Alpha-Tocopherol Counteracts the Cytotoxicity Induced by Ochratoxin A in Primary Porcine Fibroblasts


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Abbreviations
| LC50 | half-lethal concentration |
| LDH | lactate dehydrogenase |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide |
| OTA | ochratoxin A |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
1. Introduction
2. Results and Discussion
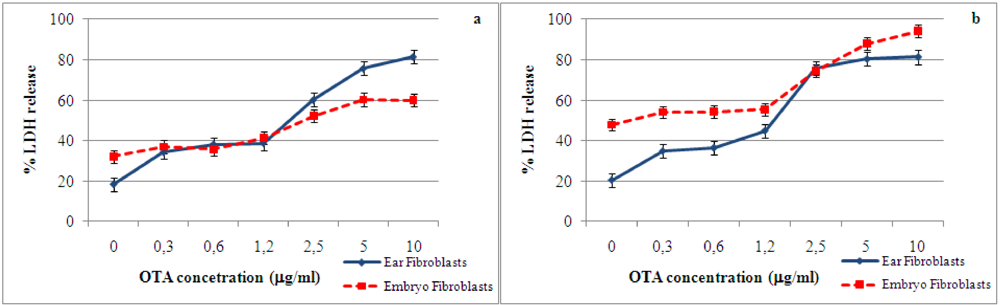
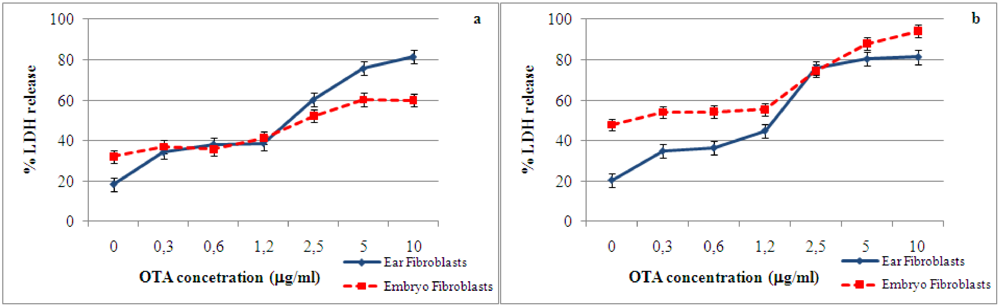
