Different Types of Cell Death Induced by Enterotoxins

Abstract
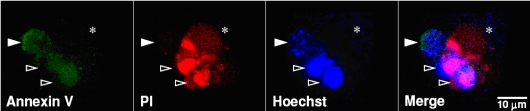
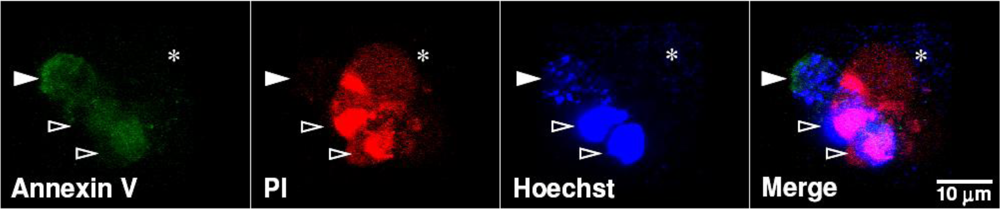
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Apoptotic Cell Death
2.1. Extrinsic (Death Receptor-mediated) and Intrinsic (Mitochondria-Regulated) Pathways of Apoptosis
2.2. Endoplasmic Reticulum Stress-Mediated Apoptosis
2.3. Lysosomal Pathway of Apoptotic Signaling
3. Necrotic Cell Death
4. Cytotoxic Enterotoxins
4.1. Cytotoxicity of S. aureus
4.2. Cytotoxicity of E. coli
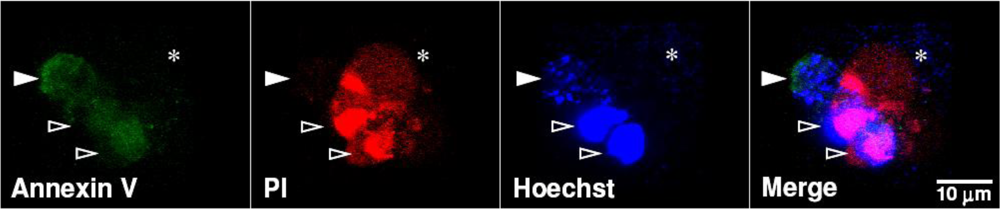
4.3. Cytotoxicity of V. cholerae
5. Concluding Remarks
Acknowledgements
References
- Balk, R.A. Severe sepsis and septic shock. Definitions, epidemiology, and clinical manifestations. Crit. Care Clin. 2000, 16, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Balk, R.A. Pathogenesis and management of multiple organ dysfunction or failure in severe sepsis and septic shock. Crit. Care Clin. 2000, 16, 337–352. [Google Scholar]
- Sessler, C.N.; Perry, J.C.; Varney, K.L. Management of severe sepsis and septic shock. Curr. Opin. Crit. Care 2004, 10, 354–363. [Google Scholar]
- Rivers, E. The outcome of patients presenting to the emergency department with severe sepsis or septic shock. Crit. Care 2006, 10, 154. [Google Scholar]
- Angus, D.C.; Wax, R.S. Epidemiology of sepsis: an update. Crit. Care Med. 2001, 29, S109–S116. [Google Scholar]
- Patel, G.P.; Gurka, D.P.; Balk, R.A. New treatment strategies for severe sepsis and septic shock. Curr. Opin. Crit. Care 2003, 9, 390–396. [Google Scholar]
- Bhakdi, S.; Bayley, H.; Valeva, A.; Walev, I.; Walker, B.; Kehoe, M.; Palmer, M. Staphylococcal alpha-toxin, streptolysin-O, and Escherichia coli hemolysin: prototypes of pore-forming bacterial cytolysins. Arch. Microbiol. 1996, 165, 73–79. [Google Scholar]
- Tesh, V.L. Virulence of enterohemorrhagic Escherichia coli: Role of molecular crosstalk. Trends Microbiol. 1998, 6, 228–233. [Google Scholar]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13 table of contents, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Sierig, G.; Cywes, C.; Wessels, M.R.; Ashbaugh, C.D. Cytotoxic effects of streptolysin o and streptolysin s enhance the virulence of poorly encapsulated group a streptococci. Infect. Immun. 2003, 71, 446–455. [Google Scholar]
- Marriott, H.M.; Dockrell, D.H. Streptococcus pneumoniae: The role of apoptosis in host defense and pathogenesis. Int. J. Biochem. Cell Biol. 2006, 38, 1848–1854. [Google Scholar]
- Katahira, J.; Sugiyama, H.; Inoue, N.; Horiguchi, Y.; Matsuda, M.; Sugimoto, N. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J. Biol. Chem. 1997, 272, 26652–26658. [Google Scholar]
- Vanden Broeck, D.; Horvath, C.; De Wolf, M.J. Vibrio cholerae: Cholera toxin. Int. J. Biochem. Cell Biol. 2007, 39, 1771–1775. [Google Scholar]
- Cherla, R.P.; Lee, S.Y.; Tesh, V.L. Shiga toxins and apoptosis. FEMS Microbiol. Lett. 2003, 228, 159–166. [Google Scholar]
- Grassme, H.; Jendrossek, V.; Gulbins, E. Molecular mechanisms of bacteria induced apoptosis. Apoptosis 2001, 6, 441–445. [Google Scholar]
- Hacker, G. Introduction: the various deaths a cell can die, and their use in microbial infections. Microbes Infect. 2009, 11, 1047–1049. [Google Scholar]
- Bohme, L.; Rudel, T. Host cell death machinery as a target for bacterial pathogens. Microbes Infect. 2009, 11, 1063–1070. [Google Scholar]
- Rosado, C.J.; Kondos, S.; Bull, T.E.; Kuiper, M.J.; Law, R.H.; Buckle, A.M.; Voskoboinik, I.; Bird, P.I.; Trapani, J.A.; Whisstock, J.C.; Dunstone, M.A. The MACPF/CDC family of pore-forming toxins. Cell Microbiol. 2008, 10, 1765–1774. [Google Scholar]
- Jin, Z.; El-Deiry, W.S. Overview of cell death signaling pathways. Cancer Biol. Ther. 2005, 4, 139–163. [Google Scholar]
- Wesche-Soldato, D.E.; Swan, R.Z.; Chung, C.S.; Ayala, A. The apoptotic pathway as a therapeutic target in sepsis. Curr. Drug Targets 2007, 8, 493–500. [Google Scholar]
