The Zinc-Dependent Protease Activity of the Botulinum Neurotoxins

Abstract
:1. Introduction
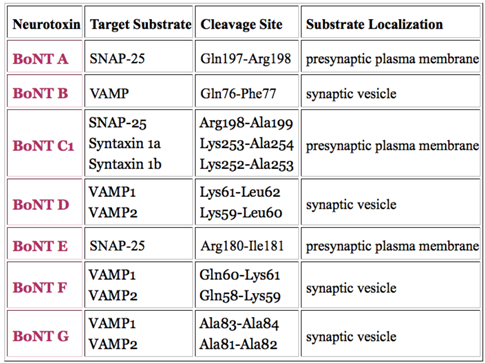
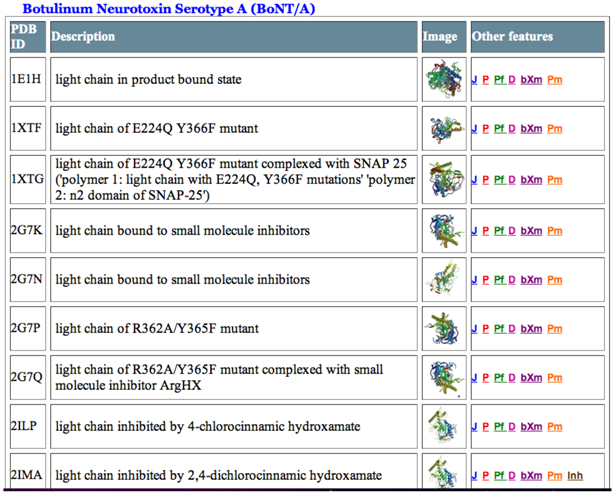
2. Proteolytic Activity

{kind=link}
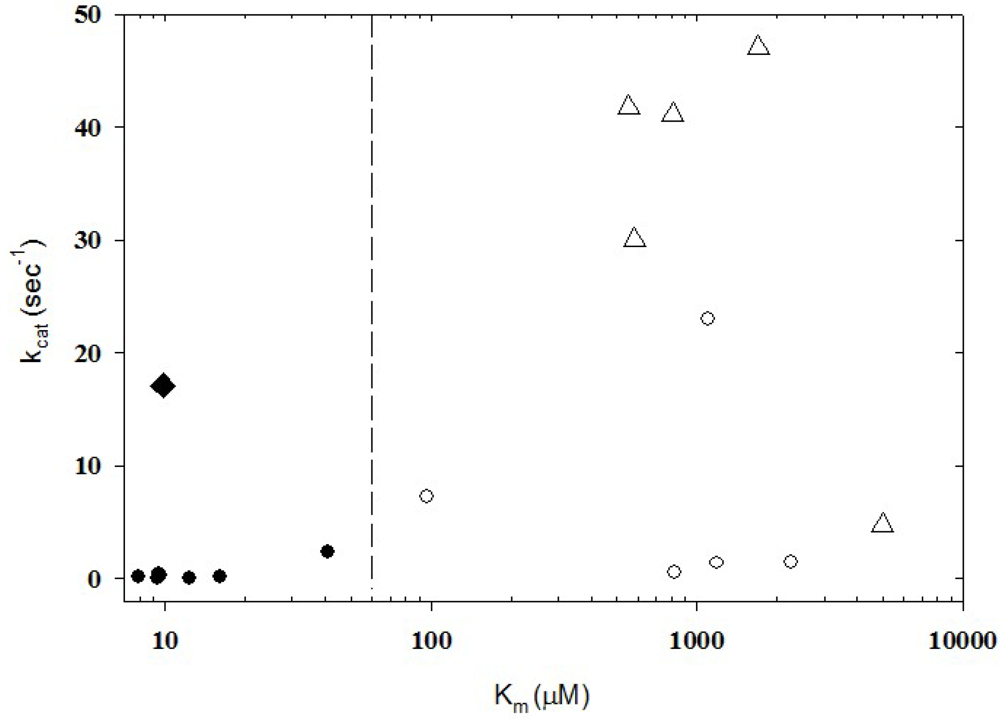{kind=link}
{kind=link}
{kind=link}
| % similarities | ||||||||
| /A | /B | /C1 | /D | /E | /F | /G | TeNT | |
| /A | - | 52 | 52 | 54 | 54 | 53 | 53 | 52 |
| /B | 31 | - | 54 | 53 | 58 | 58 | 75 | 68 |
| /C1 | 33 | 33 | - | 61 | 54 | 56 | 53 | 55 |
| /D | 34 | 33 | 56 | - | 54 | 55 | 54 | 54 |
| /E | 34 | 37 | 35 | 36 | - | 72 | 58 | 62 |
| /F | 34 | 38 | 35 | 36 | 58 | - | 59 | 62 |
| /G | 35 | 59 | 35 | 36 | 38 | 40 | - | 66 |
| TeNT | 31 | 50 | 34 | 34 | 44 | 44 | 49 | - |
| % identities | ||||||||
 |
 |
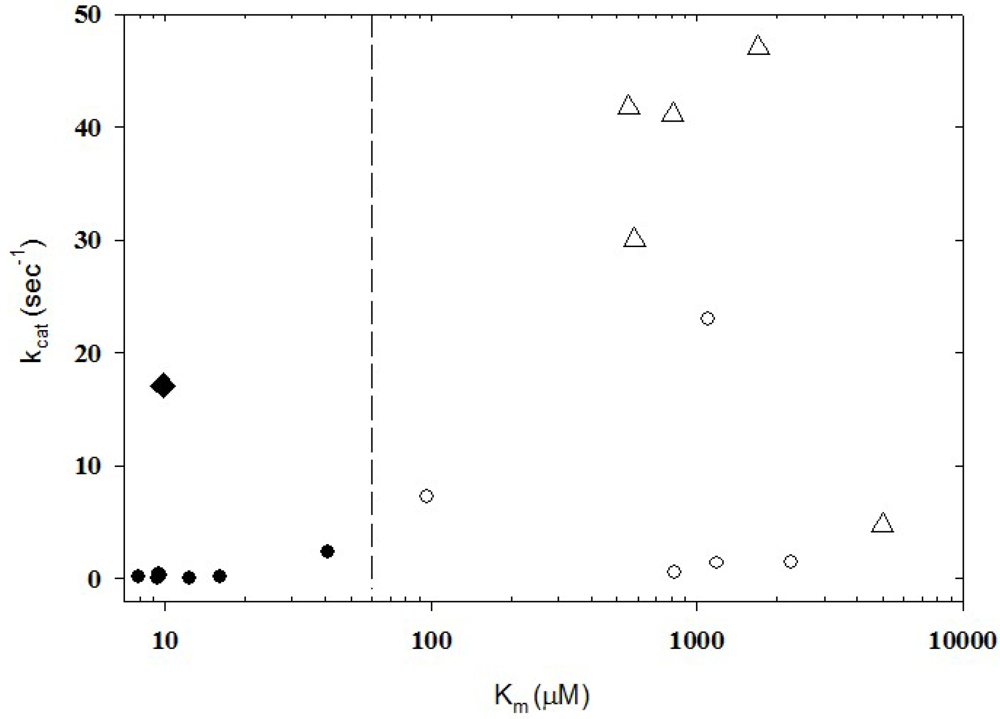
3. Enzyme Kinetics
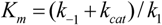 (1)
(1)
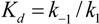 (2)
(2)
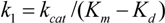 (3)
(3)
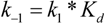 (4)
(4)
4. Inhibitors

5. Activators
6. Future Research
Acknowledgements
References
- Gomis-Ruth, F.X. Structural aspects of the metzincin clan of metalloendopeptidases. Mol. Biotechnol. 2003, 24, 157–202. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: the peptidase database. Nucleic Acids Res. 2009, 38, D227–D233. [Google Scholar]
- Singh, B.R.; Li, B.; Read, D. Botulinum versus tetanus neurotoxins: why is botulinum neurotoxin but not tetanus neurotoxin a food poison? Toxicon 1995, 33, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Rabasseda, X.; Blasi, J.; Marsal, J.; Dunant, Y.; Casanova, A.; Bizzini, B. Tetanus and botulinum toxins block the release of acetylcholine from slices of rat striatum and from the isolated electric organ of Torpedo at different concentrations. Toxicon 1988, 26, 329–336. [Google Scholar]
- Habermann, E.; Dreyer, F.; Bigalke, H. Tetanus toxin blocks the neuromuscular transmission in vitro like botulinum A toxin. Naunyn Schmiedebergs Arch. Pharmacol. 1980, 311, 33–40. [Google Scholar] [CrossRef]
- Washbourne, P.; Pellizzari, R.; Rossetto, O.; Bortoletto, N.; Tugnoli, V.; De Grandis, D.; Eleopra, R.; Montecucco, C. On the action of botulinum neurotoxins A and E at cholinergic terminals. J. Physiol. Paris 1998, 92, 135–139. [Google Scholar] [CrossRef]
- Arnon, S.S.; Schechter, R.; Inglesby, T.V.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Fine, A.D.; Hauer, J.; Layton, M.; Lillibridge, S.; Osterholm, M.T.; O'Toole, T.; Parker, G.; Perl, T.M.; Russell, P.K.; Swerdlow, D.L.; Tonat, K. Botulinum toxin as a biological weapon: medical and public health management. J. Amer. Med. Assoc. 2001, 285, 1059–1070. [Google Scholar]
- Mejia, N.I.; Vuong, K.D.; Jankovic, J. Long-term botulinum toxin efficacy, safety, and immunogenicity. Mov. Disord. 2005, 20, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, J.K.; Jankovic, J. Botulinum toxin in the treatment of tremors, dystonias, sialorrhea and other symptoms associated with Parkinson's disease. Expert Rev. Neurother. 2007, 7, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Lebeda, F.J.; Cer, R.Z.; Stephens, R.; Mudunuri, U. Temporal characteristics of botulinum neurotoxin therapy. Expert Rev. Neurother. 2010, 10, 93–103. [Google Scholar]
