Deoxynivalenol-Induced Proinflammatory Gene Expression: Mechanisms and Pathological Sequelae

Abstract
:1. Introduction
2. DON Targets Mononuclear Phagocytes
2.1. In vitro effects of DON
2.2. Mechanisms for DON inhibition of translation
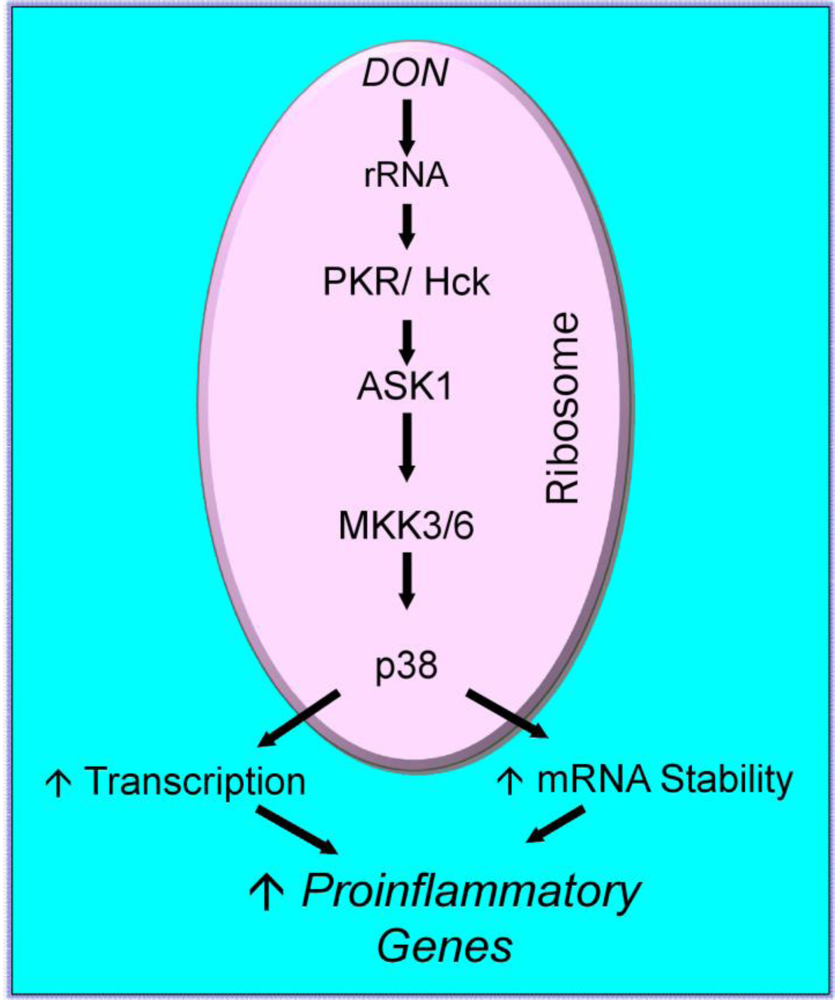
2.3. Mechanisms for DON-induced proinflammatory gene upregulation

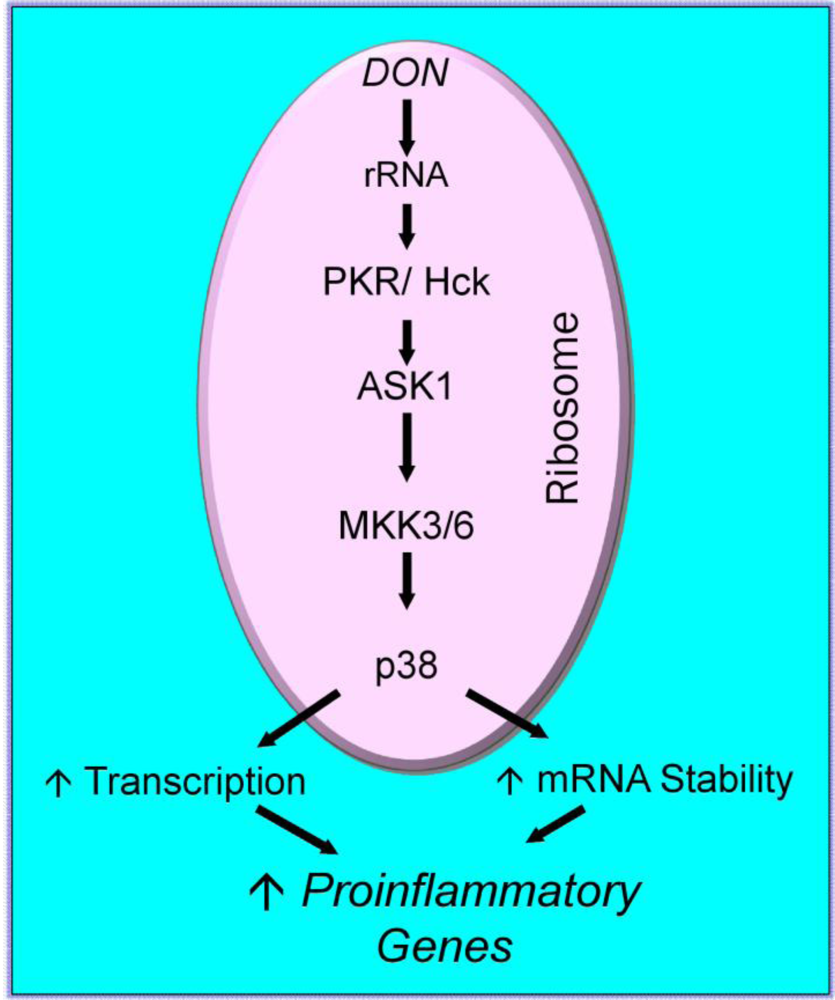{kind=link}
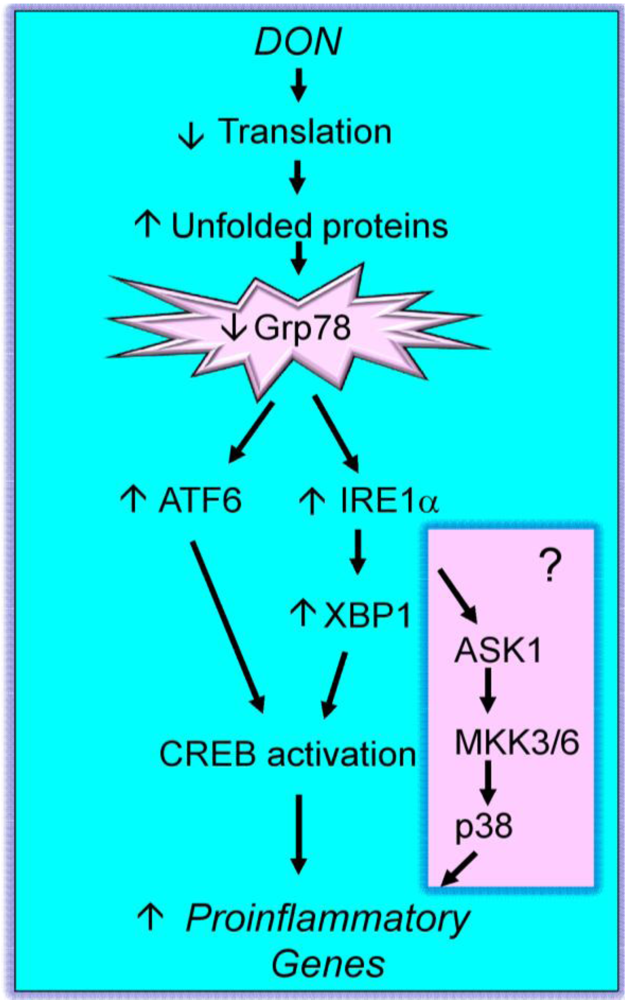{kind=link}
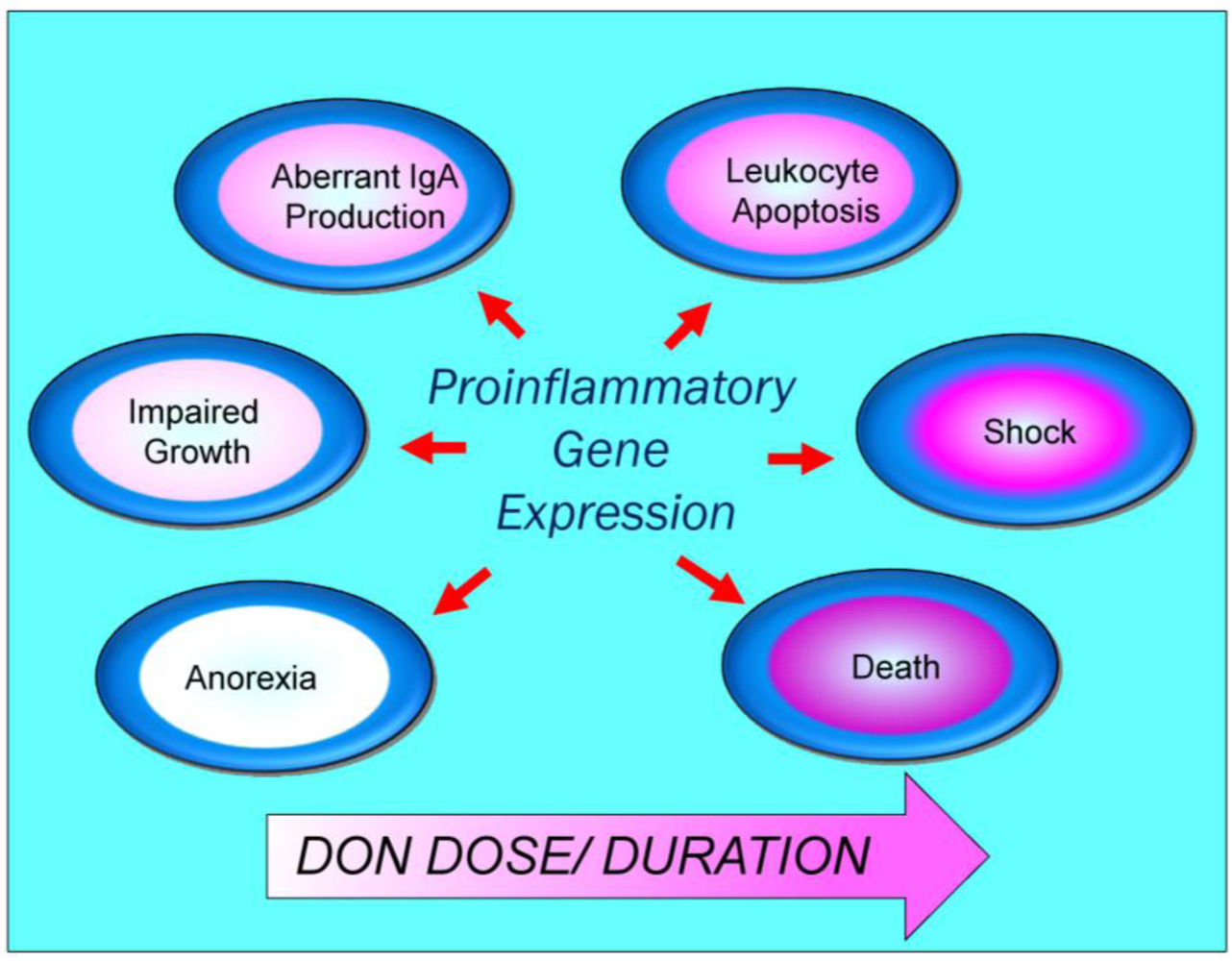{kind=link}
| Gene Family | Gene |
|---|---|
| See references [26,34,48,83,100,120]. | |
| Proinflammatory Cytokines | IL-1α, IL-1β, IL-6, IL-11, TNF-α, TGF-β |
| T Cell Cytokines | IFN-γ, IL-2 |
| Chemokines | MIP-2, MCP-1, Crg-2, CINC-1, MCP-3 |
| Transcription Factors | cFos, cJun, Fra-2, Jun-B, NR4a1 |
| Phosphatases | MKP1, CNAb, Ptpn8, Ptprj |
| Suppressors of Cytokine Signaling (SOCS) | CIS1,SOCS1, SOCS2 ,SOCS3 |
| Other | Cox-2, C3aR |
2.4. Mechanisms for DON-induced cell death
3. Initiating Events in the Ribotoxic Stress Response
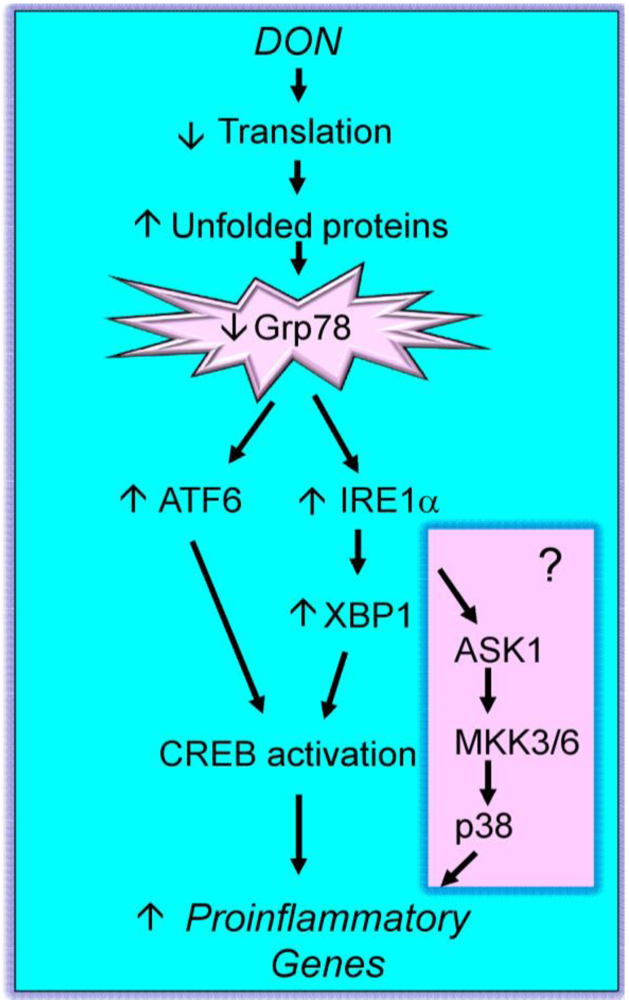
4. Pathological Sequelae to DON-Induced Innate Immune Activation
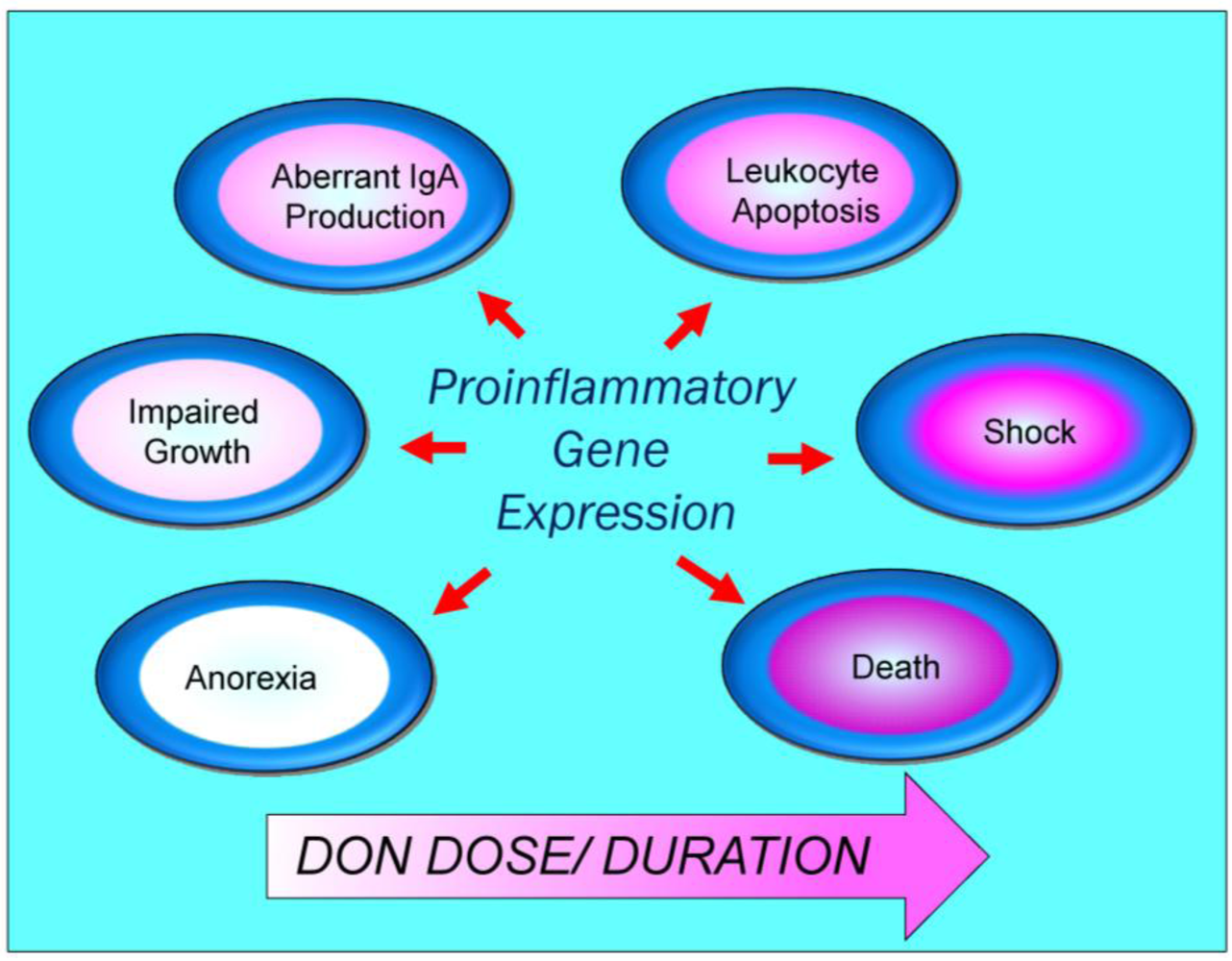
5. Conclusions
Acknowledgements
References
- Joffe, A.Z. Fusarium poae and F. sporotrichoides as principal causal agents of alimentary toxic aleukia. In Mycotoxic Fungi, Mycotoxins, Mycotoxicoses: An Encyclopedic Handbook; Wyllie, T.D., Morehouse, L.G., Eds.; Dekker: New York, NY, USA, 1978; pp. 21–86. [Google Scholar]
- Yoshizawa, T. Trichothecenes: Chemical, Biological, and Toxicological Aspects. In Developmentsin Food Science; Ueno, Y., Ed.; Kodansha Ltd.: Tokyo, Japan, 1983; pp. 195–209. [Google Scholar]
- Luo, X. Food Poisoning Caused by Fusarium Toxins; ILSI: Bangkok, Thailand, 1994; pp. 129–136. [Google Scholar]
- Bhat, R.V.; Beedu, S.R.; Ramakrishna, Y.; Munshi, K.L. Outbreak of trichothecene mycotoxicosis associated with consumption of mould-damaged wheat production in Kashmir Valley, India. Lancet 1989, 1, 35–37. [Google Scholar]
- Grove, J.F. Non-macrocyclic trichothecenes. Nat. Prod. Rep. 1988, 5, 187–209. [Google Scholar]
- Grove, J.F. Macrocyclic trichothecenes. Nat. Prod. Rep. 1993, 10, 429–448. [Google Scholar]
- Grove, J.F. Non-macrocyclic trichothecenes. Part 2. Prog. Chem. Org. Nat. Prod. 2000, 69, 1–70. [Google Scholar]
- Canady, R.A.; Coker, R.D.; Rgan, S.K.; Krska, R.; Kuiper-Goodman, T.; Olsen, M.; Pestka, J.J.; Resnik, S.; Schlatter, J. Deoxynivalenol. In Safety evaluation of certain mycotoxins in food. Fifty-sixth report of the Joint FAO/WHO Expert Committee on Food Additives; International Programme on Chemical Safety, World Health Organizatio: Geneva, Switzerland, 2001; pp. 420–555. [Google Scholar]
- Forsyth, D.M.; Yoshizawa, T.; Morooka, N.; Tuite, J. Emetic and refusal activity of deoxynivalenol to swine. Appl. Environ. Microbiol. 1977, 34, 547–552. [Google Scholar]
- Friend, D.W.; Trenholm, H.L.; Elliot, J.I.; Thompson, B.K.; Hartin, K.E. Effect of feeding vomitoxin-contaminated wheat to pigs. Can. J. Anim. Sci. 1982, 62, 1211–1222. [Google Scholar]
- Pestka, J.J.; Lin, W.S.; Miller, E.R. Emetic activity of the trichothecene 15-acetyldeoxynivalenol in swine. Food Chem. Toxicol. 1987, 25, 855–858. [Google Scholar]
- Prelusky, D.B.; Trenholm, H.L. The efficacy of various classes of anti-emetics in preventing deoxynivalenol-induced vomiting in swine. Nat. Toxins 1993, 1, 296–302. [Google Scholar]
