), 5.32–5.30 (m, 1 H, CH
2=), 3.68–3.62 (m, 6 H, CH
2), 3.59 (t, 2 H,
J 4.7 Hz, CH
2), 3.51 (dd, 2 H,
J 5.5 Hz,
J 10.8 Hz, CH
2), 3.37 (t, 2 H
J 4.8 Hz, CH
2), 1.95 (m, 3 H, CH
3);
13C-NMR (CDCl
3) δ: 168.408 (
C=O), 140.078 (
C=CH
2), 119.374 (C=
CH
2), 70.543 (CH
2O), 70.236 (CH
2O), 70.057 (CH
2O), 69.843 (CH
2O), 50.621 (
C–N
3), 39.333 (
CH
2–NH), 18.625 (
CH
3). ESI HRMS:
m/
z: calcd for C
10H
18N
4O
3Na ([M + Na]
+): 265.1271; found 265.1271.
2.6. General Procedure for Preparation of Polymeric Narrow Molecular Weight Heterobifunctional Ligands
An appropriate HPMA-AzMa copolymer (20–25 mg) and monomeric heterobifunctional ligand(5, 13 or 17, 1.5–2.0 eq) were dissolved in water (1 mL) and neutralized with dry NaHCO3 (to pH: 8), then 1 M sodium ascorbate (25 μL) and 0.05 M (50 μL) were added and the solution was left on a tumbler for 2 days. The reaction was checked by IR for the presence of azide. When no azido group could be detected the reaction mixture was diluted with water and extensively dialyzed against deionized water with the addition of EDTA, then filtered and lyophilized. Product was obtained in the form of white powder.
HPMA-16/5-A; 1H-NMR (D2O) δ: 8.10 (s, 1 H, CHtriazol), 7.80–7.50 (m, 24 H, NHHPMA), 5.62 (d, 1 H, J1,2 4.8 Hz, H-1), 5.20 (bs, 2 H, OCH2), 4.94 (d, 1 H, J1'',2'' 3.9 Hz, H-1''), 4.60–4.53 (m, 2H, NCH2), 4.52 (d, 1 H, J1',2' 7.7 Hz, H-1'), 4.42-4.35 (m, 3 H, H-3, H-6a, H-5''), 4.25–4.20 (m, 1 H, H-6b), 4.19–4.16 (m, 1 H, H-2), 4.05–4.00 (m, 3 H, H-5, H-4', H-4''), 3.96–3.56 (m, 51 H, H-4, H-2', H-3', H-5', H-6'a, H-2'', H-3'', H-6''a, H-6''b, 4 × OCH2, CHOH HPMA), 3.40–3.00 (m, 60 H, 2 × NCH2, NCH2 HPMA), 2.00–0.60 (m, 228 H, CH3, CH2 HPMA, CH3 HPMA).
HPMA-44/5-A; 1H-NMR (D2O) δ: 8.1 (s, 1 H, CHtriazol), 5.62 (d, 1 H, J1,2 4.9 Hz, H-1), 5.20 (bs, 2 H, OCH2), 4.94 (d, 1 H, J1'',2'' 3.9 Hz, H-1''), 4.60–4.54 (m, 2H, NCH2), 4.51 (d, 1 H, J1',2' 7.7 Hz, H-1'), 4.42 (m, 3 H, H-3, H-6a, H-5''), 4.22 (m, 1 H, H-6b), 4.17 (m, 1 H, H-2), 4.06–3.99 (m, 3 H, H-5, H-4', H-4''), 3.97–3.86 (m, 20 H, H-6'a, H-3'',CHOH HPMA), 3.86–3.54 (m, 16 H, H-4, H-2', H-3', H-5', H-6'b, H-2'', H-6''a, H-6''b, 4 × OCH2), 3.70–3.00 (m, 38 H, 2 × NCH2, NCH2 HPMA), 2.00–0.60 (m, 136 H, CH3, CH2, CH3 HPMA).