- Ayala, A.; Wesche-Soldato, D.E.; Perl, M.; Lomas-Neira, J.L.; Swan, R.; Chung, C.S. Blockade of apoptosis as a rational therapeutic strategy for the treatment of sepsis. Novartis Found. Symp. 2007, 280 discussion 49–52, 160–164., 37–49. [Google Scholar] [PubMed]
- Dhein, J.; Walczak, H.; Baumler, C.; Debatin, K.M.; Krammer, P.H. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature 1995, 373, 438–441. [Google Scholar]
- Tartaglia, L.A.; Rothe, M.; Hu, Y.F.; Goeddel, D.V. Tumor necrosis factor's cytotoxic activity is signaled by the p55 TNF receptor. Cell 1993, 73, 213–216. [Google Scholar]
- Kitson, J.; Raven, T.; Jiang, Y.P.; Goeddel, D.V.; Giles, K.M.; Pun, K.T.; Grinham, C.J.; Brown, R.; Farrow, S.N. A death-domain-containing receptor that mediates apoptosis. Nature 1996, 384, 372–375. [Google Scholar]
- Pan, G.; O'Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar]
- Reed, J.C. Bcl-2 family proteins. Oncogene 1998, 17, 3225–3236. [Google Scholar]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar]
- Zamzami, N.; El Hamel, C.; Maisse, C.; Brenner, C.; Munoz-Pinedo, C.; Belzacq, A.S.; Costantini, P.; Vieira, H.; Loeffler, M.; Molle, G.; Kroemer, G. Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene 2000, 19, 6342–6350. [Google Scholar]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Invest. 2005, 115, 2656–2664. [Google Scholar]
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 2001, 21, 1249–1259. [Google Scholar]
- Tan, Y.; Dourdin, N.; Wu, C.; De Veyra, T.; Elce, J.S.; Greer, P.A. Ubiquitous calpains promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 2006, 281, 16016–16024. [Google Scholar]
- Dahmer, M.K. Caspases-2, -3, and -7 are involved in thapsigargin-induced apoptosis of SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 2005, 80, 576–583. [Google Scholar]
- Cheung, H.H.; Lynn, K.N.; Liston, P.; Korneluk, R.G. Involvement of caspase-2 and caspase-9 in endoplasmic reticulum stress-induced apoptosis: A role for the IAPs. Exp. Cell Res. 2006, 312, 2347–2357. [Google Scholar]
- Upton, J.P.; Austgen, K.; Nishino, M.; Coakley, K.M.; Hagen, A.; Han, D.; Papa, F.R.; Oakes, S.A. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol. Cell. Biol. 2008, 28, 3943–3951. [Google Scholar]
- Ferri, K.F.; Kroemer, G. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 2001, 3, E255–E263. [Google Scholar]
- Guicciardi, M.E.; Leist, M.; Gores, G.J. Lysosomes in cell death. Oncogene 2004, 23, 2881–2890. [Google Scholar]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar]
- Chwieralski, C.E.; Welte, T.; Buhling, F. Cathepsin-regulated apoptosis. Apoptosis 2006, 11, 143–149. [Google Scholar]
- Duprez, L.; Wirawan, E.; Vanden Berghe, T.; Vandenabeele, P. Major cell death pathways at a glance. Microbes Infect. 2009, 11, 1050–1062. [Google Scholar]
- Vanlangenakker, N.; Vanden Berghe, T.; Krysko, D.V.; Festjens, N.; Vandenabeele, P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr. Mol. Med. 2008, 8, 207–220. [Google Scholar]
- Chautan, M.; Chazal, G.; Cecconi, F.; Gruss, P.; Golstein, P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr. Biol. 1999, 9, 967–970. [Google Scholar]
- Kawahara, A.; Ohsawa, Y.; Matsumura, H.; Uchiyama, Y.; Nagata, S. Caspase-independent cell killing by Fas-associated protein with death domain. J. Cell Biol. 1998, 143, 1353–1360. [Google Scholar]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar]
- Kalai, M.; Van Loo, G.; Vanden Berghe, T.; Meeus, A.; Burm, W.; Saelens, X.; Vandenabeele, P. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ. 2002, 9, 981–994. [Google Scholar]
- Ma, Y.; Temkin, V.; Liu, H.; Pope, R.M. NF-kappaB protects macrophages from lipopolysaccharide-induced cell death: The role of caspase 8 and receptor-interacting protein. J. Biol. Chem. 2005, 280, 41827–41834. [Google Scholar]
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar]
- Schlievert, P.M.; Case, L.C. Molecular analysis of staphylococcal superantigens. Methods Mol. Biol. 2007, 391, 113–126. [Google Scholar]
- Marrack, P.; Blackman, M.; Kushnir, E.; Kappler, J. The toxicity of staphylococcal enterotoxin B in mice is mediated by T cells. J. Exp. Med. 1990, 171, 455–464. [Google Scholar]