- Montal, M. Botulinum Neurotoxin: A Marvel of Protein Design. Annu. Rev. Biochem. 2010, 79. [Epub ahead of print]. [Google Scholar]
- Schiavo, G.; Matteoli, M.; Montecucco, C. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 2000, 80, 717–766. [Google Scholar]
- Montecucco, C.; Schiavo, G. Structure and function of tetanus and botulinum neurotoxins. Q. Rev. Biophys. 1995, 28, 423–472. [Google Scholar] [CrossRef]
- Simpson, L.L. Identification of the major steps in botulinum toxin action. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 167–193. [Google Scholar]
- Sharma, S.K.; Basavanna, U.; Shukla, H.D. Protein domain analysis of C. botulinum type A neurotoxin and its relationship with other botulinum serotypes. Toxins 2010, 2, 1–9. [Google Scholar]
- Swaminathan, S.; Eswaramoorthy, S. Structural analysis of the catalytic and binding sites of Clostridium botulinum neurotoxin B. Nat. Struct. Biol. 2000, 7, 693–699. [Google Scholar] [CrossRef]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; DasGupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998, 5, 898–902. [Google Scholar]
- Schiavo, G.; Rossetto, O.; Santucci, A.; DasGupta, B.R.; Montecucco, C. Botulinum neurotoxins are zinc proteins. J. Biol. Chem. 1992, 267, 23479–23483. [Google Scholar]
- Schiavo, G.; Poulain, B.; Rossetto, O.; Benfenati, F.; Tauc, L.; Montecucco, C. Tetanus toxin is a zinc protein and its inhibition of neurotransmitter release and protease activity depend on zinc. EMBO J. 1992, 11, 3577–3583. [Google Scholar]
- Sigrist, C.J.; Cerutti, L.; de Castro, E.; Langendijk-Genevaux, P.S.; Bulliard, V.; Bairoch, A.; Hulo, N. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2009, 38, D161–D166. [Google Scholar]
- Tonello, F.; Montecucco, C. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol. Aspects Med. 2009, 30, 431–438. [Google Scholar] [CrossRef]
- Sathyamoorthy, V.; DasGupta, B.R. Separation, purification, partial characterization and comparison of the heavy and light chains of botulinum neurotoxin types A, B, and E. J. Biol. Chem. 1985, 260, 10461–10466. [Google Scholar] [PubMed]
- Lomneth, R.; Gimenez, J.; Martin, T.F.; DasGupta, B.R. Calcium-dependent release of norepinephrine from permeabilized PC12 cells is inhibited by approximately 48 and approximately 112 kDa fragments of botulinum neurotoxin type E. Neuropharmacology 1993, 32, 285–259. [Google Scholar] [CrossRef]
- Brunger, A.T.; Breidenbach, M.A.; Jin, R.; Fischer, A.; Santos, J.S.; Montal, M. Botulinum neurotoxin heavy chain belt as an intramolecular chaperone for the light chain. PLoS Pathog. 2007, 3, 1191–1194. [Google Scholar]
- Rossetto, O.; Montecucco, C. Peculiar binding of botulinum neurotoxins. ACS Chem. Biol. 2007, 2, 96–98. [Google Scholar] [CrossRef]