HPMA-35/10-A; 1H-NMR (D2O) δ: 8.06 (s, 1 H, CHtriazol), 5.62 (d, 1 H, J1,2 4.9 Hz, H-1), 5.20 (bs, 2 H, OCH2), 4.94 (d, 1 H, J1'',2'' 4.0 Hz, H-1''), 4.60–4.53 (m, 2H, NCH2), 4.51 (d, 1 H, J1',2' 7.7 Hz, H-1'), 4.42–4.34 (m, 3 H, H-3, H-6a, H-5''), 4.22–4.20 (m, 1 H, H-6b), 4.19–4.16 (m, 1 H, H-2), 4.04–4.00 (m, 3 H, H-5, H-4', H-4''), 3.96–3.86 (m, 13 H, H-6'a, H-3'',CHOH HPMA), 3.86–3.54 (m, 16 H, H-4, H-2', H-3', H-5', H-6'b, H-2'', H-6''a, H-6''b, 4 × OCH2), 3.90–2.90 (m, 28 H, 2 × NCH2, NCH2 HPMA), 2.00–0.60 (m, 100 H, CH3, CH2, CH3 HPMA).
HPMA-20/15-A; 1H-NMR (D2O) δ: 8.10 (s, 1 H, CHtriazol), 5.62 (d, 1 H, J1,2 4.6 Hz, H-1), 5.20 (bs, 2 H, OCH2), 4.95 (d, 1 H, J1'',2'' 3.9 Hz, H-1''), 4.60–4.53 (m, 2H, NCH2), 4.52 (d, 1 H, J1',2' 7.7 Hz, H-1'), 4.42–4.36 (m, 3 H, H-3, H-6a, H-5''), 4.26–4.20 (m, 1 H, H-6b), 4.19–4.15 (m, 1 H, H-2), 4.05–4.01 (m, 3 H, H-5, H-4', H-4''), 3.96–3.86 (m, 12 H, H-6'a, H-3'',CHOH HPMA), 3.86–3.54 (m, 16 H, H-4, H-2', H-3', H-5', H-6'b, H-2'', H-6''a, H-6''b, 4 × OCH2), 3.90–3.00 (m, 26 H, 2 × NCH2, NCH2 HPMA), 2.20–0.60 (m, 90 H, CH3, CH2 HPMA, CH3 HPMA).
HPMA-37/15-A; 1H-NMR (D2O) δ: 8.10 (s, 1 H, CHtriazol), 5.62 (d, 1 H, J1,2 4.8 Hz, H-1), 5.20 (bs, 2 H, OCH2), 4.94 (d, 1 H, J1'',2'' 3.9 Hz, H-1''), 4.60–4.54 (m, 2 H, NCH2), 4.51 (d, 1 H, J1',2' 7.7 Hz, H-1'), 4.42–4.34 (m, 3 H, H-3, H-6a, H-5''), 4.24–4.19 (m, 1 H, H-6b), 4.19–4.16 (m, 1 H, H-2), 4.04–4.01 (m, 3 H, H-5, H-4', H-4''), 3.96–3.86 (m, 11 H, H-6'a, H-3'',CHOH HPMA), 3.86–3.54 (m, 16 H, H-4, H-2', H-3', H-5', H-6'b, H-2'', H-6''a, H-6''b, 4 × OCH2), 3.90–3.00 (m, 22 H, 2 × NCH2, NCH2 HPMA), 2.20–0.60 (m, 80 H, CH3, CH2 HPMA, CH3 HPMA).
HPMA-44/5-B; 1H-NMR (D2O) δ: 8.10 (s, 1 H, CHtriazol), 5.20 (bs, 2 H, OCH2), 4.94 (d, 1H, J1'',2'' 3.8 Hz, H-1''), 4.62–4.47 (m, 4 H, H-1, H-1', NCH2), 4.34 (dd, 1 H, J5'',6''a ~ J5'',6''b ~ 6.5 Hz, H-5''), 4.12–3.99 (m, 4 H, H-4', H-4'', H-4epyr, H-6epyr), 3.99–3.46 (m, 40 H, H-3, H-4, H-6a, H-6b, H-2', H-3', H-5', H-6'a, H-6'b, H-2'',H-3'', H-6''a, H-6''b, H-4apyr, H-5pyr, H-6apyr, OCH2, OCH HPMA), 3.35–2.96 (m, 45 H, H-2, NCH2 HPMA), 2.00–0.06 (m, 176 H, CH3, CH2 HPMA, CH3 HPMA).