- Nagaki, M.; Muto, Y.; Ohnishi, H.; Yasuda, S.; Sano, K.; Naito, T.; Maeda, T.; Yamada, T.; Moriwaki, H. Hepatic injury and lethal shock in galactosamine-sensitized mice induced by the superantigen staphylococcal enterotoxin B. Gastroenterology 1994, 106, 450–458. [Google Scholar]
- Florquin, S.; Amraoui, Z.; Abramowicz, D.; Goldman, M. Systemic release and protective role of IL-10 in staphylococcal enterotoxin B-induced shock in mice. J. Immunol. 1994, 153, 2618–2623. [Google Scholar]
- Jedrzkiewicz, S.; Kataeva, G.; Hogaboam, C.M.; Kunkel, S.L.; Strieter, R.M.; McKay, D.M. Superantigen immune stimulation evokes epithelial monocyte chemoattractant protein 1 and RANTES production. Infect. Immun. 1999, 67, 6198–6202. [Google Scholar]
- Faulkner, L.; Cooper, A.; Fantino, C.; Altmann, D.M.; Sriskandan, S. The mechanism of superantigen-mediated toxic shock: not a simple Th1 cytokine storm. J. Immunol. 2005, 175, 6870–6877. [Google Scholar]
- Miethke, T.; Wahl, C.; Heeg, K.; Echtenacher, B.; Krammer, P.H.; Wagner, H. T cell-mediated lethal shock triggered in mice by the superantigen staphylococcal enterotoxin B: Critical role of tumor necrosis factor. J. Exp. Med. 1992, 175, 91–98. [Google Scholar]
- Trede, N.S.; Tsytsykova, A.V.; Chatila, T.; Goldfeld, A.E.; Geha, R.S. Transcriptional activation of the human TNF-alpha promoter by superantigen in human monocytic cells: Role of NF-kappa B. J. Immunol. 1995, 155, 902–908. [Google Scholar]
- Kramer, B.; Machleidt, T.; Wiegmann, K.; Kronke, M. Superantigen-induced transcriptional activation of the human TNF gene promoter in T cells. J. Inflamm. 1995, 45, 183–192. [Google Scholar]
- Lin, Y.S.; Lei, H.Y.; Low, T.L.; Shen, C.L.; Chou, L.J.; Jan, M.S. In vivo induction of apoptosis in immature thymocytes by staphylococcal enterotoxin B. J. Immunol. 1992, 149, 1156–1163. [Google Scholar]
- Biasi, G.; Panozzo, M.; Pertile, P.; Mezzalira, S.; Facchinetti, A. Mechanism underlying superantigen-induced clonal deletion of mature T lymphocytes. Int. Immunol. 1994, 6, 983–989. [Google Scholar]
- Lin, Y.S.; Huang, Y.T.; Chen, P.S.; Lin, C.F.; Jan, M.S.; Lei, H.Y. Requirement of I-E molecule for thymocyte apoptosis induced by staphylococcal enterotoxin B in vivo. Cell Immunol. 1999, 193, 71–79. [Google Scholar]
- Facchinetti, A.; Panozzo, M.; Pertile, P.; Tessarollo, L.; Biasi, G. In vivo and in vitro death of mature T cells induced by separate signals to CD4 and alpha beta TCR. Immunobiology 1992, 185, 380–389. [Google Scholar]
- Izquierdo, M.; Grandien, A.; Criado, L.M.; Robles, S.; Leonardo, E.; Albar, J.P.; de Buitrago, G.G.; Martinez, A.C. Blocked negative selection of developing T cells in mice expressing the baculovirus p35 caspase inhibitor. EMBO J. 1999, 18, 156–166. [Google Scholar]
- Ionin, B.; Hammamieh, R.; Shupp, J.W.; Das, R.; Pontzer, C.H.; Jett, M. Staphylococcal enterotoxin B causes differential expression of Rnd3 and RhoA in renal proximal tubule epithelial cells while inducing actin stress fiber assembly and apoptosis. Microb. Pathog. 2008, 45, 303–309. [Google Scholar]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar]
- Walev, I.; Martin, E.; Jonas, D.; Mohamadzadeh, M.; Muller-Klieser, W.; Kunz, L.; Bhakdi, S. Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect. Immun. 1993, 61, 4972–4979. [Google Scholar]
- Jonas, D.; Walev, I.; Berger, T.; Liebetrau, M.; Palmer, M.; Bhakdi, S. Novel path to apoptosis: small transmembrane pores created by staphylococcal alpha-toxin in T lymphocytes evoke internucleosomal DNA degradation. Infect. Immun. 1994, 62, 1304–1312. [Google Scholar]
- Bantel, H.; Sinha, B.; Domschke, W.; Peters, G.; Schulze-Osthoff, K.; Janicke, R.U. alpha-Toxin is a mediator of Staphylococcus aureus-induced cell death and activates caspases via the intrinsic death pathway independently of death receptor signaling. J. Cell Biol. 2001, 155, 637–648. [Google Scholar]