- Lebeda, F.J.; Singh, B.R. Membrane channel activity and translocation of tetanus and botulinum neurotoxins. J. Toxicol.-Toxin Rev. 1999, 18, 45–76. [Google Scholar]
- Simpson, L.L. Kinetic studies on the interaction between botulinum toxin type A and the cholinergic neuromuscular junction. J. Pharmacol. Exp. Ther. 1980, 212, 16–21. [Google Scholar]
- Lebeda, F.J.; Adler, M.; Erickson, K.; Chushak, Y. Onset dynamics of type A botulinum neurotoxin-induced paralysis. J. Pharmacokinet. Pharmacodyn. 2008, 35, 251–267. [Google Scholar] [CrossRef]
- Foran, P.; Shone, C.C.; Dolly, J.O. Differences in the protease activities of tetanus and botulinum B toxins revealed by the cleavage of vesicle-associated membrane protein and various sized fragments. Biochemistry 1994, 33, 15365–15374. [Google Scholar]
- Osen-Sand, A.; Staple, J.K.; Naldi, E.; Schiavo, G.; Rossetto, O.; Petitpierre, S.; Malgaroli, A.; Montecucco, C.; Catsicas, S. Common and distinct fusion proteins in axonal growth and transmitter release. J. Comp. Neurol. 1996, 367, 222–234. [Google Scholar] [CrossRef]
- Williamson, L.C.; Halpern, J.L.; Montecucco, C.; Brown, J.E.; Neale, E.A. Clostridial neurotoxins and substrate proteolysis in intact neurons: botulinum neurotoxin C acts on synaptosomal-associated protein of 25 kDa. J. Biol. Chem. 1996, 271, 7694–7699. [Google Scholar]
- Foran, P.; Lawrence, G.W.; Shone, C.C.; Foster, K.A.; Dolly, J.O. Botulinum neurotoxin C1 cleaves both syntaxin and SNAP-25 in intact and permeabilized chromaffin cells: correlation with its blockade of catecholamine release. Biochemistry 1996, 35, 2630–2636. [Google Scholar]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Gaudet, P.; Lane, L.; Fey, P.; Bridge, A.; Poux, S.; Auchincloss, A.; Axelsen, K.; Braconi Quintaje, S.; Boutet, E.; Brown, P.; Coudert, E.; Datta, R.S.; de Lima, W.C.; de Oliveira Lima, T.; Duvaud, S.; Farriol-Mathis, N.; Ferro Rojas, S.; Feuermann, M.; Gateau, A.; Hinz, U.; Hulo, C.; James, J.; Jimenez, S.; Jungo, F.; Keller, G.; Lemercier, P.; Lieberherr, D.; Moinat, M.; Nikolskaya, A.; Pedruzzi, I.; Rivoire, C.; Roechert, B.; Schneider, M.; Stanley, E.; Tognolli, M.; Sjolander, K.; Bougueleret, L.; Chisholm, R.L.; Bairoch, A. Collaborative annotation of genes and proteins between UniProtKB/Swiss-Prot and dictyBase. Database (Oxford) 2009, 2009, ap016. [Google Scholar] [PubMed]
- Fairweather, N.F.; Lyness, V.A. The complete nucleotide sequence of tetanus toxin. Nucleic Acids Res. 1986, 14, 7809–7812. [Google Scholar]
- Jongeneel, C.V.; Bouvier, J.; Bairoch, A. A unique signature identifies a family of zinc-dependent metallopeptidases. FEBS Lett. 1989, 242, 211–214. [Google Scholar]
- Schiavo, G.; Poulain, B.; Rossetto, O.; Benfenati, F.; Tauc, L.; Montecucco, C. Tetanus toxin is a zinc protein and its inhibition of neurotransmitter release and protease activity depend on zinc. EMBO J. 1992, 11, 3577–3583. [Google Scholar]
- Schiavo, G.; Benfenati, F.; Poulain, B.; Rossetto, O.; Polverino de Laureto, P.; DasGupta, B.R.; Montecucco, C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 1992, 359, 832–835. [Google Scholar]
