Ochratoxin A in Roasted Coffee from French Supermarkets and Transfer in Coffee Beverages: Comparison of Analysis Methods

Abstract
:1. Introduction
2. Experimental
2.1. Chemicals
2.2. Extraction of OTA from roasted coffee
2.2.1. Extraction of OTA in acidic conditions according to the EU validated method “15141-1” [25]
2.2.2. Extraction of OTA in alkaline conditions according to the EU validated method “14132” [26]
2.2.3. Extraction of OTA according to Pittet et al. 1996 [27]
2.3. Preparation of coffee beverage and extraction of OTA

2.4. Detection of OTA and quantification
2.5. Confirmation of OTA derivatives
3. Results and Discussion
3.1. Occurrence of OTA in ground roasted coffee
3.1.1. Comparison of the recovery of OTA using the three methods

{kind=link}
{kind=link}
| Contamination level analysis method | 0.5 | 1.0 | 2.0 | 2.5 | 5.0 | 10.0 µg/kg |
|---|---|---|---|---|---|---|
| % recovery | ||||||
| EN No. 14132 [26] | 16.6–18.7 | 24.5–27.0 | 33.0–34.0 | 41.0–49.0 | 63.0–70.0 | |
| Pittet method [27] | 60–64 | 62–65 | 70–72 | 80–82 | ||
| EN No.15141-1 [25] | 50–55 | 53–55 | 57–55 | 57–57 | ||
3.1.2. OTA amount in ground roasted coffee
| Sample | CEN14132 (acidic condition) | CEN 15141-1 Entwilse, 2001 | Pittet method |
|---|---|---|---|
| 1 | 1.6 | 1 | 1.34 |
| 2 | 0.8 | 1.6 | 2 |
| 3 | 0.68 | 0.66 | NA |
| 4 | 1.1 | 0.5 | 0.65 |
| 5 | 0.4 | 0.5 | NA |
| 6 | 1.3 | 0.5 | NA |
| 7 | 1.3 | 1 | NA |
| 8 | 1 | 1 | NA |
| 9 | 1.05 | 1.35 | 1.3 |
| 10 | LOQ | LOQ | NA |
| 11 | LOQ | LOQ | NA |
| 12 | 0.93 | 0.5 | NA |
| 13 | 0.5 | 0.35 | NA |
| 14 | LOD | LOD | LOD |
| 15 | 0.8 | 0.5 | NA |
| 16 | 0.9 | 0.83 | 1.03 |
| 17 | 0.41 | 0.35 | ND |
| 18 | 1.69 | 1.25 | 1.41 |
| 19 | 0.6 | 0.5 | NA |
| 20 | 0.75 | 0.5 | NA |
| 21 | 0.5 | 0.5 | NA |
| 22 | 0.87 | 0.75 | 1.1 |
| 23 | 0.75 | 0.92 | NA |
| 24 | 11.89 | 15.08 | 10.14 |
| 25 | LOQ | LOQ | ND |
| 26 | LOQ | LOQ | ND |
| 27 | 0.76 | 0.75 | 1.2 |
| 28 | 1.37 | 1.25 | 1.6 |
| 29 | 0.7 | 0.5 | NA |
| 30 | 0.54 | 0.35 | NA |
3.2. OTA in the beverage
3.2.1. Recovery of OTA
| Contamination level analysis method | 0.5 | 1.0 | 2.0 | 2.5 | 5.0 | 10.0 |
|---|---|---|---|---|---|---|
| % recovery | ||||||
| Entwisle 14132 | 68–72 | 75–73 | 72–70 | 80–73 | 68–70 | |
| Pittet | 72–70 | 75–76 | 80–80 | |||
3.2.2. Amount of OTA in the beverage
| code | Mode of preparation | ng/L coffee | ng/kg coffee | % passing * |
|---|---|---|---|---|