- Essmann, F.; Bantel, H.; Totzke, G.; Engels, I.H.; Sinha, B.; Schulze-Osthoff, K.; Janicke, R.U. Staphylococcus aureus alpha-toxin-induced cell death: predominant necrosis despite apoptotic caspase activation. Cell Death Differ. 2003, 10, 1260–1272. [Google Scholar]
- Haslinger, B.; Strangfeld, K.; Peters, G.; Schulze-Osthoff, K.; Sinha, B. Staphylococcus aureus alpha-toxin induces apoptosis in peripheral blood mononuclear cells: Role of endogenous tumour necrosis factor-alpha and the mitochondrial death pathway. Cell Microbiol. 2003, 5, 729–741. [Google Scholar]
- Bernheimer, A.W.; Schwartz, L.L. Lysosomal disruption by bacterial toxins. J. Bacteriol. 1964, 87, 1100–1104. [Google Scholar]
- Hameed, A.; Olsen, K.J.; Lee, M.K.; Lichtenheld, M.G.; Podack, E.R. Cytolysis by Ca-permeable transmembrane channels. Pore formation causes extensive DNA degradation and cell lysis. J. Exp. Med. 1989, 169, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Gillet, Y.; Issartel, B.; Vanhems, P.; Fournet, J.C.; Lina, G.; Bes, M.; Vandenesch, F.; Piemont, Y.; Brousse, N.; Floret, D.; Etienne, J. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet 2002, 359, 753–759. [Google Scholar]
- Gillet, Y.; Issartel, B.; Vanhems, P.; Lina, G.; Vandenesch, F.; Etienne, J.; Floret, D. Severe staphylococcal pneumonia in children. Arch. Pediatr. 2001, 8 (Suppl. 4), 742s–746s. [Google Scholar] [PubMed]
- Genestier, A.L.; Michallet, M.C.; Prevost, G.; Bellot, G.; Chalabreysse, L.; Peyrol, S.; Thivolet, F.; Etienne, J.; Lina, G.; Vallette, F.M.; Vandenesch, F.; Genestier, L. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J. Clin. Invest. 2005, 115, 3117–3127. [Google Scholar]
- Labandeira-Rey, M.; Couzon, F.; Boisset, S.; Brown, E.L.; Bes, M.; Benito, Y.; Barbu, E.M.; Vazquez, V.; Hook, M.; Etienne, J.; Vandenesch, F.; Bowden, M.G. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science 2007, 315, 1130–1133. [Google Scholar]
- Loffler, B.; Hussain, M.; Grundmeier, M.; Bruck, M.; Holzinger, D.; Varga, G.; Roth, J.; Kahl, B.C.; Proctor, R.A.; Peters, G. Staphylococcus aureus panton-valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathog. 2010, 6, e1000715. [Google Scholar]
- Anderson, G.G.; Martin, S.M.; Hultgren, S.J. Host subversion by formation of intracellular bacterial communities in the urinary tract. Microbes Infect. 2004, 6, 1094–1101. [Google Scholar]
- Schilling, J.D.; Mulvey, M.A.; Hultgren, S.J. Dynamic interactions between host and pathogen during acute urinary tract infections. Urology 2001, 57, 56–61. [Google Scholar]
- Mulvey, M.A.; Lopez-Boado, Y.S.; Wilson, C.L.; Roth, R.; Parks, W.C.; Heuser, J.; Hultgren, S.J. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 1998, 282, 1494–1497. [Google Scholar]
- Guignot, J.; Breard, J.; Bernet-Camard, M.F.; Peiffer, I.; Nowicki, B.J.; Servin, A.L.; Blanc-Potard, A.B. Pyelonephritogenic diffusely adhering Escherichia coli EC7372 harboring Dr-II adhesin carries classical uropathogenic virulence genes and promotes cell lysis and apoptosis in polarized epithelial caco-2/TC7 cells. Infect. Immun. 2000, 68, 7018–7027. [Google Scholar]
- Weinrauch, Y.; Zychlinsky, A. The induction of apoptosis by bacterial pathogens. Annu. Rev. Microbiol. 1999, 53, 155–187. [Google Scholar]
- Bhakdi, S.; Mackman, N.; Nicaud, J.M.; Holland, I.B. Escherichia coli hemolysin may damage target cell membranes by generating transmembrane pores. Infect. Immun. 1986, 52, 63–69. [Google Scholar]
- Bhakdi, S.; Greulich, S.; Muhly, M.; Eberspacher, B.; Becker, H.; Thiele, A.; Hugo, F. Potent leukocidal action of Escherichia coli hemolysin mediated by permeabilization of target cell membranes. J. Exp. Med. 1989, 169, 737–754. [Google Scholar]
- Cavalieri, S.J.; Snyder, I.S. Effect of Escherichia coli alpha-hemolysin on human peripheral leukocyte viability in vitro. Infect. Immun. 1982, 36, 455–461. [Google Scholar]
- Gadeberg, O.V.; Orskov, I. In vitro cytotoxic effect of alpha-hemolytic Escherichia coli on human blood granulocytes. Infect. Immun. 1984, 45, 255–260. [Google Scholar]