- Adler, M.; Dinterman, R.E.; Wannemacher, R.W. Protection by the heavy metal chelator N,N,N',N'-tetrakis (2-pyridylmethyl)ethylenediamine (TPEN) against the lethal action of botulinum neurotoxin A and B. Toxicon 1997, 35, 1089–1100. [Google Scholar] [CrossRef]
- Hooper, N.M. Families of zinc metalloproteases. FEBS Lett. 1994, 354, 1–6. [Google Scholar]
- Lebeda, F.J.; Olson, M.A. Predictions of secondary structure and solvent accessibility of the light chain of the clostridial neurotoxins. J. Nat. Toxins 1998, 7, 227–238. [Google Scholar]
- Li, Y.; Foran, P.; Fairweather, N.F.; de Paiva, A.; Weller, U.; Dougan, G.; Dolly, J.O. A single mutation in the recombinant light chain of tetanus toxin abolishes its proteolytic activity and removes the toxicity seen after reconstitution with native heavy chain. Biochemistry 1994, 33, 7014–7020. [Google Scholar]
- Schmidt, J.J.; Bostian, K.A. Proteolysis of synthetic peptides by type A botulinum neurotoxin. J. Protein Chem. 1995, 14, 703–708. [Google Scholar] [CrossRef]
- Olson, M.A.; Armendinger, T.L. Free-energy contributions to complex formation between botulinum neurotoxin type B and synaptobrevin fragment. Protein Eng. 2002, 15, 739–743. [Google Scholar] [CrossRef]
- Lebeda, F.J.; Olson, M.A. Secondary structural predictions for the clostridial neurotoxins. Proteins 1994, 20, 293–300. [Google Scholar] [CrossRef]
- Breidenbach, M.A.; Brunger, A.T. Substrate recognition strategy for botulinum neurotoxin serotype A. Nature 2004, 432, 925–929. [Google Scholar] [CrossRef]
- Pang, Y.P.; Vummenthala, A.; Mishra, R.K.; Park, J.G.; Wang, S.; Davis, J.; Millard, C.B.; Schmidt, J.J. Potent new small-molecule inhibitor of botulinum neurotoxin serotype A endopeptidase developed by synthesis-based computer-aided molecular design. PLoS One 2009, 4, e7730. [Google Scholar]
- Sukonpan, C.; Oost, T.; Goodnough, M.; Tepp, W.; Johnson, E.A.; Rich, D.H. Synthesis of substrates and inhibitors of botulinum neurotoxin type A metalloprotease. J. Pept. Res. 2004, 63, 181–193. [Google Scholar]
- Binz, T.; Blasi, J.; Yamasaki, S.; Baumeister, A.; Link, E.; Sudhof, T.C.; Jahn, R.; Niemann, H. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J. Biol.Chem. 1994, 269, 1617–1620. [Google Scholar]
- Binz, T.; Bade, S.; Rummel, A.; Kollewe, A.; Alves, J. Arg(362) and Tyr(365) of the botulinum neurotoxin type a light chain are involved in transition state stabilization. Biochemistry 2002, 41, 1717–1723. [Google Scholar]
- Koshland, D.E. The Application and Usefulness of the Ratio kcat/KM. Bioorg. Chem. 2002, 30, 211–213. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, S.; Baldwin, M.R.; Boldt, G.E.; Crawford, A.; Janda, K.D.; Barbieri, J.T.; Kim, J.J. Light chain of botulinum neurotoxin serotype A: structural resolution of a catalytic intermediate. Biochemistry 2006, 45, 8903–8911. [Google Scholar]
- Fersht, A. Structure and Mechanism in Protein Science; W.H. Freeman and Company: New York, NY, USA, 1999. [Google Scholar]