| 1 | electric | 22 | 0.22 | 13/16 |
| piston | 32 | 0.25 | 15/19 | |
| 2 | electric | 47.2 | 0.48 | 60 |
| piston | 37.93 | 0.38 | 47.5 | |
| 4 | electric | 72.27 | 0.72 | 65/110 |
| piston | 58.75 | 0.46 | 42/71 | |
| 6 | electric | 31.18 | 0.31 | 24 |
| piston | 32.6 | 0.25 | 19 | |
| 7 | electric | 26.37 | 0.26 | 20 |
| 8 | electric | 20.68 | 0.21 | 21 |
| 9 | electric | 148.55 | 1.48 | 141/117 |
| piston | 141.93 | 1.11 | 110/88 | |
| 12 | electric | LOD | LOD | - |
| 14 | electric | 48 | 0.48 | >100 |
| piston | 43.6 | 0.34 | >100 | |
| 15 | electric | 22.42 | 0.22 | 27.5 |
| 16 | electric | 79.63 | 0.8 | 86/78 |
| piston | 85.93 | 0.67 | 69/65 | |
| 17 | electric | LOD | LOD | <LOQ |
| 18 | electric | 122.78 | 1.23 | 73/87 |
| piston | 168.65 | 1.32 | 78/94 | |
| 19 | electric | 11.87 | 0.12 | 20 |
| 22 | electric | 77.66 | 0.78 | 87/74 |
| piston | 79.88 | 0.62 | 69/59 | |
| 24 | electric | 796.03 | 7.96 | 67/78 |
| piston | 882.31 | 6.89 | 58/68 | |
| 27 | piston | 27 | 0.27 | 35.5/22 |
| 28 | electric | 161.28 | 1.6 | 117/100 |
| piston | 170.93 | 1.34 | 98/84 | |
| 29 | electric | 31 | 0.31 | 44 |
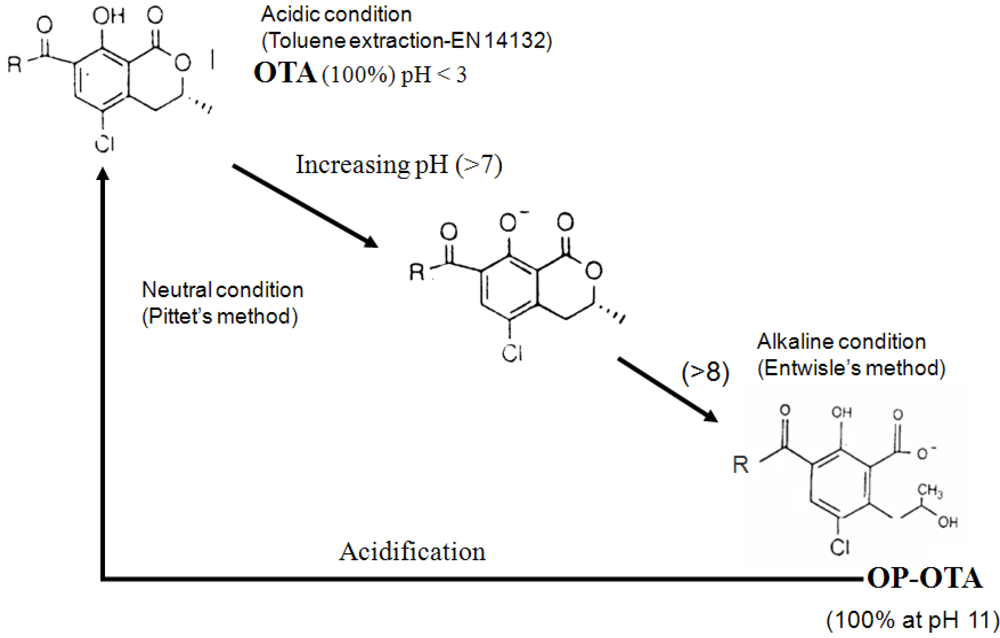
3.3. Interference of OTB and other compounds with OTA antibodies and modification of the structure of OTA during extraction explaining underestimation of OTA content in ground coffee
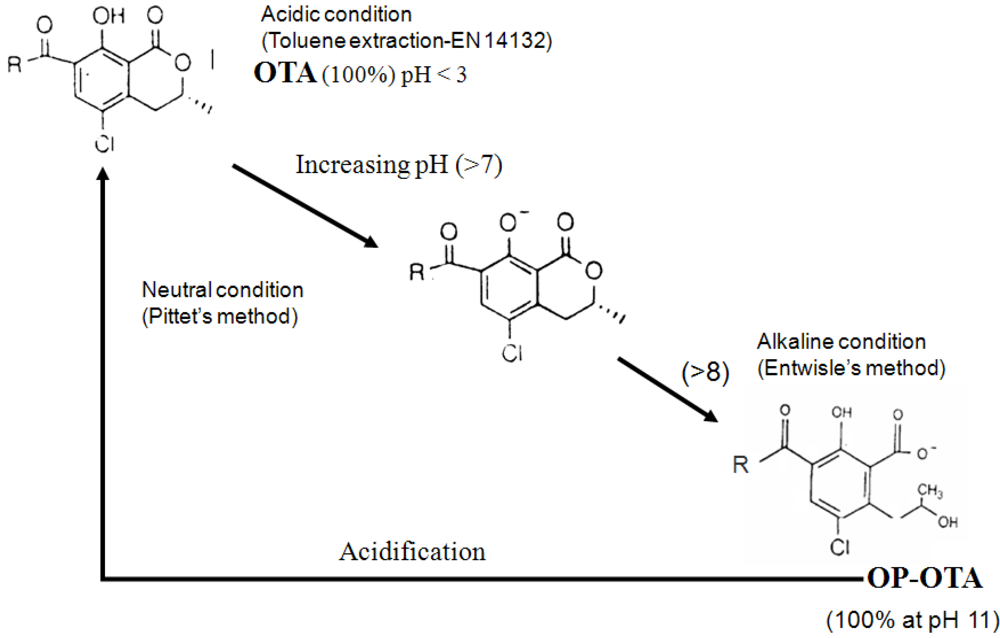
4. Conclusions
Acknowledgements
References
- Abarca, M.L.; Accensi, F.; Bargulat, M.R.; Cabanes, F.J. Current importance of ochratoxin A-producing Aspergillus spp. J. Food Prot. 2001, 64, 903–906. [Google Scholar]
- Reddy, K.R.N.; Abbas, H.K.; Abel, C.A.; Shier, W.T.; Salleh, B. Mycotoxin contamination of beverages: occurrence of Patulin in aplle juice and ochratoxin A in Coffee, Beer and Wine and their control methods. Toxins 2010, 2, 229–261. [Google Scholar]
- Jørgensen, K. Survey of pork, poultry, coffee, beer and pulses for ochratoxin A. Food Addit. Contam. 1998, 15, 550–554. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R. Review on Ochratoxin A: an overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar]
- Krug, H.P. Cafés Duros III. Relação entre a porcentagem de microrganismos e qualidade do café. Revista do Instituto do Café 1940, 27, 1827–1831. [Google Scholar]
- Krug, H.P. Cafés Duros IV. Relação entre zonas, qualidade de café e porcentagem de microrganismos. Revista do Instituto do Café 1941, 16, 288–295. [Google Scholar]