HPMA-35/10-B; 1H-NMR (D2O) δ: 8.10 (s, 1 H, CHtriazol), 5.20 (bs, 2 H, OCH2), 4.94 (d, 1H, J1'',2'' 3.8 Hz, H-1''), 4.63–4.47 (m, 4 H, H-1, H-1', NCH2), 4.34 (dd, 1 H, J5'',6''a ~ J5'',6''b ~ 6.4 Hz, H-5''), 4.12–4.00 (m, 4 H, H-4', H-4'', H-4epyr, H-6epyr), 3.98–3.46 (m, 41 H, H-3, H-4, H-6a, H-6b, H-2', H-3', H-5', H-6'a, H-6'b, H-2'',H-3'', H-6''a, H-6''b, H-4apyr, H-5pyr, H-6apyr, OCH2, OCH HPMA), 3.35–2.96 (m, 41 H, H-2, NCH2 HPMA), 2.00–0.06 (m, 160 H, CH3, CH2 HPMA, CH3 HPMA).
HPMA-44/5-C; 1H-NMR (D2O) δ: 8.10 (s, 1 H, CHtriazol), 7.90–7.40 (m, 18 H, NH HPMA), 4.94 (d, 1 H, J1'',2'' 3.9 Hz, H-1''), 4.59–4.48 (m, 3 H, H-1, H-1', OCH2), 4.34 (dd, 1 H, J5'',6''a ~ J5'',6''b ~ 6.5 Hz, H-5''), 4.23–4.16 (m, 2 H, CH2OCO), 4.14–4.06 (m, 2 H, H-4epyr, H-6epyr), 4.04–4.00 (m, 2H, H-4', H-4''), 3.98–3.49 (m, 54 H, H-3, H-4, H-5, H-6a, H-6b, H-2', H-3', H-5', H-6'a, H-6'b, H-2'', H-3'', H-6''a, H-6''b, H-4apyr, H-5pyr, H-6apyr, OCH, 6 × OCH2, OCHHPMA), 3.47–3.00 (m, 45 H, H-2, NCH2, NCH2 HPMA), 2.10–0.60 (m, 176 H, CH3, CH2 HPMA, CH3 HPMA).
HPMA-35/10-C; 1H-NMR (D2O) δ: 8.10 (s, 1 H, CHtriazol), 7.90–7.40 (m, 13 H, NH HPMA), 4.94 (d, 1 H, J1'',2'' 3.9 Hz, H-1''), 4.60–4.48 (m, 3 H, H-1, H-1', OCH2), 4.34 (dd, 1 H, J5'',6''a = J5'',6''b= 6.5 Hz, H-5''), 4.25–4.17 ( m, 2 H, CH2OCO), 4.14–4.06 (m, 2 H, H-4epyr, H-6epyr), 4.04–4.00 (m, 2 H, H-4', H-4''), 4.00–3.48 (m, 46 H, H-3, H-4, H-5, H-6a, H-6b, H-2', H-3', H-5', H-6'a, H-6'b, H-2'', H-3'', H-6''a, H-6''b, H-4apyr, H-5 pyr, H-6apyr, 6 × OCH2, OCHHPMA), 3.48–3.00 (m, 37 H, H-2, 2 × NCH2, NCH2 HPMA), 2.10–0.60 (m, 136 H, CH3, CH2 HPMA, CH3 HPMA).