2.1. Cytotoxic effect and LDH release induced by ochratoxin A
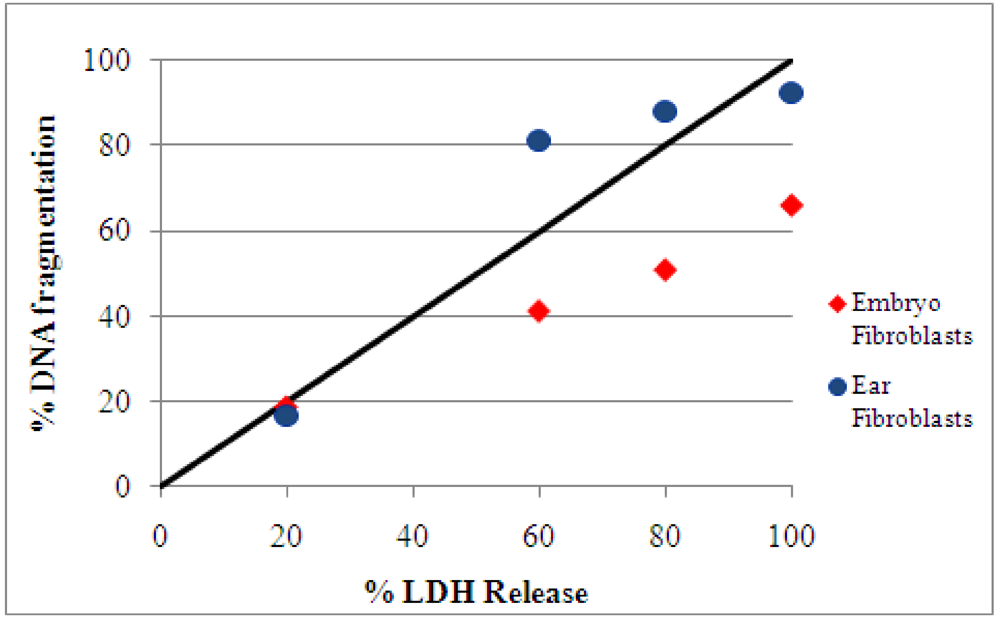
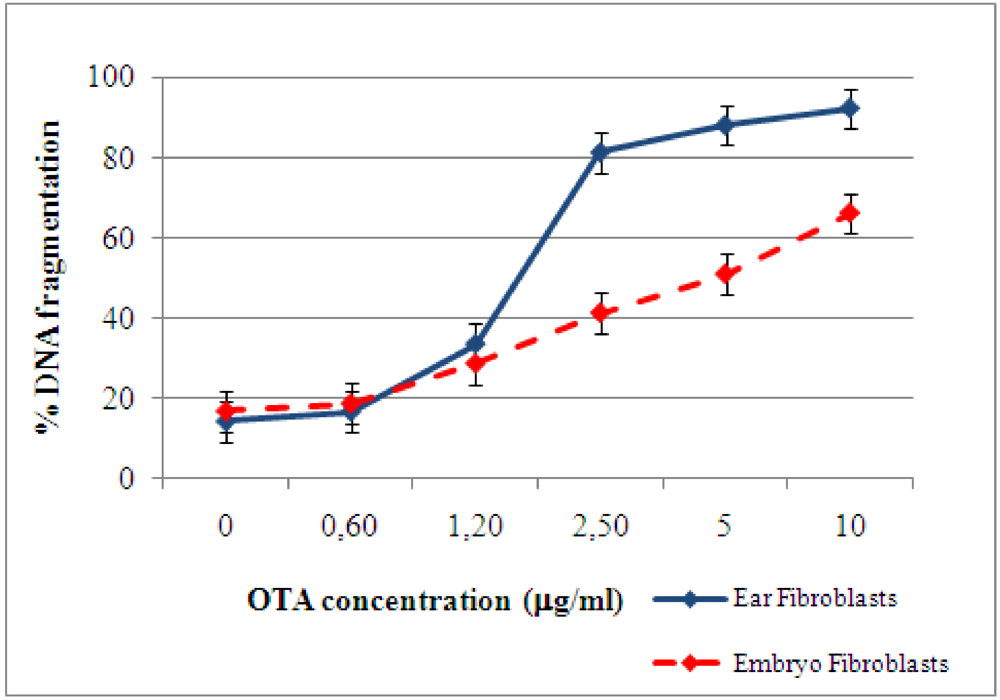
2.2. Detection and quantification of DNA damage induced by ochratoxin A
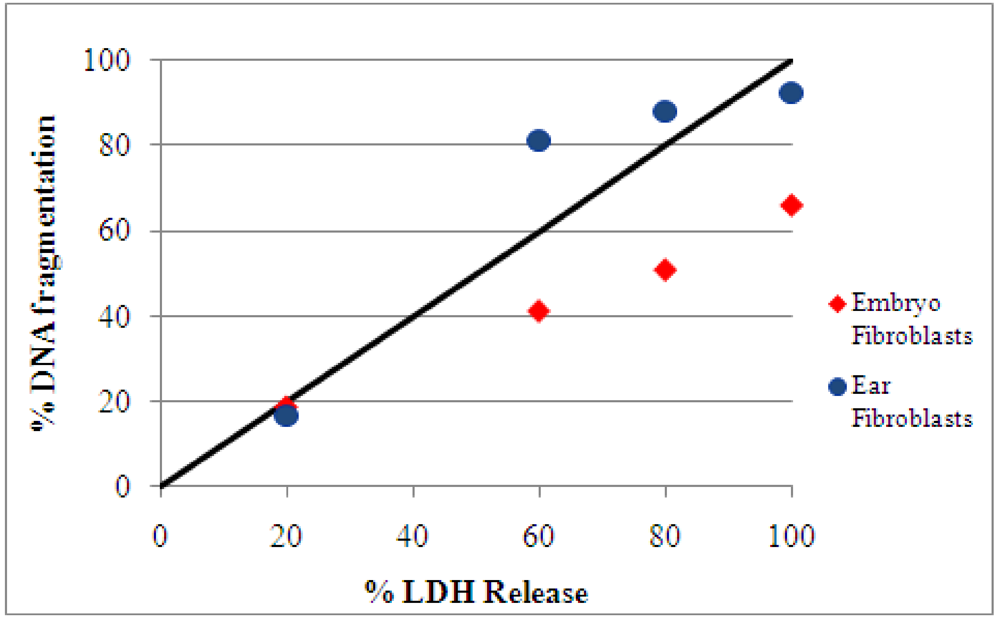
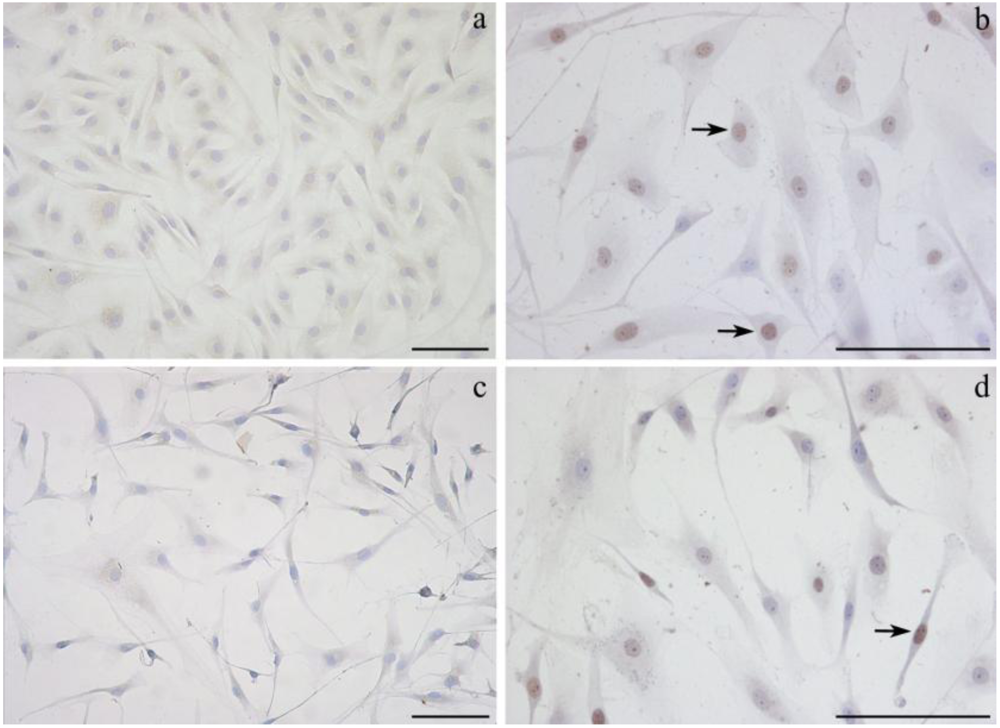
2.3. Detection of DNA damage by TUNEL assay
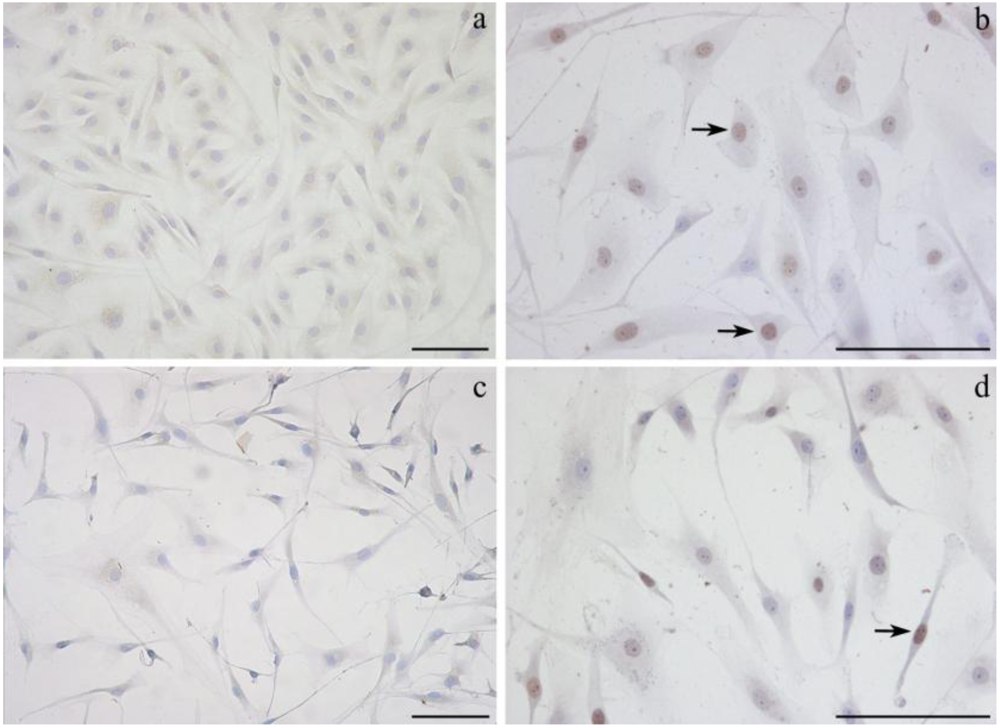
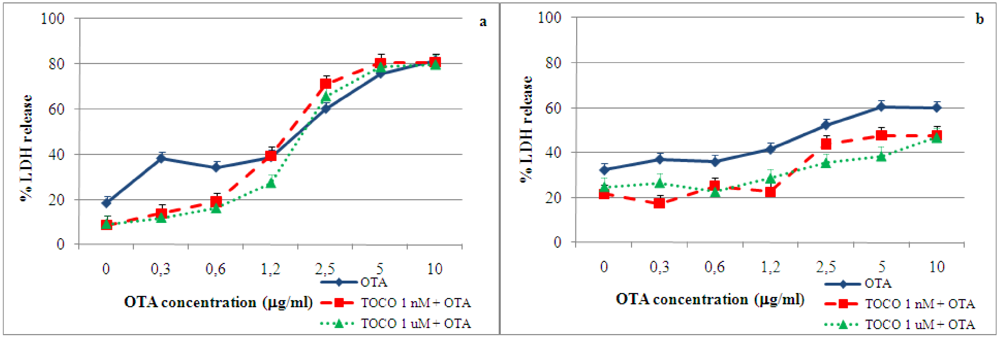
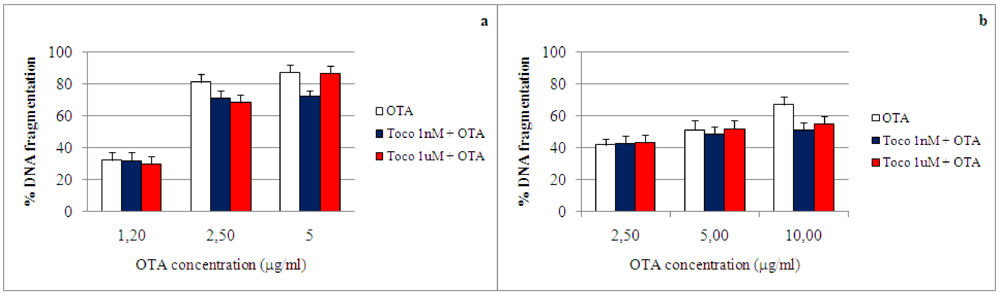
2.4. Effect of α-tocopherol on ochratoxin A–induced toxicity