- Vaidyanathan, V.V.; Yoshino, K.; Jahnz, M.; Dorries, C.; Bade, S.; Nauenburg, S.; Niemann, H.; Binz, T. Proteolysis of SNAP-25 isoforms by botulinum neurotoxin types A, C, and E: domains and amino acid residues controlling the formation of enzyme-substrate complexes and cleavage. J. Neurochem. 1999, 72, 327–337. [Google Scholar] [PubMed]
- Sanders, D.; Habermann, E. Evidence for a link between specific proteolysis and inhibition of [3H]-noradrenaline release by the light chain of tetanus toxin. Naunyn Schmiedebergs Arch. Pharmacol. 1992, 346, 358–361. [Google Scholar]
- Adler, M.; Nicholson, J.D.; Starks, D.F.; Kane, C.T.; Cornille, F.; Hackley, B.E., Jr. Evaluation of phosphoramidon and three synthetic phosphonates for inhibition of botulinum neurotoxin B catalytic activity. J. Appl. Toxicol. 1999, 19 (Suppl. 1), S5–S11. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.S.; Sheridan, R.E.; Adler, M. A study of zinc-dependent metalloendopeptidase inhibitors as pharmacological antagonists in botulinum neurotoxin poisoning. Toxicon 1995, 33, 551–557. [Google Scholar] [CrossRef]
- Adler, M.; Deshpande, S.S.; Sheridan, R.E.; Lebeda, F.J. Evaluation of captopril and other potentially therapeutic compounds in antagonizing botulinum toxin-induced muscle paralysis. In Therapy with Botulinum Toxin; Jankovic, J., Hallett, M., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1994; pp. 63–70. [Google Scholar]
- Bakry, N.; Kamata, Y.; Simpson, L.L. Lectins from Triticum vulgaris and Limax flavus are universal antagonists of botulinum neurotoxin and tetanus toxin. J. Pharmacol. Exp. Ther. 1991, 258, 830–836. [Google Scholar]
- Simpson, L.L. The interaction between aminoquinolines and presynaptically acting neurotoxins. J. Pharmacol. Exp. Ther. 1982, 222, 43–48. [Google Scholar]
- Deshpande, S.S.; Sheridan, R.E.; Adler, M. Efficacy of certain quinolines as pharmacological antagonists in botulinum neurotoxin poisoning. Toxicon 1997, 35, 433–445. [Google Scholar] [CrossRef]
- Simpson, L.L. Ammonium chloride and methylamine hydrochloride antagonize clostridial neurotoxins. J. Pharmacol. Exp. Ther. 1983, 225, 546–552. [Google Scholar]
- Simpson, L.L.; Coffield, J.A.; Bakry, N. Inhibition of vacuolar adenosine triphosphatase antagonizes the effects of clostridial neurotoxins but not phospholipase A2 neurotoxins. J. Pharmacol. Exp. Ther. 1994, 269, 256–262. [Google Scholar]
- Simpson, L.L. A preclinical evaluation of aminopyridines as putative therapeutic agents in the treatment of botulism. Infect. Immun. 1986, 52, 858–862. [Google Scholar]
- Simpson, L.L. Use of pharmacologic antagonists to deduce commonalities of biologic activity among clostridial neurotoxins. J. Pharmacol. Exp. Ther. 1988, 245, 867–872. [Google Scholar]
- Adler, M.; Scovill, J.; Parker, G.; Lebeda, F.J.; Piotrowski, J.; Deshpande, S.S. Antagonism of botulinum toxin-induced muscle weakness by 3,4-diaminopyridine in rat phrenic nerve-hemidiaphragm preparations. Toxicon 1995, 33, 527–537. [Google Scholar]
- Simpson, L.L. Pharmacological studies on the subcellular site of action of botulinum toxin type A. J. Pharmacol. Exp. Ther. 1978, 206, 661–669. [Google Scholar]
