Escherichia coli Cytotoxic Necrotizing Factor 1 (CNF1): Toxin Biology, in Vivo Applications and Therapeutic Potential




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Cytotoxic Necrotizing Factor 1: In Vitro Effects
2.1. CNF1 structure and endocytosis
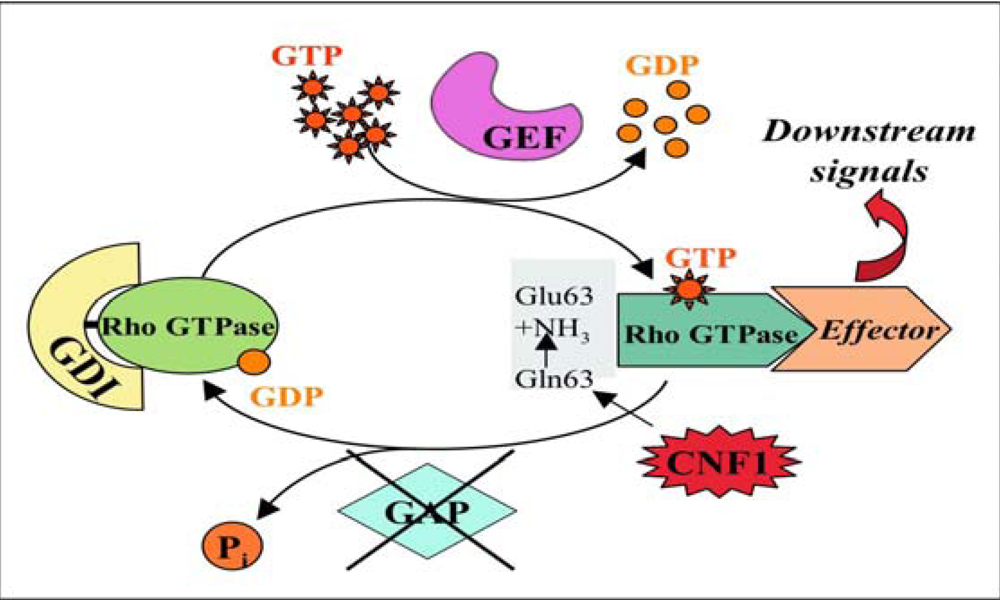
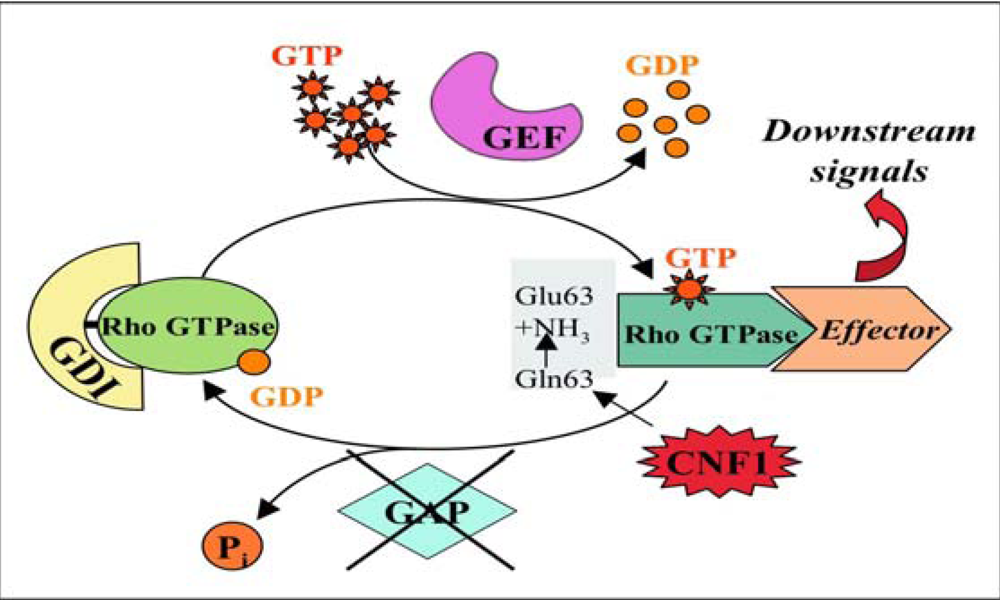
2.2. CNF1 enzymatic activity
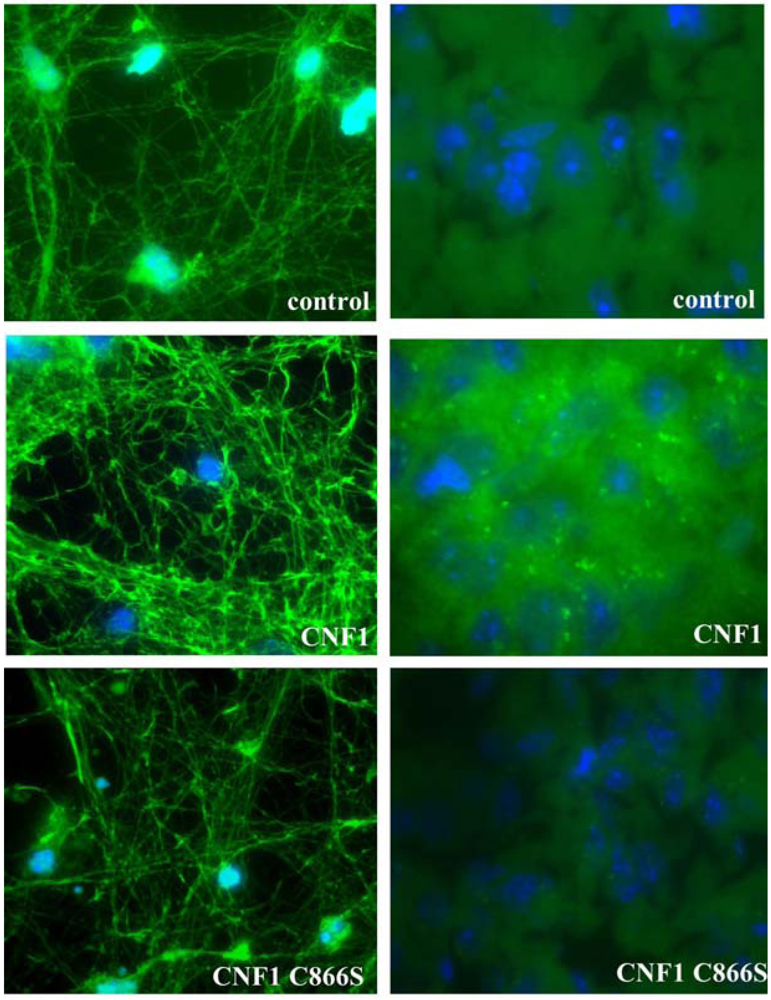
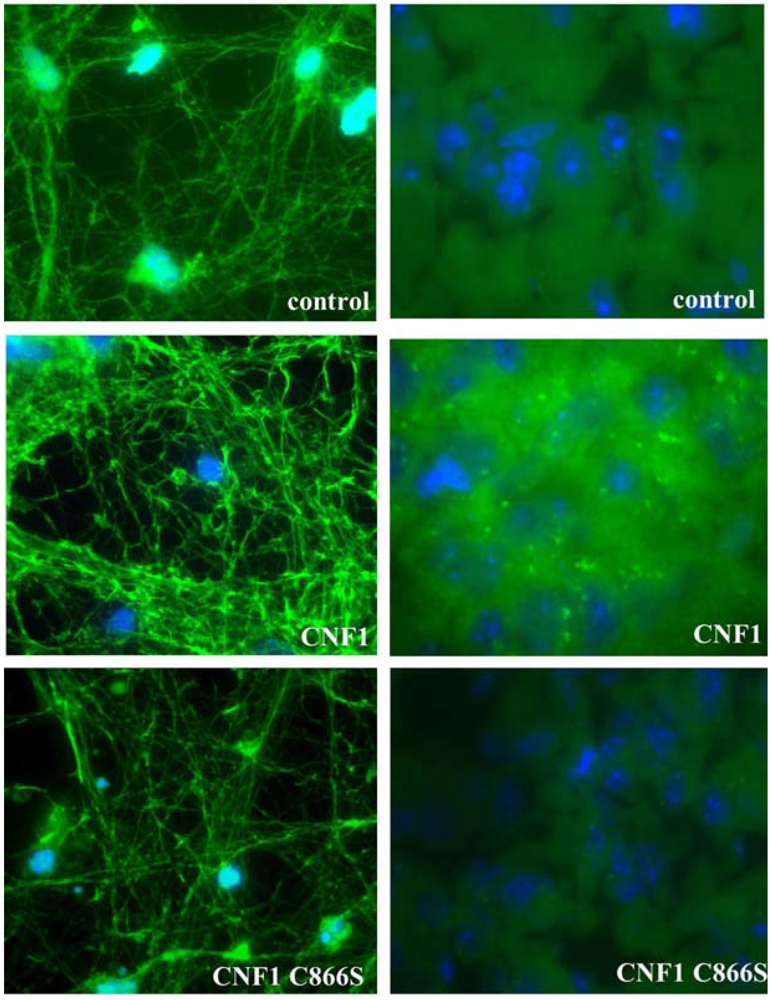
2.3. Effects of CNF1 on eukaryotic cultured cells
3. The Cytotoxic Necrotizing Factor 1: In Vivo Effects
3.1. Learning and memory
3.2. CNF1 as adjuvant or drug delivery agent
3.3. Analgesic activity of CNF1
4. Conclusions
Acknowledgements
References and Notes
- Fabbri, A.; Travaglione, S.; Falzano, L.; Fiorentini, C. Bacterial protein toxins: current and potential clinical use. Curr Med. Chem. 2008, 15, 1116–1125. [Google Scholar] [PubMed]
- Hackett, R.; Kam, P.C. Botulinum toxin: pharmacology and clinical developments: a literature review. Med. Chem. 2007, 3, 333–345. [Google Scholar] [PubMed]
- Boquet, P. The cytotoxic necrotizing factor 1 (CNF1) from Escherichia coli. Toxicon 2001, 39, 1673–1680. [Google Scholar] [PubMed]
- Elliott, S.J.; Srinivas, S.; Albert, M.J.; Alam, K.; Robins-Browne, R.M.; Gunzburg, S.T.; Mee, B.J.; Chang, B.J. Characterization of the roles of hemolysin and other toxins in enteropathy caused by alpha-hemolytic Escherichia coli linked to human diarrhea. Infect. Immun. 1998, 66, 2040–2051. [Google Scholar] [PubMed]
- Okeke, I.N.; Fayinka, S.T.; Lamikanra, A. Antibiotic resistance in Escherichia coli from Nigerian students, 1986-1998. Emerg. Infect. Dis. 2000, 6, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Paciorek, J. Virulence properties of Escherichia coli faecal strains isolated in Poland from healthy children and strains belonging to serogroups O18, O26, O44, O86, O126 and O127 isolated from children with diarrhoea. J. Med. Microbiol. 2002, 51, 548–556. [Google Scholar] [PubMed]
- De Rycke, J.; Milon, A.; Oswald, E. Necrotoxic Escherichia coli (NTEC): two emerging categories of human and animal pathogens. Vet. Res. 1999, 30, 221–233. [Google Scholar] [PubMed]