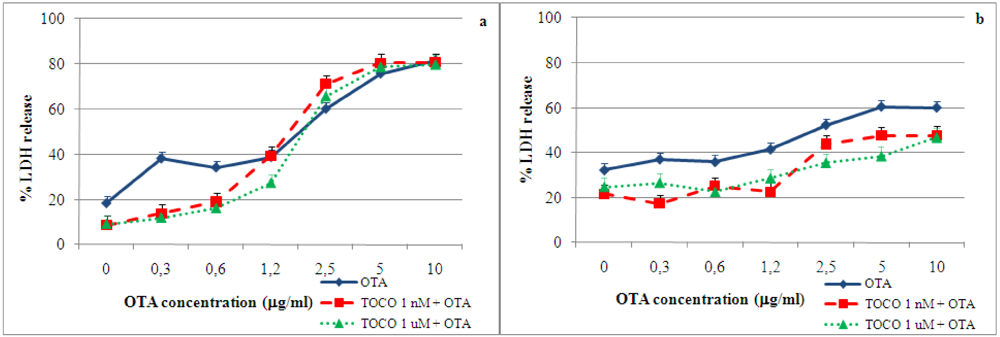
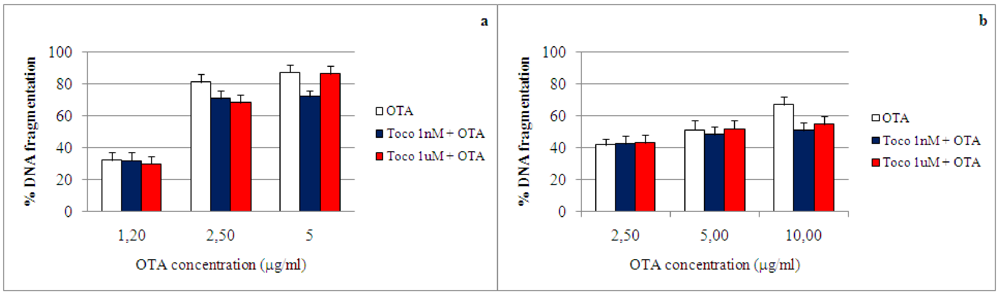
3. Materials and Methods
3.1. Chemicals
3.2. Cell culture
3.3. Determination of the half-lethal concentration and LDH release induced by ochratoxin A
3.4. Detection and quantification of DNA damage induced by ochratoxin A
3.5. Detection of DNA damage induced by ochratoxin A byTUNEL (TdT-mediated dUTP nick end labeling) assay
3.6. Determination of the effect of α-tocopherol against OTA-induced toxicity
3.7. Statistical analysis
4. Conclusions
Acknowledgements
References
- Hussein, H.S.; Brasel, J.M. Toxicity, metabolism, and impact of mycotoxins on humans and animals. Toxicology 2001, 167, 101–134. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51 (1), 61–99. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer World Health Organization, Some Naturally Occurring Substances, Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer World Health Organization: Lyon, France, 1993; 56, pp. 26–32.
- European Food Safety Authority. Opinion of the scientific panel on contaminants in the food chain on a request from the commission related to Ochratoxin A in food. Question N° EFSA-Q-2005-154. EFSA J. 2006, 365, 1–56.
- O’Brien, E.; Dietrich, D.R. Ochratoxin A: the continuing enigma. Crit. Rev. Toxicol. 2005, 35, 33–60. [Google Scholar]
- Council for Agricultural Science and Technology, Mycotoxins: Risks in Plant, Animal and Human System. Task Force Report, Council for Agricultural Science and Technology, No. 139; Council for Agricultural Science & Technology: Ames, IA, USA, 2003.