- Rotter, B.A.; Prelusky, D.B.; Pestka, J.J. Toxicology of deoxynivalenol (vomitoxin). J. Toxicol. Environ. Health 1996, 48, 1–34. [Google Scholar]
- Young, L.G.; McGirr, L.; Valli, V.E.; Lumsden, J.H.; Lun, A. Vomitoxin in corn fed to young pigs. J. Anim. Sci. 1983, 57, 655–664. [Google Scholar]
- Pestka, J.J. Mechanisms of deoxynivalenol-induced gene expression and apoptosis. Food Addit. Contam. 2008, 25, 1128–1140. [Google Scholar]
- Turner, P.C.; Rothwell, J.A.; White, K.L.M.; Gong, Y.; Cade, J.E.; Wild, C.P. Urinary deoxynivalenol is correlated with cereal intake in individuals from the United Kingdom. Environ. Health Perspec. 2008, 116, 21–25. [Google Scholar]
- Turner, P.C.; Taylor, E.F.; White, K.L.M.; Cade, J.E.; Wild, C.P. A comparison of 24 h urinary deoxynivalenol with recent v. average cereal consumption for UK adults. British J. Nutri. 2009, 102, 1276–1284. [Google Scholar] [CrossRef]
- Shi, Y.H.; Porter, K.; Parameswaran, N.; Bae, H.K.; Pestka, J.J. Role of GRP78/BiP Degradation and ER Stress in Deoxynivalenol-Induced Interleukin-6 Upregulation in the Macrophage (vol 109, pg 247, 2009). Toxicol. Sci. 2009, 110, 249–250. [Google Scholar] [CrossRef]
- Ueno, Y. Trichothecenes: Chemical, Biological, and Toxicological Aspects. In Trichothecenes; Ueno, Y., Ed.; Elsevier Press: Amsterdam, The Netherlands, 1983; pp. 135–146. [Google Scholar]
- Shifrin, V.I.; Anderson, P. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J. Biol. Chem. 1999, 274, 13985–13992. [Google Scholar]
- Li, M.; Pestka, J.J. Comparative induction of 28S ribosomal RNA cleavage by ricin and the trichothecenes deoxynivalenol and T-2 toxin in the macrophage. Toxicol. Sci. 2008, 105, 67–78. [Google Scholar]
- Zhou, H.R.; Lau, A.S.; Pestka, J.J. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol. Sci. 2003, 74, 335–344. [Google Scholar]
- Williams, B.R. Signal integration via PKR. Sci. STKE 2001, 89, re2. [Google Scholar]
- He, K.; Vines, L.; Pestka, J.J. Deoxynivalenol-induced modulation of microRNA expression in RAW 264.7 macrophages-A potential novel mechanism for translational inhibition. Toxicologist (Toxicol.Sci. Suppl.) 2010, 114, 310. [Google Scholar]
- Chung, Y.J.; Zhou, H.R.; Pestka, J.J. Transcriptional and posttranscriptional roles for p38 mitogen-activated protein kinase in upregulation of TNF-alpha expression by deoxynivalenol (vomitoxin). Toxicol. Appl. Pharmacol. 2003, 193, 188–201. [Google Scholar]
- Jia, Q.; Zhou, H.R.; Bennink, M.; Pestka, J.J. Docosahexaenoic acid attenuates mycotoxin-induced immunoglobulin a nephropathy, interleukin-6 transcription, and mitogen-activated protein kinase phosphorylation in mice. J. Nutr. 2004, 134, 3343–3349. [Google Scholar] [PubMed]
- Gray, J.S.; Bae, H.K.; Li, J.C.B.; Lau, A.S.; Pestka, J.J. Double-stranded RNA-activated protein kinase (PKR) mediates induction of IL-8 expression by deoxynivalenol, Shiga toxin 1 and ricin in monocytes. Toxicol. Sci. 2008, 105, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.S.; Pestka, J.J. Transcriptional regulation of deoxynivalenol-induced IL-8 expression in human monocytes. Toxicol. Sci. 2007, 99, 502–511. [Google Scholar]
- Li, S.; Ouyang, Y.; Yang, G.H.; Pestka, J.J. Modulation of transcription factor AP-1 activity in murine EL-4 thymoma cells by vomitoxin (deoxynivalenol). Toxicol. Appl. Pharmacol. 2000, 163, 17–25. [Google Scholar]
- Li, S.G.; Ouyang, Y.L.; Dong, W.M.; Pestka, J.J. Superinduction of IL-2 gene expression by vomitoxin (deoxynivalenol) involves increased mRNA stability. Toxicol. Appl. Pharmacol. 1997, 147, 331–342. [Google Scholar]