- Shone, C.C.; Quinn, C.P.; Wait, R.; Hallis, B.; Fooks, S.G.; Hambleton, P. Proteolytic cleavage of synthetic fragments of vesicle-associated membrane protein, isoform-2 by botulinum type B neurotoxin. Eur. J. Biochem. 1993, 217, 965–971. [Google Scholar] [CrossRef]
- Yamasaki, S.; Baumeister, A.; Binz, T.; Blasi, J.; Link, E.; Cornille, F.; Roques, B.; Fykse, E.M.; Sudhof, T.C.; Jahn, R.; et al. Cleavage of members of the synaptobrevin/VAMP family by types D and F botulinal neurotoxins and tetanus toxin. J. Biol. Chem. 1994, 269, 12764–12772. [Google Scholar] [PubMed]
- Cornille, F.; Goudreau, N.; Ficheux, D.; Niemann, H.; Roques, B.P. Solid-phase synthesis, conformational analysis and in vitro cleavage of synthetic human synaptobrevin II 1-93 by tetanus toxin L chain. Eur. J. Biochem. 1994, 222, 173–181. [Google Scholar] [CrossRef]
- Shone, C.C.; Roberts, A.K. Peptide substrate specificity and properties of the zinc-endopeptidase activity of botulinum type B neurotoxin. Eur. J. Biochem. 1994, 225, 263–270. [Google Scholar] [CrossRef]
- Cornille, F.; Deloye, F.; Fournie-Zaluski, M.C.; Roques, B.P.; Poulain, B. Inhibition of neurotransmitter release by synthetic proline-rich peptides shows that the N-terminal domain of vesicle-associated membrane protein/synaptobrevin is critical for neuro-exocytosis. J. Biol. Chem. 1995, 270, 16826–16832. [Google Scholar]
- Zuniga, J.E.; Schmidt, J.J.; Fenn, T.; Burnett, J.C.; Arac, D.; Gussio, R.; Stafford, R.G.; Badie, S.S.; Bavari, S.; Brunger, A.T. A potent peptidomimetic inhibitor of botulinum neurotoxin serotype A has a very different conformation than SNAP-25 substrate. Structure 2008, 16, 1588–1597. [Google Scholar]
- Eswaramoorthy, S.; Kumaran, D.; Swaminathan, S. A novel mechanism for Clostridium botulinum neurotoxin inhibition. Biochemistry 2002, 41, 9795–9802. [Google Scholar] [CrossRef]
- Martin, L.; Cornille, F.; Turcaud, S.; Meudal, H.; Roques, B.P.; Fournie-Zaluski, M.C. Metallopeptidase inhibitors of tetanus toxin: A combinatorial approach. J. Med. Chem. 1999, 42, 515–525. [Google Scholar]
- Boldt, G.E.; Eubanks, L.M.; Janda, K.D. Identification of a botulinum neurotoxin A protease inhibitor displaying efficacy in a cellular model. Chem. Commun. (camb) 2006, 3063–3065. [Google Scholar] [PubMed]
- Burnett, J.C.; Opsenica, D.; Sriraghavan, K.; Panchal, R.G.; Ruthel, G.; Hermone, A.R.; Nguyen, T.L.; Kenny, T.A.; Lane, D.J.; McGrath, C.F.; Schmidt, J.J.; Vennerstrom, J.L.; Gussio, R.; Solaja, B.A.; Bavari, S. A refined pharmacophore identifies potent 4-amino-7-chloroquinoline-based inhibitors of the botulinum neurotoxin serotype A metalloprotease. J. Med. Chem. 2007, 50, 2127–2136. [Google Scholar]
- Zuniga, J.E.; Schmidt, J.J.; Fenn, T.; Burnett, J.C.; Arac, D.; Gussio, R.; Stafford, R.G.; Badie, S.S.; Bavari, S.; Brunger, A.T. A potent peptidomimetic inhibitor of botulinum neurotoxin serotype A has a very different conformation than SNAP-25 substrate. Structure 2008, 16, 1588–1597. [Google Scholar] [CrossRef]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, W441–W445. [Google Scholar]