- Cavalieri, S.J.; Snyder, I.S. Effect of Escherichia coli alpha-hemolysin on human peripheral leukocyte function in vitro. Infect. Immun. 1982, 37, 966–974. [Google Scholar]
- Bhakdi, S.; Muhly, M.; Korom, S.; Schmidt, G. Effects of Escherichia coli hemolysin on human monocytes. Cytocidal action and stimulation of interleukin 1 release. J. Clin. Invest. 1990, 85, 1746–1753. [Google Scholar] [CrossRef] [PubMed]
- Suttorp, N.; Floer, B.; Schnittler, H.; Seeger, W.; Bhakdi, S. Effects of Escherichia coli hemolysin on endothelial cell function. Infect. Immun. 1990, 58, 3796–3801. [Google Scholar]
- Keane, W.F.; Welch, R.; Gekker, G.; Peterson, P.K. Mechanism of Escherichia coli alpha-hemolysin-induced injury to isolated renal tubular cells. Am. J. Pathol. 1987, 126, 350–357. [Google Scholar]
- Jonas, D.; Schultheis, B.; Klas, C.; Krammer, P.H.; Bhakdi, S. Cytocidal effects of Escherichia coli hemolysin on human T lymphocytes. Infect. Immun. 1993, 61, 1715–1721. [Google Scholar]
- Fernandez-Prada, C.; Tall, B.D.; Elliott, S.E.; Hoover, D.L.; Nataro, J.P.; Venkatesan, M.M. Hemolysin-positive enteroaggregative and cell-detaching Escherichia coli strains cause oncosis of human monocyte-derived macrophages and apoptosis of murine J774 cells. Infect. Immun. 1998, 66, 3918–3924. [Google Scholar]
- Russo, T.A.; Davidson, B.A.; Genagon, S.A.; Warholic, N.M.; Macdonald, U.; Pawlicki, P.D.; Beanan, J.M.; Olson, R.; Holm, B.A.; Knight, P.R., 3rd. E. coli virulence factor hemolysin induces neutrophil apoptosis and necrosis/lysis in vitro and necrosis/lysis and lung injury in a rat pneumonia model. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L207–L216. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.L.; Islur, A.; Haq, R.; Mascarenhas, M.; Karmali, M.A.; Perdue, M.H.; Zanke, B.W.; Sherman, P.M. Escherichia coli Shiga toxins induce apoptosis in epithelial cells that is regulated by the Bcl-2 family. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G811–G819. [Google Scholar]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar]
- Kiyokawa, N.; Taguchi, T.; Mori, T.; Uchida, H.; Sato, N.; Takeda, T.; Fujimoto, J. Induction of apoptosis in normal human renal tubular epithelial cells by Escherichia coli Shiga toxins 1 and 2. J. Infect. Dis. 1998, 178, 178–184. [Google Scholar]
- Uchida, H.; Kiyokawa, N.; Taguchi, T.; Horie, H.; Fujimoto, J.; Takeda, T. Shiga toxins induce apoptosis in pulmonary epithelium-derived cells. J. Infect. Dis. 1999, 180, 1902–1911. [Google Scholar]
- Erwert, R.D.; Eiting, K.T.; Tupper, J.C.; Winn, R.K.; Harlan, J.M.; Bannerman, D.D. Shiga toxin induces decreased expression of the anti-apoptotic protein Mcl-1 concomitant with the onset of endothelial apoptosis. Microb. Pathog. 2003, 35, 87–93. [Google Scholar]
- Fujii, J.; Wood, K.; Matsuda, F.; Carneiro-Filho, B.A.; Schlegel, K.H.; Yutsudo, T.; Binnington-Boyd, B.; Lingwood, C.A.; Obata, F.; Kim, K.S.; Yoshida, S.; Obrig, T. Shiga toxin 2 causes apoptosis in human brain microvascular endothelial cells via C/EBP homologous protein. Infect. Immun. 2008, 76, 3679–3689. [Google Scholar]
- Tesh, V.L.; Samuel, J.E.; Perera, L.P.; Sharefkin, J.B.; O'Brien, A.D. Evaluation of the role of Shiga and Shiga-like toxins in mediating direct damage to human vascular endothelial cells. J. Infect. Dis. 1991, 164, 344–352. [Google Scholar]
- Yoshida, T.; Fukada, M.; Koide, N.; Ikeda, H.; Sugiyama, T.; Kato, Y.; Ishikawa, N.; Yokochi, T. Primary cultures of human endothelial cells are susceptible to low doses of Shiga toxins and undergo apoptosis. J. Infect. Dis. 1999, 180, 2048–2052. [Google Scholar]
- Lee, S.Y.; Cherla, R.P.; Caliskan, I.; Tesh, V.L. Shiga toxin 1 induces apoptosis in the human myelogenous leukemia cell line THP-1 by a caspase-8-dependent, tumor necrosis factor receptor-independent mechanism. Infect. Immun. 2005, 73, 5115–5126. [Google Scholar]
- Lee, M.S.; Cherla, R.P.; Leyva-Illades, D.; Tesh, V.L. Bcl-2 regulates the onset of shiga toxin 1-induced apoptosis in THP-1 cells. Infect. Immun. 2009, 77, 5233–5244. [Google Scholar]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008, 10, 770–780. [Google Scholar]