- Korcheva, V.; Wong, J.; Lindauer, M.; Jacoby, D.B.; Iordanov, M.S.; Magun, B. Role of apoptotic signaling pathways in regulation of inflammatory responses to ricin in primary murine macrophages. Mol. Immunol. 2007, 44, 2761–2771. [Google Scholar]
- Cherla, R.P.; Lee, S.Y.; Mees, P.L.; Tesh, V.L. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J. Leukoc. Biol. 2006, 79, 397–407. [Google Scholar]
- Leyva-Illades, D.; Cherla, R.P.; Galindo, C.L.; Chopra, A.K.; Tesh, V.L. Global transcriptional response of macrophage-like THP-1 cells to Shiga toxin type 1. Infec. Immunity 2010, 78, 2454–2465. [Google Scholar]
- Moon, Y.; Pestka, J.J. Vomitoxin-induced cyclooxygenase-2 gene expression in macrophages mediated by activation of ERK and p38 but not JNK mitogen-activated protein kinases. Toxicol. Sci. 2002, 69, 373–382. [Google Scholar]
- Moon, Y.; Pestka, J.J. Deoxynivalenol-induced mitogen-activated protein kinase phosphorylation and IL-6 expression in mice suppressed by fish oil. J. Nutr. Biochem. 2003, 14, 717–726. [Google Scholar]
- Moon, Y.; Pestka, J.J. Cyclooxygenase-2 mediates interleukin-6 upregulation by vomitoxin (deoxynivalenol) in vitro and in vivo. Toxicol. Appl. Pharmacol. 2003, 187, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Kinser, S.; Li, M.; Jia, Q.; Pestka, J.J. Truncated deoxynivalenol-induced splenic immediate early gene response in mice consuming (n-3) polyunsaturated fatty acids. J. Nutr. Biochem. 2005, 16, 88–95. [Google Scholar]
- Nielsen, C.; Lippke, H.; Didier, A.; Dietrich, R.; Martlbauer, E. Potential of deoxynivalenol to induce transcription factors in human hepatoma cells. Molec. Nutri. Food Res. 2009, 53, 479–491. [Google Scholar]
- Choi, H.J.; Yang, H.; Park, S.H.; Moon, Y. HuR/ELAVL1 RNA binding protein modulates interleukin-8 induction by muco-active ribotoxin deoxynivalenol. Toxicol. Appl. Pharmacol. 2009, 240, 46–54. [Google Scholar]
- Ouyang, Y.L.; Li, S.; Pestka, J.J. Effects of vomitoxin (deoxynivalenol) on transcription factor NF-kappa B/Rel binding activity in murine EL-4 thymoma and primary CD4+ T cells. Toxicol. Appl. Pharmacol. 1996, 140, 328–336. [Google Scholar]
- Wong, S.S.; Zhou, H.R.; Pestka, J.J. Effects of vomitoxin (deoxynivalenol) on the binding of transcription factors AP-1, NF-kappaB, and NF-IL6 in raw 26.7 macrophage cells. J. Toxicol. Environ. Health A 2002, 65, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; Jia, Q.; Pestka, J.J. Ribotoxic stress response to the trichothecene deoxynivalenol in the macrophage involves the SRC family kinase Hck. Toxicol. Sci. 2005, 85, 916–926. [Google Scholar]
- Moon, Y.; Uzarski, R.; Pestka, J.J. Relationship of trichothecene structure to COX-2 induction in the macrophage: selective action of type B (8-keto) trichothecenes. J. Toxicol. Environ. Health A 2003, 66, 1967–1983. [Google Scholar]
- Wong, S.S.; Schwartz, R.C.; Pestka, J.J. Superinduction of TNF-alpha and IL-6 in macrophages by vomitoxin (deoxynivalenol) modulated by mRNA stabilization. Toxicology 2001, 161, 139–149. [Google Scholar]
- Jia, Q.; Zhou, H.R.; Shi, Y.; Pestka, J.J. Docosahexaenoic acid consumption inhibits deoxynivalenol-induced CREB/ATF1 activation and IL-6 gene transcription in mouse macrophages. J. Nutr. 2006, 136, 366–372. [Google Scholar]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell Biol. 1997, 17, 3373–3381. [Google Scholar]