- Capkova, K.; Hixon, M.S.; Pellett, S.; Barbieri, J.T.; Johnson, E.A.; Janda, K.D. Benzylidene cyclopentenediones: First irreversible inhibitors against botulinum neurotoxin A's zinc endopeptidase. Bioorg. Med. Chem. Lett. 2010, 20, 206–208. [Google Scholar]
- Silhar, P.; Capkova, K.; Salzameda, N.T.; Barbieri, J.T.; Hixon, M.S.; Janda, K.D. Botulinum neurotoxin A protease: discovery of natural product exosite inhibitors. J. Am. Chem. Soc. 2010, 132, 2868–2869. [Google Scholar]
- Eubanks, L.M.; Hixon, M.S.; Jin, W.; Hong, S.; Clancy, C.M.; Tepp, W.H.; Baldwin, M.R.; Malizio, C.J.; Goodnough, M.C.; Barbieri, J.T.; Johnson, E.A.; Boger, D.L.; Dickerson, T.J.; Janda, K.D. An in vitro and in vivo disconnect uncovered through high-throughput identification of botulinum neurotoxin A antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 2602–2607. [Google Scholar]
- Schmidt, J.J.; Bostian, K.A. Endoproteinase activity of type A botulinum neurotoxin: substrate requirements and activation by serum albumin. J. Protein Chem. 1997, 16, 19–26. [Google Scholar] [CrossRef]
- Costa, S.A.; Tzanov, T.; Ana Filipa Carneiro, A.F.; Paar, A.; Gubitz, G.M.; Cavaco-Paulo, A. Studies of stabilization of native catalase using additives. Enz. Microb. Technol. 2002, 30, 387–391. [Google Scholar] [CrossRef] [Green Version]
- McAllister, L.A.; Hixon, M.S.; Kennedy, J.P.; Dickerson, T.J.; Janda, K.D. Superactivation of the Botulinum Neurotoxin Serotype A Light Chain Metalloprotease: A New Wrinkle in Botulinum Neurotoxin. J. Am. Chem. Soc. 2006, 128, 4176–4177. [Google Scholar]
- Botts, J.; Morales, M. Analytical description of the effects of modifiers and of enzyme multivalency upon the steady state catalyzed reaction rate. Trans. Faraday Soc. 1953, 49, 696–707. [Google Scholar]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics; Butterworths: London, UK, 1979. [Google Scholar]
- Verderio, C.; Rossetto, O.; Grumelli, C.; Frassoni, C.; Montecucco, C.; Matteoli, M. Entering neurons: botulinum toxins and synaptic vesicle recycling. EMBO Rep. 2006, 7, 995–999. [Google Scholar] [CrossRef]
- Marcus, S.M. Reflections on the care of a patient severely poisoned by 'rogue' botulinum toxin and rendered paralysed for a protracted hospital stay. Botulinum J. 2009, 1, 318–339. [Google Scholar]
- Adler, M.; Keller, J.E.; Sheridan, R.E.; Deshpande, S.S. Persistence of botulinum neurotoxin A demonstrated by sequential administration of serotypes A and E in rat EDL muscle. Toxicon 2001, 39, 233–243. [Google Scholar]
- Keller, J.E.; Neale, E.A.; Oyler, G.; Adler, M. Persistence of botulinum neurotoxin action in cultured spinal cord cells. FEBS Lett. 1999, 456, 137–142. [Google Scholar] [CrossRef]
- Cai, S.; Sarkar, H.K.; Singh, B.R. Enhancement of the endopeptidase activity of botulinum neurotoxin by its associated proteins and dithiothreitol. Biochemistry 1999, 38, 6903–6910. [Google Scholar]