- Carvalho, V.D.; Chalfoun, S.M. Aspectos qualitativos do café. Informe Agropecuário 1985, 11, 79–92. [Google Scholar]
- Micco, C.; Grossi, M.; Miraglia, M.; Brera, C. A study of the contamination by ochratoxin A in green coffee and roasted coffee beans. Food Addit. Contam. 1989, 6, 333–339. [Google Scholar]
- Nakajima, M.; Tsubouchi, H.; Miyabe, M.; Ueno, Y. Survey of aflatoxin B1 and ochratoxin A in commercial green coffee beans by high-performance liquid chromatography linked with immunoaffinity chromatography. Food Agric. Immunol. 1997, 9, 77–83. [Google Scholar]
- Trucksess, M.; Giler, J.; Young, K.; White, K.D.; Page, S.W. Determination and survey of ochratoxin A in wheat, barley and coffee 1997. J. AOAC Int. 1999, 82, 85–89. [Google Scholar]
- Romani, S.; Sacchetti, G.; Chaves, C.C.; Pinnavaia, G.G.; Dalla, M. Screening on the occurrence of ochratoxin A in green coffee beans of different origins and types. J. Agric. Food Chem. 2000, 48, 3616–3619. [Google Scholar]
- Taniwaki, M.H.; Pitt, J.I.; Teixeira, A.A.; Iamanaka, B.T. The source of ochratoxin A in Brazilian coffee and its formation in relation to processing methods. Int. J. Food Microbiol. 2003, 82, 173–179. [Google Scholar]
- Martins, M.L.; Martins, H.M.; Gimeno, A. Incidence of microflora and of ochratoxin A in green coffee beans (Coffea arabica). Food Addit. Contam. 2003, 20, 1127–1131. [Google Scholar]
- Tsubouchi, H.; Yamamoto, K.; Hisada, K.; Sakabe, Y.; Udagawa, S. Effect of roasting on ochratoxin A level in green coffee beans inoculated with Aspergillus ochraceus. Mycopathologia 1987, 97, 111–115. [Google Scholar]
- Studer-Rohr, I.; Dietrich, D.R.; Schlatter, J.; Schlatter, C. The occurrence of ochratoxin A in coffee. Food Chem. Toxicol. 1995, 33, 341–355. [Google Scholar]
- Stegen, G.V.D.; Jorissen, U.; Pittet, A.; Saccon, M.; Steines, W.; Vincenzi, M.; Winkler, M.; Zapp, J.; Schlatter, C. Screening of European coffee final products for occurrence of ochratoxin A. Food Addit. Contam. 1997, 14, 211–216. [Google Scholar]
- Bresch, H.; Urbanek, M.; Hell, K. Ochratoxin A in coffee, tea and beer. Archiv fur Lebensmittelhygiene 2000, 51, 89–94. [Google Scholar]
- Perez de Obamos, A.; Gonzalez-Penas, E.; Lopez de Cerain, A. Influence of roasting and brew preparation on the ochratoxin A content in coffee infusion. Food Addit. Contam. 2005, 22, 463–471. [Google Scholar]