- Ringot, D.; Chango, A.; Scheneider, Y.-J.; Larondelle, Y. Toxicokinetics and toxicodynamics of ochratoxin A, an update. Chem. Biol. Interact. 2006, 159, 18–46. [Google Scholar]
- Kamp, H.G.; Eisenbrand, G.; Janzowski, C.; Kiossev, J.; Latendresse, J.R.; Schlatter, J.; Turesky, R.J. Ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Mol. Nutr. Food Res. 2005, 49 (12), 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Baudrimont, I.; Ahouandjivo, R.; Creppy, E.E. Prevention of lipid peroxidation induced by ochratoxin A in Vero cells in culture by several agents. Chem. Biol. Interact. 1997, 104, 29–40. [Google Scholar]
- Petrik, J.; Zanić-Grubisić, T.; Barisić, K.; Pepeljnjak, S.; Radić, B.; Ferencić, Z.; Cepelak, I. Apoptosis and oxidative stress induced by ochratoxin A in rat kidney. Arch. Toxicol. 2003, 77 (12), 685–693. [Google Scholar] [CrossRef] [PubMed]
- Pfohl-Leszkowicz, A.; Bartsch, H.; Azémar, B.; Mohr, U.; Estéve, J.; Castegnaro, M. MESNA protects rats against nephrotoxicity but not carcinogenicity induced by ochratoxin A, implicating two separate pathways. Facta Univ. Ser. Med. Biol. 2002, 9, 57–63. [Google Scholar]
- Azzi, A.; Breyer, I.; Feher, M.; Pastori, M.; Ricciarelli, R.; Spycher., S.; Staffieri, M.; Stocker, A.; Zimmer, S.; Zingg, J.M. Specific cellular responses to alpha-tocopherol. J. Nutr. 2000, 130 (7), 1649–1652. [Google Scholar] [PubMed]
- Baldi, A. Vitamin E in dairy cows. Liv. Prod. Sci. 2005, 98 (1–2), 117–122. [Google Scholar] [CrossRef]
- Azzi, A. Molecular mechanism of α-tocopherol action. Free Rad. Biol. Med. 2007, 43, 16–21. [Google Scholar] [CrossRef]
- Wang, X.; Quinn, P.J. Vitamin E and its function in membranes. Progr. Lipid Res. 1999, 38, 309–336. [Google Scholar]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Rad. Biol. Med. 2007, 43, 4–15. [Google Scholar]
- Rimbach, G.; Minihane, A.M.; Majewicz, J.; Fischer, A.; Pallauf, J.; Virgli, F.; Weinberg, P.D. Regulation of cell signaling by vitamin E. Proc. Nutr. Soc. 2002, 61 (4), 415–425. [Google Scholar] [CrossRef] [PubMed]
- Baldi, A.; Losio, M.N.; Cheli, F.; Rebucci, R.; Sangalli, L.; Fusi, E.; Bertasi, B.; Pavoni, E.; Carli, S.; Politis, I. Evaluation of the protective effects of α-tocopherol and retinol against ochratoxin A cytotoxicity. Brit. J. Nutr. 2004, 91, 507–512. [Google Scholar] [CrossRef]
- Russo, A.; La Fauci, L.; Acquaviva, R.; Campisi, A.; Raciti, G.; Scifo, C.; Renis, M.; Galvano, G.; Vanella, A.; Galvano, F. Ochratoxin A-induced DNA damage in human fibroblast: protective effect of cyanidin 3-O-beta-d-glucoside. J. Nutr. Biochem. 2005, 16 (1), 31–37. [Google Scholar] [CrossRef] [PubMed]
- Grosse, Y.; Chekir-Ghedira, L.; Huc, A.; Obrecht-Pflumio, S.; Dirheimer, G.; Bacha, H.; Pfohl-Leszkowicz, A. Retinol, ascorbic acid and alpha-tocopherol prevent DNA adduct formation in mice treated with the mycotoxins ochratoxin A and zearalenone. Cancer Lett. 1997, 114 (1–2), 225–229. [Google Scholar] [CrossRef] [PubMed]
- Fusi, E.; Rebucci, R.; Pecorini, C.; Rossi, L.; D’Ambrosio, F.; Baldi, A. Evaluation of the damage induced by ochratoxin A and the protective role of a-tocopherol in cultured bovine mammary epithelial cells. Vet. Res. Comm. 2008, 32 (1), 343–345. [Google Scholar] [CrossRef]
- Schwerdt, G.; Holzinger, H.; Sauvant, C.; Königs, M.; Humpf, H.U.; Gekle, M. Long-term effects of ochratoxin A on fibrosis and cell death in human proximal tubule or fibroblast cells in primary culture. Toxicology 2007, 232 (1–2), 57–67. [Google Scholar] [CrossRef] [PubMed]