- Takita, T.; Aono, T.; Sakurama, H.; Itoh, T.; Wada, T.; Minoda, M.; Yasukawa, K.; Inouye, K. Effects of introducing negative charges into the molecular surface of thermolysin by site-directed mutagenesis on its activity and stability. Biochim. Biophys. Acta 2008, 1784, 481–488. [Google Scholar]
- Schulte-Baukloh, H.; Zurawski, T.H.; Knispel, H.H.; Miller, K.; Haferkamp, A.; Dolly, J.O. Persistence of the synaptosomal-associated protein-25 cleavage product after intradetrusor botulinum toxin A injections in patients with myelomeningocele showing an inadequate response to treatment. BJU Int. 2007, 100, 1075–1080. [Google Scholar]
- Meunier, F.A.; Lisk, G.; Sesardic, D.; Dolly, J.O. Dynamics of motor nerve terminal remodeling unveiled using SNARE-cleaving botulinum toxins: the extent and duration are dictated by the sites of SNAP-25 truncation. Mol. Cell. Neurosci. 2003, 22, 454–466. [Google Scholar]
- O'Sullivan, G.A.; Mohammed, N.; Foran, P.G.; Lawrence, G.W.; Dolly, J.O. Rescue of exocytosis in botulinum toxin A-poisoned chromaffin cells by expression of cleavage-resistant SNAP-25. Identification of the minimal essential C-terminal residues. J. Biol. Chem. 1999, 274, 36897–36904. [Google Scholar] [PubMed]
- Hayashi, T.; Yamasaki, S.; Nauenburg, S.; Binz, T.; Niemann, H. Disassembly of the reconstituted synaptic vesicle membrane fusion complex in vitro. EMBO J. 1995, 14, 2317–2325. [Google Scholar]
- Poulain, B.; Popoff, M.R.; Molgó, J. How do the Botulinum Neurotoxins block neurotransmitter release: from botulism to the molecular mechanism of action. Botulinum J. 2008, 1, 14–87. [Google Scholar] [CrossRef]
- Fernandez-Salas, E.; Steward, L.E.; Ho, H.; Garay, P.E.; Sun, S.W.; Gilmore, M.A.; Ordas, J.V.; Wang, J.; Francis, J.; Aoki, K.R. Plasma membrane localization signals in the light chain of botulinum neurotoxin. Proc. Natl. Acad. Sci. USA 2004, 101, 3208–3213. [Google Scholar]
- Ferrer-Montiel, A.V.; Canaves, J.M.; DasGupta, B.R.; Wilson, M.C.; Montal, M. Tyrosine phosphorylation modulates the activity of clostridial neurotoxins. J. Biol. Chem. 1996, 271, 18322–18325. [Google Scholar]
- Ibanez, C.; Blanes-Mira, C.; Fernandez-Ballester, G.; Planells-Cases, R.; Ferrer-Montiel, A. Modulation of botulinum neurotoxin A catalytic domain stability by tyrosine phosphorylation. FEBS Lett. 2004, 578, 121–127. [Google Scholar] [CrossRef]
- Ellis, R.J. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lebeda, F.J.; Cer, R.Z.; Mudunuri, U.; Stephens, R.; Singh, B.R.; Adler, M. The Zinc-Dependent Protease Activity of the Botulinum Neurotoxins. Toxins 2010, 2, 978-997. https://doi.org/10.3390/toxins2050978
Lebeda FJ, Cer RZ, Mudunuri U, Stephens R, Singh BR, Adler M. The Zinc-Dependent Protease Activity of the Botulinum Neurotoxins. Toxins. 2010; 2(5):978-997. https://doi.org/10.3390/toxins2050978
Chicago/Turabian StyleLebeda, Frank J., Regina Z. Cer, Uma Mudunuri, Robert Stephens, Bal Ram Singh, and Michael Adler. 2010. "The Zinc-Dependent Protease Activity of the Botulinum Neurotoxins" Toxins 2, no. 5: 978-997. https://doi.org/10.3390/toxins2050978