- Marcato, P.; Mulvey, G.; Armstrong, G.D. Cloned Shiga toxin 2 B subunit induces apoptosis in Ramos Burkitt's lymphoma B cells. Infect. Immun. 2002, 70, 1279–1286. [Google Scholar]
- Takahashi, K.; Funata, N.; Ikuta, F.; Sato, S. Neuronal apoptosis and inflammatory responses in the central nervous system of a rabbit treated with Shiga toxin-2. J. Neuroinflammation 2008, 5, 11. [Google Scholar]
- Ray, P.E.; Liu, X.H. Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr. Nephrol. 2001, 16, 823–839. [Google Scholar]
- Griffin, P.M.; Tauxe, R.V. The epidemiology of infections caused by Escherichia coli O157:H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol. Rev. 1991, 13, 60–98. [Google Scholar] [PubMed]
- Pijpers, A.H.; van Setten, P.A.; van den Heuvel, L.P.; Assmann, K.J.; Dijkman, H.B.; Pennings, A.H.; Monnens, L.A.; van Hinsbergh, V.W. Verocytotoxin-induced apoptosis of human microvascular endothelial cells. J. Am. Soc. Nephrol. 2001, 12, 767–778. [Google Scholar]
- Johannes, L.; Romer, W. Shiga toxins—From cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar]
- Fujii, J.; Matsui, T.; Heatherly, D.P.; Schlegel, K.H.; Lobo, P.I.; Yutsudo, T.; Ciraolo, G.M.; Morris, R.E.; Obrig, T. Rapid apoptosis induced by Shiga toxin in HeLa cells. Infect. Immun. 2003, 71, 2724–2735. [Google Scholar]
- Kojio, S.; Zhang, H.; Ohmura, M.; Gondaira, F.; Kobayashi, N.; Yamamoto, T. Caspase-3 activation and apoptosis induction coupled with the retrograde transport of shiga toxin: inhibition by brefeldin A. FEMS Immunol. Med. Microbiol. 2000, 29, 275–281. [Google Scholar]
- Suzuki, A.; Doi, H.; Matsuzawa, F.; Aikawa, S.; Takiguchi, K.; Kawano, H.; Hayashida, M.; Ohno, S. Bcl-2 antiapoptotic protein mediates verotoxin II-induced cell death: Possible association between bcl-2 and tissue failure by E. coli O157:H7. Genes Dev. 2000, 14, 1734–1740. [Google Scholar] [PubMed]
- Johannes, L.; Decaudin, D. Protein toxins: intracellular trafficking for targeted therapy. Gene Ther. 2005, 12, 1360–1368. [Google Scholar]
- Sandvig, K.; van Deurs, B. Delivery into cells: lessons learned from plant and bacterial toxins. Gene Ther. 2005, 12, 865–872. [Google Scholar]
- Stechmann, B.; Bai, S.K.; Gobbo, E.; Lopez, R.; Merer, G.; Pinchard, S.; Panigai, L.; Tenza, D.; Raposo, G.; Beaumelle, B.; Sauvaire, D.; Gillet, D.; Johannes, L.; Barbier, J. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010, 141, 231–242. [Google Scholar]
- De Rycke, J.; Gonzalez, E.A.; Blanco, J.; Oswald, E.; Blanco, M.; Boivin, R. Evidence for two types of cytotoxic necrotizing factor in human and animal clinical isolates of Escherichia coli. J. Clin. Microbiol. 1990, 28, 694–699. [Google Scholar]
- Fabbri, A.; Falzano, L.; Travaglione, S.; Stringaro, A.; Malorni, W.; Fais, S.; Fiorentini, C. Rho-activating Escherichia coli cytotoxic necrotizing factor 1: Macropinocytosis of apoptotic bodies in human epithelial cells. Int. J. Med. Microbiol. 2002, 291, 551–554. [Google Scholar]
- Fiorentini, C.; Falzano, L.; Fabbri, A.; Stringaro, A.; Logozzi, M.; Travaglione, S.; Contamin, S.; Arancia, G.; Malorni, W.; Fais, S. Activation of rho GTPases by cytotoxic necrotizing factor 1 induces macropinocytosis and scavenging activity in epithelial cells. Mol. Biol. Cell 2001, 12, 2061–2073. [Google Scholar]
- Donelli, G.; Fiorentini, C. Cell injury and death caused by bacterial protein toxins. Toxicol. Lett. 1992, 64–65, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Mills, M.; Meysick, K.C.; O'Brien, A.D. Cytotoxic necrotizing factor type 1 of uropathogenic Escherichia coli kills cultured human uroepithelial 5637 cells by an apoptotic mechanism. Infect. Immun. 2000, 68, 5869–5880. [Google Scholar]
- Malorni, W.; Fiorentini, C. Is the Rac GTPase-activating toxin CNF1 a smart hijacker of host cell fate? FASEB J. 2006, 20, 606–609. [Google Scholar]
- Miraglia, A.G.; Travaglione, S.; Meschini, S.; Falzano, L.; Matarrese, P.; Quaranta, M.G.; Viora, M.; Fiorentini, C.; Fabbri, A. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IkappaB kinase pathway: role of nuclear factor-kappaB and Bcl-2. Mol. Biol. Cell 2007, 18, 2735–2744. [Google Scholar]