- Landraud, L.; Gauthier, M.; Fosse, T.; Boquet, P. Frequency of Escherichia coli strains producing the cytotoxic necrotizing factor (CNF1) in nosocomial urinary tract infections. Lett. Appl. Microbiol. 2000, 30, 213–216. [Google Scholar] [PubMed]
- Blanco, J.; Alonso, M.P.; González, E.A.; Blanco, M.; Garabal, J.I. Virulence factors of bacteraemic Escherichia coli with particular reference to production of cytotoxic necrotising factor (CNF) by P-fimbriate strains. J. Med. Microbiol. 1990, 31, 175–183. [Google Scholar] [PubMed]
- Houdouin, V.; Bonacorsi, S.; Brahimi, N.; Clermont, O.; Nassif, X.; Bingen, E. A uropathogenicity island contributes to the pathogenicity of Escherichia coli strains that cause neonatal meningitis. Infect. Immun. 2002, 70, 5865–5869. [Google Scholar] [PubMed]
- Lemonnier, M.; Landraud, L.; Lemichez, E. Rho GTPase-activating bacterial toxins: from bacterial virulence regulation to eukaryotic cell biology. FEMS Microbiol. Rev. 2007, 31, 515–534. [Google Scholar] [PubMed]
- Caprioli, A.; Falbo, V.; Roda, L.G.; Ruggeri, F.M.; Zona, C. Partial purification and characterization of an Escherichia coli toxic factor that induces morphological cell alterations. Infect. Immun. 1983, 39, 1300–1306. [Google Scholar] [PubMed]
- Caprioli, A.; Donelli, G.; Falbo, V.; Possenti, R.; Roda, L.G.; Roscetti, G.; Ruggeri, F.M. A cell division-active protein from E. coli. Biochem. Biophys. Res. Commun. 1984, 118, 587–593. [Google Scholar] [CrossRef]
- Buetow, L.; Flatau, G.; Chiu, K.; Boquet, P.; Ghosh, P. Structure of the Rho-activating domain of Escherichia coli cytotoxic necrotizing factor 1. Nat. Struct. Biol. 2001, 8, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Lemichez, E.; Flatau, G.; Bruzzone, M.; Boquet, P.; Gauthier, M. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol. Microbiol. 1997, 24, 1061–1070. [Google Scholar] [PubMed]
- Pei, S.; Doye, A.; Boquet, P. Mutation of specific acidic residues of the CNF1 T domain into lysine alters cell membrane translocation of the toxin. Mol Microbiol. 2001, 41, 1237–1247. [Google Scholar] [PubMed]
- Contamin, S.; Galmiche, A.; Doye, A.; Flatau, G.; Benmerah, A.; Boquet, P. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol. Biol. Cell 2000, 11, 1775–1787. [Google Scholar] [PubMed]