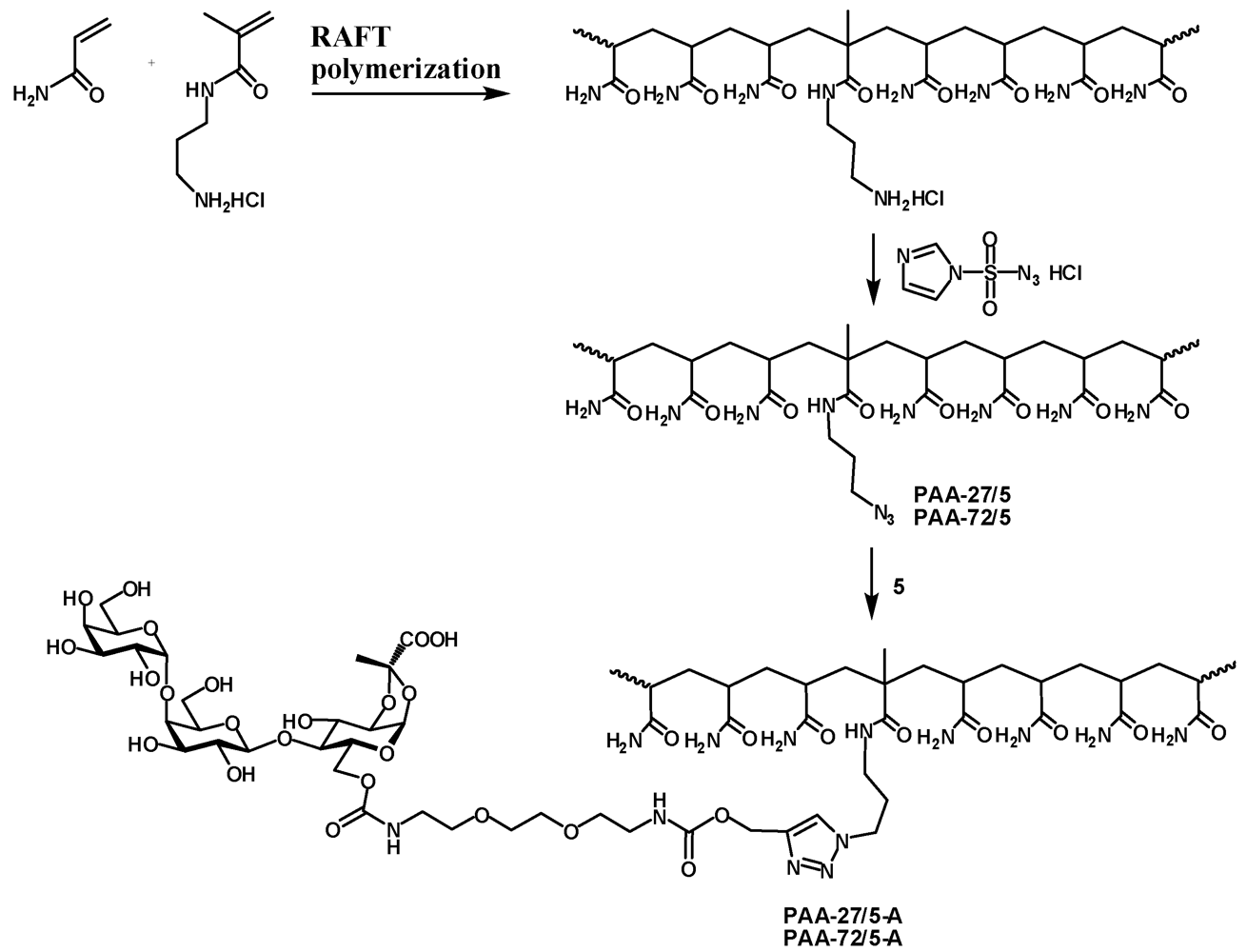
2.7. Typical RAFT Polymerization of Acrylamide Monomers
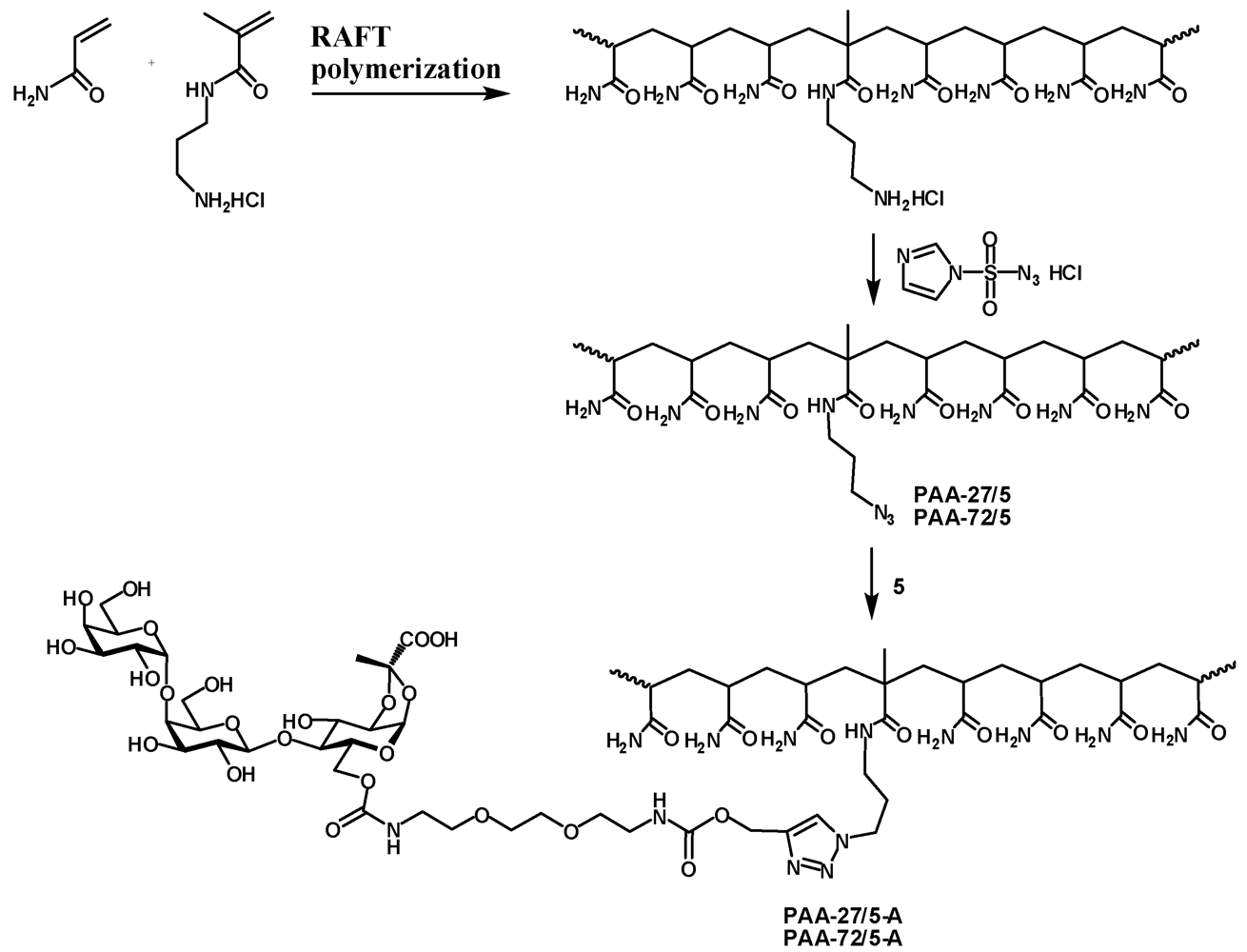
The statistical copolymerization of 3-aminopropyl methacrylamide (APMA) and acrylamide (AA) was performed at 70 °C in the presence of 4-cyanopentanoic acid dithiobenzoate (CTP) and 4,4'-azobis(4-cyanovaleric acid) (ACVA) as chain transfer agent and initiator, respectively. In a typical protocol AA (2 g, 28 mmol) and APMA (0.25 g, 1.4 mmol) was dissolved in dimethylsulfoxide (DMSO) (10 mL) followed by the addition of CTP (8 mg, 29 µmol) and ACVA (2 mg, 7 µmol) (targeted DPn 977, Targeted molecular weight 70 kDa). The mixture was purged with nitrogen for 45 minutes and flask was placed in oil bath for polymerization under nitrogen for 24 h. The polymerization reaction mixture was quenched using liquid nitrogen and polymer was precipitated in acetone. The molecular weight and molecular weight distributions of copolymers were determined using aqueous gel permeation chromatography.
PAA-27/5. Poly[acrylamide-co-(3-aminopropylmethacrylamide hydrochloride)] (M.W.: 27000; 5% of amine; 210 mg, 141 μmol) was dissolved in water (2.5 mL) and imidazole-1-sulfonyl azide [
17] (118 mg, 563 μmol) was added followed by 0.4 M solution of CuSO
4/H
2O (12.5 μL) and pH of the solution was adjusted to 10 by addition of 5 M NaOH (200 μL). The reaction mixture was stirred at room temperature for 6 h, then dialyzed, filtered and lyophilized to provide the product as an off white powder (177 mg). IR: 2103.0 cm
−1,
1H-NMR (D
2O) δ: 3.44–3.35 (m, 2 H, NCH
2), 3.32–3.12 (m, 2 H, NCH
2), 2.38–1.98 (m, 16 H, CH
2 linker, CH), 1.84–1.34 (m, 32 H, CH
2), 1.2–1.08 (m, 3 H, CH
3).