- Gray, J.S.; Bae, H.K.; Li, J.C.; Lau, A.S.; Pestka, J.J. Double-stranded RNA-activated protein kinase mediates induction of interleukin-8 expression by deoxynivalenol, Shiga toxin 1, and ricin in monocyte. Toxicol. Sci. 2008, 105, 322–330. [Google Scholar]
- Islam, Z.; Gray, J.S.; Pestka, J.J. p38 Mitogen-activated protein kinase mediates IL-8 induction by the ribotoxin deoxynivalenol in human monocytes. Toxicol. Appl. Pharmacol. 2006, 213, 235–244. [Google Scholar]
- Zhou, H.R.; Islam, Z.; Pestka, J.J. Rapid, sequential activation of mitogen-activated protein kinases and transcription factors precedes proinflammatory cytokine mRNA expression in spleens of mice exposed to the trichothecene vomitoxin. Toxicol. Sci. 2003, 72, 130–142. [Google Scholar]
- Zhou, H.R.; Jia, Q.; Pestka, J.J. Ribotoxic stress response to the trichothecene deoxynivalenol in the macrophage involves the SRC family kinase Hck. Toxicol. Sci. 2005, 85, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Tsygankov, A.Y.; Shore, S.K. Src: regulation, role in human carcinogenesis and pharmacological inhibitors. Curr. Pharm. Des. 2004, 10, 1745–1756. [Google Scholar]
- English, B.K.; Ihle, J.N.; Myracle, A.; Yi, T. Hck tyrosine kinase activity modulates tumor necrosis factor production by murine macrophages. J. Exp. Med. 1993, 178, 1017–1022. [Google Scholar]
- Yang, G.H.; Jarvis, B.B.; Chung, Y.J.; Pestka, J.J. Apoptosis induction by the satratoxins and other trichothecene mycotoxins: relationship to ERK, p38 MAPK, and SAPK/JNK activation. Toxicol. Appl. Pharmacol. 2000, 164, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; Islam, Z.; Pestka, J.J. Induction of competing apoptotic and survival signaling pathways in the macrophage by the ribotoxic trichothecene deoxynivalenol. Toxicol. Sci. 2005, 87, 113–122. [Google Scholar]
- Islam, Z.; Nagase, M.; Ota, A.; Ueda, S.; Yoshizawa, T.; Sakato, N. Structure-function relationship of T-2 toxin and its metabolites in inducing thymic apoptosis in vivo in mice. Biosci. Biotechnol. Biochem. 1998, 62, 1492–1497. [Google Scholar] [PubMed]
- Islam, Z.; Nagase, M.; Yoshizawa, T.; Yamauchi, K.E.; Sakato, N. T-2 toxin induces thymic apoptosis in vivo in mice. Toxicol. Appl. Pharmacol. 1998, 148, 205–214. [Google Scholar]
- Le Drean, G.; Auffret, M.; Batina, P.; Arnold, F.; Sibiril, Y.; Arzur, D.; Parent-Massin, D. Myelotoxicity of trichothecenes and apoptosis: An in vitro study on human cord blood CD34(+) hematopoietic progenitor. Toxicol. Vitro 2005, 19, 1015–1024. [Google Scholar]
- Miura, K.; Nakajima, Y.; Yamanaka, N.; Terao, K.; Shibato, T.; Ishino, S. Induction of apoptosis with fusarenon-X in mouse thymocytes. Toxicology 1998, 127, 195–206. [Google Scholar]
- Nagase, M.; Alam, M.M.; Tsushima, A.; Yoshizawa, T.; Sakato, N. Apoptosis induction by T-2 toxin: Activation of caspase-9, caspase-3, and DFF-40/CAD through cytosolic release of cytochrome c in HL-60 cells. Biosci. Biotechnol. Biochem. 2001, 65, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Poapolathep, A.; Kumagai, S.; Suzuki, H.; Doi, K. Development of early apoptosis and changes in T-cell subsets in mouse thymocyte primary cultures treated with nivalenol. Exp. Mol. Pathol. 2004, 77, 149–152. [Google Scholar]
- Yoshino, N.; Takizawa, M.; Akiba, H.; Okumura, H.; Tashiro, F.; Honda, M.; Ueno, Y. Transient elevation of intracellular calcium ion levels as an early event in T-2 toxin-induced apoptosis in human promyelotic cell line HL-60. Nat. Toxins 1996, 4, 234–241. [Google Scholar]