- Gekle, M.; Sauvant, C.; Schwerdt, G. Ochratoxin A at nanomolar concentrations: a signal modulator in renal cells. Mol. Nutr. Food Res. 2005, 49 (2), 118–130. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Heussner, A.H.; Dietrich, D.R. Species-, sex-, and cell type-specific effects of Ochratoxin A and B. Toxicol. Sci. 2001, 63, 256–264. [Google Scholar]
- Gekle, M.; Schwerdt, G.; Freudinger, R.; Mildeberger, S.; Wilflingseder, D.; Pollack, V.; Dander, M.; Schramek, H. Ochratoxin A induces JNK activation and apoptosis in MDCK-C7 cells at nanomolar concentrations. J. Pharmacol. Exp. Ther. 2000, 293, 837–844. [Google Scholar]
- Zeiss, C.J. The apoptosis-necrosis continuum: insights from genetically altered mice. Vet. Pathol. 2003, 40, 481–495. [Google Scholar]
- Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007, 35 (4), 495–516. [Google Scholar] [CrossRef] [PubMed]
- Manderville, R.A.; Pfohl-Leszkowicz, A. Bioactivation and DNA adduction as a rationale for Ochratoxin A carcinogenesis. World Mycotoxin J. 2008, 1 (3), 357–367. [Google Scholar] [CrossRef]
- Schaaf, G.J.; Nijmeijer, S.M.; Maas, R.F.; Roestenberg, P.; de Groene, E.M.; Fink-Gremmels, J. The role of oxidative stress in the ochratoxin A-mediated toxicity in proximal tubular cells. Biochim. Biophys. Acta 2002, 1588 (2), 149–158. [Google Scholar] [PubMed]
- Fazzio, A.; Marillet, D.; Azzi, A. The effect of alpha-tocopherol and beta-tocopherol on proliferation, protein kinase C activity and gene expression in different cell lines. Biochem. Mol. Biol. Int. 1997, 41, 93–101. [Google Scholar]
- Baldi, A.; Fusi, E.; Rebucci, R.; Pinotti, L.; Cheli, F. In vitro cytotoxicity of aflatoxin B1, M1 and ochratoxin A and protective effects of antioxidants. J. Dairy Sci. 2002, 85, 29. [Google Scholar]
- Cheli, F.; Politis, I.; Rossi, L.; Fusi, E.; Baldi, A. Effects of retinoids on proliferation and plasminogen activator expression in a bovine mammary epithelial cell line. J. Dairy Res. 2003, 70, 367–372. [Google Scholar]
- Sandau, K.; Pfeilschifter, J.; Brüne, B. Nitric oxide and superoxide induced p53 and Bax accumulation during mesangial cell apoptosis. Kidney Int. 1997, 52, 378–386. [Google Scholar]
- SAS, User's Guide: Statistics, Release 9.1; SAS Inst. Inc.: Cary, NC, USA, 2001.
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fusi, E.; Rebucci, R.; Pecorini, C.; Campagnoli, A.; Pinotti, L.; Saccone, F.; Cheli, F.; Purup, S.; Sejrsen, K.; Baldi, A. Alpha-Tocopherol Counteracts the Cytotoxicity Induced by Ochratoxin A in Primary Porcine Fibroblasts. Toxins 2010, 2, 1265-1278. https://doi.org/10.3390/toxins2061265
Fusi E, Rebucci R, Pecorini C, Campagnoli A, Pinotti L, Saccone F, Cheli F, Purup S, Sejrsen K, Baldi A. Alpha-Tocopherol Counteracts the Cytotoxicity Induced by Ochratoxin A in Primary Porcine Fibroblasts. Toxins. 2010; 2(6):1265-1278. https://doi.org/10.3390/toxins2061265
Chicago/Turabian StyleFusi, Eleonora, Raffaella Rebucci, Chiara Pecorini, Anna Campagnoli, Luciano Pinotti, Francesca Saccone, Federica Cheli, Stig Purup, Kristen Sejrsen, and Antonella Baldi. 2010. "Alpha-Tocopherol Counteracts the Cytotoxicity Induced by Ochratoxin A in Primary Porcine Fibroblasts" Toxins 2, no. 6: 1265-1278. https://doi.org/10.3390/toxins2061265