- Fleckenstein, J.M.; Hardwidge, P.R.; Munson, G.P.; Rasko, D.A.; Sommerfelt, H.; Steinsland, H. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect. 2010, 12, 89–98. [Google Scholar]
- Holmes, R.; Jobling, M.G.; Gonnell, T. Cholera toxin and related enterotoxins of gram-negative bacteria. In Bacterial Toxins and Virulence Factors in Disease. Handbook of Natural Toxins; Moss, J., Iglewski, B., Vaughn, M., Tu, A.T., Eds.; CRC Press: New York, NY, USA., 1995; pp. 225–255. [Google Scholar]
- Sixma, T.K.; Kalk, K.H.; van Zanten, B.A.; Dauter, Z.; Kingma, J.; Witholt, B.; Hol, W.G. Refined structure of Escherichia coli heat-labile enterotoxin, a close relative of cholera toxin. J. Mol. Biol. 1993, 230, 890–918. [Google Scholar]
- Nashar, T.O.; Webb, H.M.; Eaglestone, S.; Williams, N.A.; Hirst, T.R. Potent immunogenicity of the B subunits of Escherichia coli heat-labile enterotoxin: receptor binding is essential and induces differential modulation of lymphocyte subsets. Proc. Natl. Acad. Sci. USA 1996, 93, 226–230. [Google Scholar]
- Nashar, T.O.; Williams, N.A.; Hirst, T.R. Cross-linking of cell surface ganglioside GM1 induces the selective apoptosis of mature CD8+ T lymphocytes. Int. Immunol. 1996, 8, 731–736. [Google Scholar]
- Truitt, R.L.; Hanke, C.; Radke, J.; Mueller, R.; Barbieri, J.T. Glycosphingolipids as novel targets for T-cell suppression by the B subunit of recombinant heat-labile enterotoxin. Infect. Immun. 1998, 66, 1299–1308. [Google Scholar]
- Williams, N.A.; Hirst, T.R.; Nashar, T.O. Immune modulation by the cholera-like enterotoxins: from adjuvant to therapeutic. Immunol. Today 1999, 20, 95–101. [Google Scholar]
- Liang, S.; Hajishengallis, G. Heat-labile enterotoxins as adjuvants or anti-inflammatory agents. Immunol. Invest. 2010, 39, 449–467. [Google Scholar]
- Connell, T.D. Cholera toxin, LT-I, LT-IIa and LT-IIb: The critical role of ganglioside binding in immunomodulation by type I and type II heat-labile enterotoxins. Expert Rev. Vaccines 2007, 6, 821–834. [Google Scholar]
- Arce, S.; Nawar, H.F.; Russell, M.W.; Connell, T.D. Differential binding of Escherichia coli enterotoxins LT-IIa and LT-IIb and of cholera toxin elicits differences in apoptosis, proliferation, and activation of lymphoid cells. Infect. Immun. 2005, 73, 2718–2727. [Google Scholar]
- Tamayo, E.; Merino, R.; Gonzalez-Rojas, J.; Marquina, R.; Santiuste, I.; Amado, J.A.; Rappuoli, R.; Del Giudice, G.; Merino, J. The Escherichia coli heat-labile enterotoxin induces apoptosis of immature lymphocytes in vivo via a glucocorticoid-dependent pathway. Eur. J. Immunol. 2005, 35, 3505–3515. [Google Scholar]
- Tamayo, E.; Postigo, J.; Del Giudice, G.; Rappuoli, R.; Benito, A.; Yagita, H.; Merino, R.; Merino, J. Involvement of the intrinsic and extrinsic cell-death pathways in the induction of apoptosis of mature lymphocytes by the Escherichia coli heat-labile enterotoxin. Eur. J. Immunol. 2009, 39, 439–446. [Google Scholar]
- Soriani, M.; Williams, N.A.; Hirst, T.R. Escherichia coli enterotoxin B subunit triggers apoptosis of CD8(+) T cells by activating transcription factor c-myc. Infect. Immun. 2001, 69, 4923–4930. [Google Scholar]
- Salmond, R.J.; Pitman, R.S.; Jimi, E.; Soriani, M.; Hirst, T.R.; Ghosh, S.; Rincon, M.; Williams, N.A. CD8+ T cell apoptosis induced by Escherichia coli heat-labile enterotoxin B subunit occurs via a novel pathway involving NF-kappaB-dependent caspase activation. Eur. J. Immunol. 2002, 32, 1737–1747. [Google Scholar]
- Salmond, R.J.; Williams, R.; Hirst, T.R.; Williams, N.A. Selective induction of CD8+CD4- thymocyte apoptosis mediated by the B-subunit of Escherichia coli heat-labile enterotoxin. Immunol. Lett. 2003, 88, 43–46. [Google Scholar]
- Salmond, R.J.; Williams, R.; Hirst, T.R.; Williams, N.A. The B subunit of Escherichia coli heat-labile enterotoxin induces both caspase-dependent and -independent cell death pathways in CD8+ T cells. Infect. Immun. 2004, 72, 5850–5857. [Google Scholar]