- Fabbri, A.; Gauthier, M.; Boquet, P. The 5' region of cnf1 harbours a translational regulatory mechanism for CNF1 synthesis and encodes the cell-binding domain of the toxin. Mol. Microbiol. 1999, 33, 108–118. [Google Scholar] [PubMed]
- Chung, J.W.; Hong, S.J.; Kim, K.J.; Goti, D.; Stins, M.F.; Shin, S.; Dawson, V.L.; Dawson, T.M.; Kim, K.S. 37-kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J. Biol. Chem. 2003, 278, 16857–16862. [Google Scholar] [PubMed]
- Kim, K.J.; Chung, J.W.; Kim, K.S. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 2005, 280, 1360–1368. [Google Scholar] [PubMed]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The Cytotoxic Necrotizing Factors from Yersinia pseudotuberculosis and from Escherichia coli Bind to Different Cellular Receptors but Take the Same Route to the Cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [PubMed]
- McNichol, B.A.; Rasmussen, S.B.; Carvalho, H.M.; Meysick, K.C.; O'Brien, A.D. Two domains of cytotoxic necrotizing factor type 1 bind the cellular receptor, laminin receptor precursor protein. Infect. Immun. 2007, 75, 5095–5104. [Google Scholar] [CrossRef] [PubMed]
- Knust, Z.; Blumenthal, B.; Aktories, K.; Schmidt, G. Cleavage of Escherichia coli cytotoxic necrotizing factor 1 is required for full biologic activity. Infect. Immun. 2009, 77, 1835–1841. [Google Scholar] [PubMed]
- Lemichez, E.; Flatau, G.; Bruzzone, M.; Boquet, P.; Gauthier, M. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol. Microbiol. 1997, 24, 1061–1070. [Google Scholar] [PubMed]
- Schmidt, G.; Selzer, J.; Lerm, M.; Aktories, K. The Rho-deamidating cytotoxic necrotizing factor 1 from Escherichia coli possesses transglutaminase activity. Cysteine 866 and histidine 881 are essential for enzyme activity. J. Biol. Chem. 1998, 273, 13669–13674. [Google Scholar] [PubMed]
- DerMardirossian, C.; Bokoch, G.M. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell. Biol. 2005, 15, 356–363. [Google Scholar] [PubMed]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [PubMed]
- Schmidt, A.; Hall, A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002, 16, 1587–1609. [Google Scholar] [PubMed]
- Moon, S.; Zheng, Y. Rho GTPase-activating proteins in cell regulation. Trends Cell. Biol. 2003, 13, 13–22. [Google Scholar] [PubMed]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. J. Biol. Chem. 1997, 272, 19532–19537. [Google Scholar] [PubMed]