- Blanc, M.; Pittet, A.; Munoz-Box, R.; Viani, R. Behavior of Ochratoxin A during Green Coffee Roasting and Soluble Coffee Manufacture. J. Agric. Food Chem. 1998, 46, 673–675. [Google Scholar]
- Boudra, H.; Bars, P.L.; Bars, J.L. Thermostability of Ochratoxin A in Wheat under Two Moisture Conditions. Appl. Environ. Microbiol. 1995, 61, 1156–1158. [Google Scholar]
- Stegen, G.H.D.; Essens, P.J.M.; Lijn, J. Effect of Roasting Conditions on Reduction of Ochratoxin A in Coffee. J. Agric. Food Chem. 2001, 49, 4713–4715. [Google Scholar]
- International Coffee Organization, Coffee Statistics 1999; London, UK, 2000; Nr16, ISSN 1364-9086.
- European Union. Règlement (CE) No 1881/2006 de la Commission du 19 décembre 2006 portant fixation de teneurs maximales pour certains contaminants dans les denrées alimentaires. Journal Officiel de l’Union Européenne 2006, L364, 5–24.
- Turner, N.W.; Subrahmanyam, S.; Piletsky, S.A. Analytical methods for determination of mycotoxins: A review. Anal. Chim. Acta 2009, 632, 168–180. [Google Scholar]
- Organisation Internationale de Normalisation, Produits alimentaires–Dosage de l'ochratoxine A dans les céréales et produits dérivés–Partie 1: méthode par chromatographie liquide haute performance comprenant une étape d'extraction par chromatographie sur gel de silice. ISO: Genève, Suisse, 12 1998; NF-EN-ISO 15141-1.
- Entwisle, A.C.; Williams, A.C.; Mann, P.J.; Russell, J.; Slack, P.T.; Gilbert, J. Combined phenyl silane and immunoaffinity column clean-up with liquid chromatography for determination of ochratoxin A in roasted coffee: collaborative study. J. AOAC Int. 2001, 84, 444–450. [Google Scholar]
- Pittet, A.; Tornare, D.; Huggett, A.; Viani, R. Liquid Chromatographic Determination of Ochratoxin A in Pure and Adulterated Soluble Coffee Using an Immunoaffinity Column Cleanup Procedure. J. Agric. Food Chem. 1996, 44, 3564–3569. [Google Scholar]
- Xiao, H.; Madhyastha, S.; Marquardt, R.R.; Li, S.; Vodela, J.K.; Frohlich, A.; Kemppainen, A. Toxicity of ochratoxin A, its opened lactone form and several of its analogs : structure activity relationships. Toxicol. Appl. Pharmacol. 1996, 137, 182–192. [Google Scholar]
- Organisation Internationale de Normalisation, Produits alimentaires - Dosage de l'ochratoxine A dans l'orge et dans le café torréfié - Méthode par purification sur colonne d'immunoaffinité suivie d'une analyse par CLHP. ISO: Genève, Suisse, 10 2003; NF-EN-ISO 14132.