- Zhou, H.R.; Harkema, J.R.; Hotchkiss, J.A.; Yan, D.; Roth, R.A.; Pestka, J.J. Lipopolysaccharide and the trichothecene vomitoxin (deoxynivalenol) synergistically induce apoptosis in murine lymphoid organs. Toxicol. Sci. 2000, 53, 253–263. [Google Scholar]
- Islam, Z.; Pestka, J.J. Role of IL-1 beta in LPS potentiation of deoxynivalenol-induced leukocyte apoptosis in mice. Toxicol. Sci. 2003, 72, 1605. [Google Scholar]
- Islam, Z.; Pestka, J.J. LPS priming potentiates and prolongs proinflammatory cytokine response to the trichothecene deoxynivalenol in the mouse. Toxicol. Appl. Pharmacol. 2006, 211, 53–63. [Google Scholar]
- Yang, G.H.; Li, S.; Pestka, J.J. Down-regulation of the endoplasmic reticulum chaperone GRP78/BiP by vomitoxin (Deoxynivalenol). Toxicol. Appl. Pharmacol. 2000, 162, 207–217. [Google Scholar]
- Pestka, J.J.; Uzarski, R.L.; Islam, Z. Induction of apoptosis and cytokine production in the Jurkat human T cells by deoxynivalenol: role of mitogen-activated protein kinases and comparison to other 8-ketotrichothecenes. Toxicology 2005, 206, 207–219. [Google Scholar]
- Uzarski, R.L.; Pestka, J.J. Comparative susceptibility of B cells with different lineages to cytotoxicity and apoptosis induction by translational inhibitors. J. Toxicol. Environ. Health A 2003, 66, 2105–2118. [Google Scholar]
- Uzarski, R.L.; Islam, Z.; Pestka, J.J. Potentiation of trichothecene-induced leukocyte cytotoxicity and apoptosis by TNF-alpha and Fas activation. Chem. Biol. Interact. 2003, 146, 105–119. [Google Scholar]
- Pestka, J.J.; Yan, D.; King, L.E. Flow cytometric analysis of the effects of in-vitro exposure to vomitoxin (deoxynivalenol) on apoptosis in murine T-cells, B-cells and IgA(+)-cells. Food Chem. Toxicol. 1994, 32, 1125–1136. [Google Scholar]
- Shi, Y.; Porter, K.; Parameswaran, N.; Bae, H.K.; Pestka, J.J. Role of GRP78/BiP Degradation and ER Stress in Deoxynivalenol-Induced Interleukin-6 Upregulation in the Macrophage. Toxicol. Sci. 2009, 109, 247–255. [Google Scholar]
- Parent-Massin, D. Haematotoxicity of trichothecenes. Toxicol. Lett. 2004, 153, 75–81. [Google Scholar]
- Bae, H.K.; Shinozuka, J.; Islam, Z.; Pestka, J.J. Satratoxin G interaction with 40S and 60S ribosomal subunits precedes apoptosis in the macrophage. Toxicol. Appl. Pharmacol. 2009, 237, 137–145. [Google Scholar]
- Ting, J.P.Y.; Duncan, J.A.; Lei, Y. How the Noninflammasome NLRs Function in the Innate Immune System. Science 2010, 327, 286–290. [Google Scholar]
- Bae, H.K.; Pestka, J.J. Deoxynivalenol induces p38 interaction with the ribosome in monocytes and macrophages. Toxicol. Sci. 2008, 105, 59–66. [Google Scholar]
- Bae, H.; Gray, J.S.; Li, M.; Vines, L.; Kim, J.; Pestka, J.J. Hematopoetic Cell Kinase Associates with the 40S Ribosomal Subunit and Mediates the Ribotoxic Stress Response to Deoxynivalenol in Mononuclear Phagocytes. Toxicol. Sci. 2010, 115, 444–452. [Google Scholar]
- Li, M.X.; Pestka, J.J. Comparative induction of 28S ribosomal RNA cleavage by ricin and the trichothecenes deoxynivalenol and T-2 toxin in the macrophage. Toxicol. Sci. 2008, 105, 67–78. [Google Scholar]
- Yorimitsu, T.; Klionsky, D.J. Eating the endoplasmic reticulum: quality control by autophagy. Trends Cell. Biol. 2007, 17, 279–285. [Google Scholar]