- Schmidt, G.; Sher, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [PubMed]
- Lerm, M.; Selzer, J.; Hoffmeyer, A.; Rapp, U.R.; Aktories, K.; Schmidt, G. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of the C-Jun N-terminal kinase in HeLa cells. Infect. Immun. 1999, 67, 496–503. [Google Scholar] [PubMed]
- Rittinger, K.; Walker, P.A.; Eccleston, J.F.; Nurmahomed, K.; Owen, D.; Laue, E.; Gamblin, S.J.; Smerdon, S.J. Crystal structure of a small G protein in complex with the GTPase-activating protein rhoGAP. Nature 1997, 388, 693–697. [Google Scholar] [PubMed]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clément, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar] [PubMed]
- Fu, Y.; Galán, J.E. A Salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature 1999, 401, 293–297. [Google Scholar] [PubMed]
- Schlumberger, M.C.; Hardt, W.D. Triggered phagocytosis by Salmonella: bacterial molecular mimicry of RhoGTPase activation/deactivation. Curr. Top. Microbiol. Immunol. 2005, 291, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [PubMed]
- Fiorentini, C.; Arancia, G.; Caprioli, A.; Falbo, V.; Ruggeri, F.M.; Donelli, G. Cytoskeletal changes induced in HEp-2 cells by the cytotoxic necrotizing factor of Escherichia coli. Toxicon 1988, 26, 1047–1056. [Google Scholar] [PubMed]
- Fiorentini, C.; Fabbri, A.; Flatau, G.; Donelli, G.; Matarrese, P.; Lemichez, E.; Falzano, L.; Boquet, P. Escherichia coli cytotoxic necrotizing factor 1 (CNF1), a toxin that activates the Rho GTPase. J. Biol. Chem. 1997, 272, 19532–19537. [Google Scholar] [PubMed]
- Falzano, L.; Fiorentini, C.; Donelli, G.; Michel, E.; Kocks, C.; Cossart, P.; Cabanié, L.; Oswald, E.; Boquet, P. Induction of phagocytic behaviour in human epithelial cells by Escherichia coli cytotoxic necrotizing factor type 1. Mol. Microbiol. 1993, 9, 1247–1254. [Google Scholar] [PubMed]
- Fiorentini, C.; Falzano, L.; Fabbri, A.; Stringaro, A.; Logozzi, M.; Travaglione, S.; Contamin, S.; Arancia, G.; Malorni, W.; Fais, S. Activation of rho GTPases by cytotoxic necrotizing factor 1 induces macropinocytosis and scavenging activity in epithelial cells. Mol. Biol. Cell. 2001, 12, 2061–2273. [Google Scholar] [PubMed]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [PubMed]