- Tsuji, T.; Asano, Y.; Handa, T.; Honma, Y.; Ichinose, Y.; Yokochi, T. Induction of apoptosis in lymphoid tissues of mice after intramuscular injection of enterotoxigenic Escherichia coli enterotoxin. Immunobiology 2000, 201, 377–390. [Google Scholar]
- Colwell, R.R. Global climate and infectious disease: The cholera paradigm. Science 1996, 274, 2025–2031. [Google Scholar]
- Islam, M.S.; Drasar, B.S.; Sack, R.B. The aquatic flora and fauna as reservoirs of Vibrio cholerae: a review. J. Diarrhoeal Dis. Res. 1994, 12, 87–96. [Google Scholar]
- Sack, D.A.; Sack, R.B.; Nair, G.B.; Siddique, A.K. Cholera. Lancet 2004, 363, 223–233. [Google Scholar]
- Kaper, J.B.; Morris, J.G., Jr.; Levine, M.M. Cholera. Clin. Microbiol. Rev. 1995, 8, 48–86. [Google Scholar]
- Levine, M.M.; Kaper, J.B.; Black, R.E.; Clements, M.L. New knowledge on pathogenesis of bacterial enteric infections as applied to vaccine development. Microbiol. Rev. 1983, 47, 510–550. [Google Scholar]
- Zhang, R.G.; Scott, D.L.; Westbrook, M.L.; Nance, S.; Spangler, B.D.; Shipley, G.G.; Westbrook, E.M. The three-dimensional crystal structure of cholera toxin. J. Mol. Biol. 1995, 251, 563–573. [Google Scholar]
- Pierce, N.F. Differential inhibitory effects of cholera toxoids and ganglioside on the enterotoxins of Vibrio cholerae and Escherichia coli. J. Exp. Med. 1973, 137, 1009–1023. [Google Scholar]
- King, C.A.; Van Heyningen, W.E. Deactivation of cholera toxin by a sialidase-resistant monosialosylganglioside. J. Infect. Dis. 1973, 127, 639–647. [Google Scholar]
- Cassel, D.; Selinger, Z. Mechanism of adenylate cyclase activation by cholera toxin: inhibition of GTP hydrolysis at the regulatory site. Proc. Natl. Acad. Sci. USA 1977, 74, 3307–3311. [Google Scholar]
- Gill, D.M. Mechanism of action of cholera toxin. Adv. Cyclic Nucleotide Res. 1977, 8, 85–118. [Google Scholar]
- Woogen, S.D.; Ealding, W.; Elson, C.O. Inhibition of murine lymphocyte proliferation by the B subunit of cholera toxin. J. Immunol. 1987, 139, 3764–3770. [Google Scholar]
- Yankelevich, B.; Soldatenkov, V.A.; Hodgson, J.; Polotsky, A.J.; Creswell, K.; Mazumder, A. Differential induction of programmed cell death in CD8+ and CD4+ T cells by the B subunit of cholera toxin. Cell Immunol. 1996, 168, 229–234. [Google Scholar]
- Sun, J.B.; Czerkinsky, C.; Holmgren, J. Sublingual 'oral tolerance' induction with antigen conjugated to cholera toxin B subunit generates regulatory T cells that induce apoptosis and depletion of effector T cells. Scand. J. Immunol. 2007, 66, 278–286. [Google Scholar]
- Allam, M.; Bertrand, R.; Zhang-Sun, G.; Pappas, J.; Viallet, J. Cholera toxin triggers apoptosis in human lung cancer cell lines. Cancer Res. 1997, 57, 2615–2618. [Google Scholar]
- Pessina, A.; Croera, C.; Savalli, N.; Bonomi, A.; Cavicchini, L.; Turlizzi, E.; Guizzardi, F.; Guido, L.; Daprai, L.; Neri, M.G. Bcl-2 down modulation in WEHI-3B/CTRES cells resistant to Cholera Toxin (CT)-induced apoptosis. Cell Res. 2006, 16, 306–312. [Google Scholar]
- Rosch, J.W.; Boyd, A.R.; Hinojosa, E.; Pestina, T.; Hu, Y.; Persons, D.A.; Orihuela, C.J.; Tuomanen, E.I. Statins protect against fulminant pneumococcal infection and cytolysin toxicity in a mouse model of sickle cell disease. J. Clin. Invest. 2010, 120, 627–635. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lin, C.-F.; Chen, C.-L.; Huang, W.-C.; Cheng, Y.-L.; Hsieh, C.-Y.; Wang, C.-Y.; Hong, M.-Y. Different Types of Cell Death Induced by Enterotoxins. Toxins 2010, 2, 2158-2176. https://doi.org/10.3390/toxins2082158
Lin C-F, Chen C-L, Huang W-C, Cheng Y-L, Hsieh C-Y, Wang C-Y, Hong M-Y. Different Types of Cell Death Induced by Enterotoxins. Toxins. 2010; 2(8):2158-2176. https://doi.org/10.3390/toxins2082158
Chicago/Turabian StyleLin, Chiou-Feng, Chia-Ling Chen, Wei-Ching Huang, Yi-Lin Cheng, Chia-Yuan Hsieh, Chi-Yun Wang, and Ming-Yuan Hong. 2010. "Different Types of Cell Death Induced by Enterotoxins" Toxins 2, no. 8: 2158-2176. https://doi.org/10.3390/toxins2082158