- Boddu, J; Cho, S.H.; Muehlbauer, G.J. Transcriptome analysis of trichothecene-induced gene expression in barley. Mol. Plant-Microbe Interact. 2007, 20, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.M.; Pennings, J.L.A.; Blokland, M.; Peijnenburg, A.; van Loveren, H. Protein expression profiling of mouse thymoma cells upon exposure to the trichothecene deoxynivalenol (DON): Implications for its mechanism of action. J. Immunotoxicol. 2009. [Google Scholar]
- Matsukawa, J.; Matsuzawa, A.; Takeda, K.; Ichijo, H. The ASK1-MAP Kinase Cascades in Mammalian Stress Response. J. Biochem. 2004, 136, 261–265. [Google Scholar]
- Iordanov, M.S.; Magun, B.E. Loss of cellular K+ mimics ribotoxic stress. Inhibition of protein synthesis and activation of the stress kinases SEK1/MKK4, stress-activated protein kinase/c-Jun NH2-terminal kinase 1, and p38/HOG1 by palytoxin. J. Biol. Chem. 1998, 273, 3528–3534. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Keough, R.A.; Gonda, T.J.; Ishii, S. Ribosomal stress induces processing of Mybbp1a and its translocation from the nucleolus to the nucleoplasm. Genes Cells 2008, 13, 27–39. [Google Scholar] [PubMed]
- Azcona Olivera, J.I.; Ouyang, Y.; Murtha, J.; Chu, F.S.; Pestka, J.J. Induction of cytokine mRNAs in mice after oral exposure to the trichothecene vomitoxin (deoxynivalenol): relationship to toxin distribution and protein synthesis inhibition. Toxicol. Appl. Pharmacol. 1995, 133, 109–120. [Google Scholar]
- Pestka, J.J.; Amuzie, C.J. Tissue distribution and proinflammatory cytokine gene expression following acute oral exposure to deoxynivalenol: comparison of weanling and adult mice. Food Chem. Toxicol. 2008, 46, 2826–2831. [Google Scholar]
- Amuzie, C.J.; Harkema, J.R.; Pestka, J.J. Tissue distribution and proinflammatory cytokine induction by the trichothecene deoxynivalenol in the mouse: comparison of nasal vs. oral exposure. Toxicology 2008, 248, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kinser, S.; Jia, Q.; Li, M.; Laughter, A.; Cornwell, P.; Corton, J.C.; Pestka, J.J. Gene expression profiling in spleens of deoxynivalenol-exposed mice: immediate early genes as primary targets. J. Toxicol. Environ. Health A 2004, 67, 1423–1441. [Google Scholar]
- Zhou, H.R.; Yan, D.; Pestka, J.J. Induction of cytokine gene expression in mice after repeated and subchronic oral exposure to vomitoxin (Deoxynivalenol): differential toxin-induced hyporesponsiveness and recovery. Toxicol. Appl. Pharmacol. 1998, 151, 347–358. [Google Scholar]
- Zhou, H.R.; Harkema, J.R.; Yan, D.; Pestka, J.J. Amplified proinflammatory cytokine expression and toxicity in mice coexposed to lipopolysaccharide and the trichothecene vomitoxin (deoxynivalenol). J. Toxicol. Environ. Health 1999, 57, 115–136. [Google Scholar]
- Zhou, H.R.; Islam, Z.; Pestka, J.J. Kinetics of lipopolysaccharide-induced transcription factor activation/inactivation and relation to proinflammatory gene expression in the murine spleen. Toxicol. Appl. Pharmacol. 2003, 187, 147–161. [Google Scholar]
- Clark, I.A. The advent of the cytokine storm. Immunol. Cell. Biol. 2007, 85, 271–273. [Google Scholar]
- Roth, R.A.; Harkema, J.R.; Pestka, J.J.; Ganey, P.E. Is exposure to bacterial endotoxin a determinant of susceptibility to intoxication from xenobiotic agents? Toxicol. Appl. Pharmacol. 1997, 147, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Islam, Z.; King, L.E.; Fraker, P.J.; Pestka, J.J. Differential induction of glucocorticoid-dependent apoptosis in murine lymphoid subpopulations in vivo following coexposure to lipopolysaccharide and vomitoxin (deoxynivalenol). Toxicol. Appl. Pharmacol. 2003, 187, 69–79. [Google Scholar]