- Lacerda, H.M.; Pullinger, G.D.; Lax, A.J.; Rozengurt, E. Cytotoxic necrotizing factor 1 from Escherichia coli and dermonecrotic toxin from Bordetella bronchiseptica induce p21(rho)-dependent tyrosine phosphorylation of focal adhesion kinase and paxillin in Swiss 3T3 cells. J. Biol. Chem. 1997, 272, 9587–9596. [Google Scholar] [PubMed]
- Boyer, L.; Travaglione, S.; Falzano, L.; Gauthier, N.C.; Popoff, M.R.; Lemichez, E.; Fiorentini, C.; Fabbri, A. Rac GTPase instructs Nuclear Factor-kB activating by conveying the SCF complex and IkB-alpha to the ruffling membranes. Mol. Biol. Cell. 2004, 15, 1124–1133. [Google Scholar] [PubMed]
- Falzano, L.; Filippini, P.; Travaglione, S.; Miraglia, A.G.; Fabbri, A.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 blocks cell cycle G2/M transition in uroepithelial cells. Infect. Immun. 2006, 74, 3765–3772. [Google Scholar] [PubMed]
- Travaglione, S.; Messina, G.; Fabbri, A.; Falzano, L.; Giammarioli, A.M.; Grossi, M.; Rufini, S.; Fiorentini, C. Cytotoxic necrotizing factor 1 hinders skeletal muscle differentiation in vitro by perturbing the activation/deactivation balance of Rho GTPases. Cell Death Differ. 2005, 12, 78–86. [Google Scholar] [PubMed]
- Falzano, L.; Quaranta, M.G.; Travaglione, S.; Filippini, P.; Fabbri, A.; Viora, M.; Donelli, G.; Fiorentini, C. Cytotoxic necrotizing factor 1 enhances reactive oxygen species-dependent transcription and secretion of proinflammatory cytokines in human uroepithelial cells. Infect Immun. 2003, 71, 4178–4181. [Google Scholar] [PubMed]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Donelli, G.; Boquet, P.; Malorni, W. Rho-dependent cell spreading activated by E. coli cytotoxic necrotizing factor 1 hinders apoptosis in epithelial cells. Cell Death Differ. 1998, 5, 720–728. [Google Scholar] [PubMed]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Fabbri, A.; Donelli, G.; Cossarizza, A.; Boquet, P.; Malorni, W. Toxin-induced activation of Rho GTP-binding protein increases Bcl-2 expression and influences mitochondrial homeostasis. Exp. Cell. Res. 1998, 242, 341–350. [Google Scholar] [PubMed]