- Horwitz, W.; Albert, R. The Horwitz Ratio (HorRat): A Useful Index of Method Performance with Respect to Precision. J. AOAC Int. 2006, 89, 1095–1109. [Google Scholar]
- Pfohl-Leszkowicz, A.; Molinié, A. ; Castegnaro, M. Presence of ochratoxin a, citrinin and fumonisin b1 in breakfast cereals collected in french market. Comparison of OTA analysis using or not immunoaffinity clean-up before HPLC. Revista Mexicana de Micologia 2004, 19, 7–15. [Google Scholar]
- Pfohl-Leszkowicz, A.; Molinié, A.; Castegnaro, M. Underestimation of fumonisin B1 and ochratoxin A, from complex matrices by use of immunoaffinity columns. In Mycotoxins and Phycotoxins, Proceeding of the XIth International IUPAC Symposium on Mycotoxins and Phycotoxins, Bethesda, MD, USA, 17–21 May 2004; Njapau, H., Trujillio, S., Van Egmond, H.P., Park, D.L., Eds.; Wageningen Academic: Bethesda, MD, USA, 2006; pp. 83–89. [Google Scholar]
- Molinié, A.; Faucet, V.; Castegnaro, M.; Pfohl-Leszkowicz, A. Analysis of some breakfast cereals collected on the French market for their content in ochratoxin A, citrinin and fumonisin B1. Development of a new method for simultaneous extraction of ochratoxin A and citrinin. Food Chem. 2005, 92, 391–400. [Google Scholar] [CrossRef]
- Castegnaro, M.; Tozlovanu, M.; Wild, C.; Molinié, A.; Sylla, A.; Pfohl-Leszkowicz, A. Advantages and drawbacks of immunoaffinity columns in analysis of mycotoxins in food. Mol. Nutr. Food Res. 2006, 50, 480–481. [Google Scholar]
- Cantafora, A.; Grossi, M.; Miraglia, M.; Benelli, L. Determination of ochratoxin A in coffee beans using reversed-phase high performance liquid chromatography. La Rivista della Societa Italiana di Scienza dell’Alimentazione 1983, 12, 103–108. [Google Scholar]
- Leoni, L.A.B.; Soares, L.M.V.; Oliveira, P.L.C. Ochratoxin A in Brazilian roasted and instant coffees. Food Addit. Contam. 2000, 17, 867–870. [Google Scholar]
- Batista, L.R.; Chalfoun, S.M.; Prado, G.; Schwan, R.F.; Wheals, A.E. Toxigenic fungi associated with processed coffee beans (coffea arabica L). Int. J. Food Microbiol. 2003, 85, 293–300. [Google Scholar]
- Pardo, E.; Marim, S.; Ramos, A.J.; Sanchis, V. Occurrence of ochratoxigenic fungi and ochratoxin A in green coffee from different origins. Food Sci. Technol. Int. 2004, 10, 45–50. [Google Scholar]
- Gopinandhan, T.N.; Velmourougane, K.; Panneerselvam, P.; Keshamma, E.; Raghuramulu, Y. Occurrence of ochratoxin A in green and commercial coffee samples. J. Food Sci. Technol. 2007, 44, 247–249. [Google Scholar]
- Gopinandhan, T.N.; Kannan, G.S.; Panneerselvam, P.; Velmourougane, K.; Raghuramulu, Y.; Jayarama, J. Survey on ochratoxin A in Indian green coffee destined for export. Food Addit. Contam. B 2008, 1, 51–57. [Google Scholar]
- De Moraes, M.H.P.; Luchese, R.H. Ochratoxin A on green coffee: influence of harvest and drying processing procedures. J. Agric. Food Chem. 2003, 51, 5824–5828. [Google Scholar]
- Batista, L.R.; Chalfoun, S.M.; Silva, C.F.; Cirillo, M.; Varga, E.A.; Schwan, R.F. Ochratoxin A in coffee beans (Coffea arabica L.) processed by dry and wet methods. Food Control 2009, 20, 784–790. [Google Scholar] [CrossRef]
- Joint Expert Committee Food Additives (JECFA), Safety Evaluation of Certain Food Additives and Contaminants. In WHO Food Additives Series No. 59; WHO: Geneva, Switzerland, 2008; pp. 357–417.