- Islam, Z.; Moon, Y.S.; Zhou, H.R.; King, L.E.; Fraker, P.J.; Pestka, J.J. Endotoxin potentiation of trichothecene-induced lymphocyte apoptosis is mediated by up-regulation of glucocorticoids. Toxicol. Appl. Pharmacol. 2002, 180, 43–55. [Google Scholar]
- Islam, Z.; Pestka, J.J. Role of IL-1 beta in endotoxin potentiation of deoxynivalenol-induced corticosterone response and leukocyte apoptosis in mice. Toxicol. Sci. 2003, 74, 93–102. [Google Scholar]
- Islam, Z.; Pestka, J.J. LPS priming potentiates and prolongs proinflammatory cytokine response to the trichothecene deoxynivalenol in the mouse. Toxicol. Appl. Pharmacol. 2005, 211, 53–63. [Google Scholar]
- Kolf-Clauw, M.; Castellote, J.; Joly, B.; Bourges-Abella, N.; Raymond-Letron, I.; Pinton, P.; Oswald, I.P. Development of a pig jejunal explant culture for studying the gastrointestinal toxicity of the mycotoxin deoxynivalenol: Histopathological analysis. Toxicol. Vitro 2009, 23, 1580–1584. [Google Scholar]
- Maresca, M.; Yahi, N.; Younes-Sakr, L.; Boyron, M.; Caporiccio, B.; Fantini, J. Both direct and indirect effects account for the pro-inflammatory activity of enteropathogenic mycotoxins on the human intestinal epithelium: Stimulation of interleukin-8 secretion, potentiation of interleukin-1 beta effect and increase in the transepithelial passage of commensal bacteria. Toxicol. Appl. Pharmacol. 2008, 228, 84–92. [Google Scholar]
- Sergent, T.; Parys, M.; Garsou, S.; Pussemier, L.; Schneider, Y.J.; Larondelle, Y. Deoxynivalenol transport across human intestinal Caco-2 cells and its effects on cellular metabolism at realistic intestinal concentrations. Toxicol. Lett. 2006, 164, 167–176. [Google Scholar]
- Pinton, P.; Nougayrede, J.P.; Del Rio, J.C.; Moreno, C.; Marin, D.E.; Ferrier, L.; Bracarense, A.P.; Kolf-Clauw, M.; Oswald, I.P. The food contaminant deoxynivalenol decreases intestinal barrier permeability and reduces claudin expression. Toxicol. Appl. Pharmacol. 2009, 237, 41–48. [Google Scholar]
- Amuzie, C.J.; Shinozuka, J.; Pestka, J.J. Induction of suppressors of cytokine signaling by the trichothecene deoxynivalenol in the mouse. Toxicol. Sci. 2009, 111, 277–287. [Google Scholar]
- Konsman, J.P.; Parnet, P.; Dantzer, R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002, 25, 154–159. [Google Scholar]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin.Cell. Dev. Biol. 2008, 19, 414–422. [Google Scholar]
- Amuzie, C.J.; Pestka, J.J. Suppression of Insulin-Like Growth Factor Acid-Labile Subunit Expression-A Novel Mechanism for Deoxynivalenol-Induced Growth Retardation. Toxicol. Sci. 2010, 113, 412–421. [Google Scholar]
- Voss, K.A. A New Perspective on Deoxynivalenol and Growth Suppression. Toxicol. Sci. 2010, 113, 281–283. [Google Scholar]
- Pestka, J.J.; Moorman, M.A.; Warner, R.L. Dysregulation of IgA production and IgA nephropathy induced by the trichothecene vomitoxin. Food Chem.Toxicol. 1989, 27, 361–368. [Google Scholar]
- Pestka, J.J. Deoxynivalenol-induced IgA production and IgA nephropathy-aberrant mucosal immune response with systemic repercussions. Toxicol. Lett. 2003, 140–141, 287–295. [Google Scholar] [CrossRef] [PubMed]
- D'Amico, G. The commonest glomerulonephritis in the world: IgA nephropathy. Q. J. Med. 1987, 64, 709–727. [Google Scholar]