PAA-72/5. The title product was obtained from poly[acrylamide-co-(3-aminopropylmethacrylamide hydrochloride)] as described for preparation of PAA-27/5 (M.W.: 72000; 5% of amine; 210 mg, 141 μmol) as described for PAA-27/5. The title product obtained as off white powder (156 mg). IR: 2103.9 cm−1. 1H-NMR (D2O) δ: 3.44–3.34 (m, 2 H, NCH2), 3.30–3.12 (m, 2 H, NCH2), 2.38–1.98 (m, 16 H, CH2 linker, CH), 1.82–1.34 (m, 34 H, CH2), 1.2–1.08 (m, 3 H, CH3).
PAA-27/5-A. Azide PAA-27/5 (21.8 mg, 14.3 μmol) and 5 (23.5 mg, 28.3 μmol) were dissolved in water (1 mL) and pH adjusted to 8. Then 1 M sodium ascorbate (25 μL) and 0.05 M copper sulfate (50 μL). The reaction mixture was left on a tumbler for 2 days. Then it was dialyzed, filtered and freeze-dried to afford the polymer PAA-27/5-A as a white powder (35 mg). 1H-NMR (D2O) δ: 8.06 (s, 1 H, CHtriazol), 5.62 (d, 1 H, J1,2 4.9 Hz, H-1), 5.18 (bs, 2 H, OCH2), 4.94 (d, 1 H, J1'',2'' 3.9 Hz, H-1''), 4.51 (d, 1 H, J1',2' 7.8 Hz, H-1'), 4.50–4.35 (m, 5 H, J6a,6b 12.0 Hz, H-3, H-6a, H-5'', NCH2), 4.23 (dd, 1 H, J5,6b 5.1 Hz, H-6b), 4.18 (dd, 1 H, J2,3 3.9 Hz, H-2), 4.04–4.01 (m, 3 H, H-5, H-4', H-4''), 3.94–3.89 (m, 2 H, H-6'a, H-3''), 3.85–3.60 (m, 16 H, H-4, H-2', H-3', H-5', H-6'b, H-2'', H-6''a, H-6''b, 4 × OCH2), 3.34–3.29 (m, 4 H, 2 × NCH2), 3.20–3.02 (m, 2 H, NCH2), 2.40–2.00 (m,16 H, CH2 , CH PAA), 1.80–1.30 (m, 31 H, CH3, CH2 PAA), 1.20–1.00 (m, 3 H, CH3 MAA).
PAA-72/5-A. Azide PAA-72/5 (21.6 mg, 14.4 μmol) and 5 (24.3 mg, 29 μmol) were dissolved in water (1 mL) and reaction and work-up were performed as described for PAA-27/5-A. The tile PAA-72/5-A was obtained as a white powder (35.3 mg). 1H-NMR (D2O) δ: 8.10 (s, 1 H, CHtriazol), 5.62 (d, 1 H, J1,2 4.9 Hz, H-1), 5.18 (bs, 2 H, OCH2), 4.94 (d, 1 H, J1'',2'' 3.9 Hz, H-1''), 4.51 (d, 1 H, J1',2' 7.7 Hz, H-1'), 4.49–4.34 (m, 5 H, J6a,6b 11.8 Hz, H-3, H-6a, H-5'', NCH2), 4.23 (dd, 1 H, J5,6b 5.1 Hz, H-6b), 4.18 (dd, 1 H, J2,3 4.0 Hz, H-2), 4.05–4.00 (m, 3 H, H-5, H-4', H-4''), 3.94–3.89 (m, 2 H, H-6'a, H-3''), 3.85–3.60 (m, 16 H, H-4, H-2', H-3', H-5', H-6'b, H-2'', H-6''a, H-6''b, 4 × OCH2), 3.34–3.28 (m, 4 H, 2 × NCH2), 3.20–3.02 (m, 2 H, NCH2), 2.40–1.90 (m,19 H, CH2, CH PAA), 1.80–1.30 (m, 36 H, CH3, CH2 PAA), 1.20–1.00 (m, 3 H, CH3 MAA).


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}