- Kuiper-Goodman, T.; Scott, P.M. Risk assessment of the mycotoxin ochratoxin A. Biomed. Environ. Sci. 1989, 2, 179–248. [Google Scholar]
- Hartge, P.; Hoover, R.; West, D.W.; Lyon, J.L. Coffee drinking and risk of bladder cancer. J. Natl. Cancer Inst. 1983, 70, 1021–1026. [Google Scholar]
- Kurashi, N.; Inoue, M.; Iwasaki, S.; Sasazuki, S.; Tsugane, S. Coffee, green tea and caffeine consumption and subsequent risk of bladder cancer in relation to smoking status: a prospective study in Japan. Cancer Sci. 2009, 100, 284–291. [Google Scholar]
- Covolo, L.; Placide, D.; Gelatti, U.; Carta, A.; Di Carlo, A.S.; Lodetti, P.; Picciche, A.; Orizi, G.; Campagna, M.; Arici, C.; Dorru, S. Bladder cancer, GSTs, NAT1, NAT2, sult 1A1, XRRC1, XRRC3, XPD genetic polymorphisms and coffee consumption: a case control study. Eur. J. Epidemiol. 2008, 23, 355–362. [Google Scholar]
- Pfohl-Leszkowicz, A.; Bartsch, H.; Azémar, B.; Mohr, U.; Estève, J.; Castegnaro, M. MESNA protects rats against nephrotoxicity but not carcinogenicity induced by ochratoxin A, implicating two separate pathways. Facta Universitatis, Series Medicine & Biology 2002, 9, 57–63. [Google Scholar]
- Faucet-Marquis, V.; Pont, F.; Størmer, F.; Rizk, T.; Castegnaro, M.; Pfohl-Leszkowicz, A. Evidence of a new dechlorinated OTA derivative formed in opossum kidney cell cultures after pre-treatment by modulators of glutathione pathways. Correlation with DNA adducts formation. Mol. Nutr. Food Res. 2006, 50, 531–542. [Google Scholar]
- Lebrun, S.; Golka, K.; Schulze, H.; Follman, W. Glutathione S-transferase polymorphisms and ochratoxin A toxicity in primary human urothelial cells. Toxicology 2006, 224, 81–90. [Google Scholar]
- Faucet, V.; Pfohl-Leszkowicz, A.; Dai, J.; Castegnaro, M.; Manderville, R. Evidence for Covalent DNA Adduction by Ochratoxin A Following Chronic Exposure to Rat and Subacute Exposure to Pig. Chem. Res. Toxicol. 2004, 17, 1289–1296. [Google Scholar]
- Mantle, P.; Faucet-Marquis, V.; Manderville, R.; Sciqualli, B.; Pfohl-Leszkowicz, A. Structures of covalent adducts between DNA and ochratoxin A: a new factor in debate about genotoxicity and human risk assessment. Chem. Res. Toxicol. 2010, 23, 89–98. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tozlovanu, M.; Pfohl-Leszkowicz, A. Ochratoxin A in Roasted Coffee from French Supermarkets and Transfer in Coffee Beverages: Comparison of Analysis Methods. Toxins 2010, 2, 1928-1942. https://doi.org/10.3390/toxins2081928
Tozlovanu M, Pfohl-Leszkowicz A. Ochratoxin A in Roasted Coffee from French Supermarkets and Transfer in Coffee Beverages: Comparison of Analysis Methods. Toxins. 2010; 2(8):1928-1942. https://doi.org/10.3390/toxins2081928
Chicago/Turabian StyleTozlovanu, Mariana, and Annie Pfohl-Leszkowicz. 2010. "Ochratoxin A in Roasted Coffee from French Supermarkets and Transfer in Coffee Beverages: Comparison of Analysis Methods" Toxins 2, no. 8: 1928-1942. https://doi.org/10.3390/toxins2081928
APA StyleTozlovanu, M., & Pfohl-Leszkowicz, A. (2010). Ochratoxin A in Roasted Coffee from French Supermarkets and Transfer in Coffee Beverages: Comparison of Analysis Methods. Toxins, 2(8), 1928-1942. https://doi.org/10.3390/toxins2081928