- Dong, W.; Pestka, J.J. Persistent dysregulation of IgA production and IgA nephropathy in the B6C3F1 mouse following withdrawal of dietary vomitoxin (deoxynivalenol). Fundam. Appl. Toxicol. 1993, 20, 38–47. [Google Scholar]
- Pestka, J.J.; Dong, W.; Warner, R.L.; Rasooly, L.; Bondy, G.S.; Brooks, K.H. Elevated membrane IgA+ and CD4+ (T helper) populations in murine Peyer's patch and splenic lymphocytes during dietary administration of the trichothecene vomitoxin (deoxynivalenol). Food Chem. Toxicol. 1990, 28, 409–420. [Google Scholar]
- Pestka, J.J.; Dong, W.; Warner, R.L.; Rasooly, L.; Bondy, G.S. Effect of dietary administration of the trichothecene vomitoxin (deoxynivalenol) on IgA and IgG secretion by Peyer's patch and splenic lymphocytes. Food Chem. Toxicol. 1990, 28, 693–699. [Google Scholar]
- Pestka, J.J.; Moorman, M.A.; Warner, R.L. Altered serum immunoglobulin response to model intestinal antigens during dietary exposure to vomitoxin (deoxynivalenol). Toxicol. Lett. 1990, 50, 75–84. [Google Scholar]
- Rasooly, L.; Abouzied, M.M.; Brooks, K.H.; Pestka, J.J. Polyspecific and autoreactive IgA secreted by hybridomas derived from Peyer's patches of vomitoxin-fed mice: characterization and possible pathogenic role in IgA nephropathy. Food Chem.Toxicol. 1994, 32, 337–348. [Google Scholar]
- Rasooly, L.; Pestka, J.J. Vomitoxin-induced dysregulation of serum IgA, IgM and IgG reactive with gut bacterial and self antigens. Food Chem.Toxicol. 1992, 30, 499–504. [Google Scholar]
- Rasooly, L.; Pestka, J.J. Polyclonal autoreactive IgA increase and mesangial deposition during vomitoxin-induced IgA nephropathy in the BALB/c mouse. Food Chem.Toxicol. 1994, 32, 329–336. [Google Scholar]
- Zhou, H.R.; Yan, D.; Pestka, J.J. Differential cytokine mRNA expression in mice after oral exposure to the trichothecene vomitoxin (deoxynivalenol): dose response and time course. Toxicol. Appl. Pharmacol. 1997, 144, 294–305. [Google Scholar]
- Beagley, K.W.; Elson, C.O. Cells and cytokines in mucosal immunity and inflammation. Gastroenterol. Clin. North Am. 1992, 21, 347–366. [Google Scholar]
- Yan, D.; Zhou, H.R.; Brooks, K.H.; Pestka, J.J. Role of macrophages in elevated IgA and IL-6 production by Peyer's patch cultures following acute oral vomitoxin exposure. Toxicol. Appl. Pharmacol. 1998, 148, 261–273. [Google Scholar]
- Pestka, J.J.; Zhou, H.R. Interleukin-6-deficient mice refractory to IgA dysregulation but not anorexia induction by vomitoxin (deoxynivalenol) ingestion. Food Chem. Toxicol. 2000, 38, 565–575. [Google Scholar]
- Turner, P.C.; Burley, V.J.; Rothwell, J.A.; White, K.L.M.; Cade, J.E.; Wild, C.P. Deoxynivalenol: Rationale for development and application of a urinary biomarker. Food Addit. Contam. 2008, 25, 864–871. [Google Scholar]
- Chung, Y.J.; Zhou, H.R.; Pestka, J.J. Up-regulation of macrophage inflammatory protein-2 and complement 3A receptor by the trichothecenes deoxynivalenol and satratoxin G. Toxicology 2003, 186, 51–65. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pestka, J.J. Deoxynivalenol-Induced Proinflammatory Gene Expression: Mechanisms and Pathological Sequelae. Toxins 2010, 2, 1300-1317. https://doi.org/10.3390/toxins2061300
Pestka JJ. Deoxynivalenol-Induced Proinflammatory Gene Expression: Mechanisms and Pathological Sequelae. Toxins. 2010; 2(6):1300-1317. https://doi.org/10.3390/toxins2061300
Chicago/Turabian StylePestka, James J. 2010. "Deoxynivalenol-Induced Proinflammatory Gene Expression: Mechanisms and Pathological Sequelae" Toxins 2, no. 6: 1300-1317. https://doi.org/10.3390/toxins2061300