- Giamboi Miraglia, A.; Travaglione, S.; Meschini, S.; Falzano, L.; Matarrese, P.; Quaranta, M.G.; Viora, M.; Fiorentini, C.; Fabbri, A. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IkappaB kinase pathway: role of nuclear factor-kappaB and Bcl-2. Mol. Biol. Cell 2007, 18, 2735–2744. [Google Scholar] [PubMed]
- Munro, P.; Flatau, G.; Doye, A.; Boyer, L.; Oregioni, O.; Mege, J.L.; Landraud, L.; Lemichez, E. Activation and proteasomal degradation of rho GTPases by cytotoxic necrotizing factor-1 elicit a controlled inflammatory response. J. Biol. Chem. 2004, 279, 35849–35857. [Google Scholar] [PubMed]
- Thomas, W.; Ascott, Z.K.; Harmey, D.; Slice, L.W.; Rozengurt, E.; Lax, A.J. Cytotoxic necrotizing factor from Escherichia coli induces RhoA-dependent expression of the cyclooxygenase-2 Gene. Infect. Immun. 2001, 69, 6839–6845. [Google Scholar] [PubMed]
- Falzano, L.; Rivabene, R.; Santini, M.T.; Fabbri, A.; Fiorentini, C. An Escherichia coli cytotoxin increases superoxide anion generation via rac in epithelial cells. Biochem. Biophys. Res. Commun. 2001, 283, 1026–1030. [Google Scholar] [PubMed]
- Zito, K.; Knott, G.; Shepherd, G.M.; Shenolikar, S.; Svoboda, K. Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron 2004, 44, 321–334. [Google Scholar] [PubMed]
- Fukazawa, Y.; Saitoh, Y.; Ozawa, F.; Ohta, Y.; Mizuno, K.; Inokuchi, K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 2003, 38, 447–446. [Google Scholar] [PubMed]
- Diana, G.; Valentini, G.; Travaglione, S.; Falzano, L.; Pieri, M.; Zona, C.; Meschini, S.; Fabbri, A.; Fiorentini, C. Enhancement of learning and memory after activation of cerebral Rho GTPases. Proc. Natl. Acad. Sci. USA 2007, 104, 636–641. [Google Scholar]
- Kamiya, N.; Asano, I.; Yoshino, J.; Sasaki, K.; Honma, Y.; Kawase, H.; Yokochi, T.; Shiraki, K.; Tsuji, T. Long-term persistence of cellular immunity to Oka vaccine virus induced by pernasal co-administration with Escherichia coli enterotoxin in mice. Vaccine 2001, 19, 3131–3136. [Google Scholar] [PubMed]
- Vajdy, M.; Lycke, N. Presence of antigen-specific long-term memory cells in systemic lymphoid tissues as well as locally in the gut lamina propria following oral immunization with cholera toxin adjuvant. Adv. Exp. Med. Biol. 1995, 371B, 1495–1500. [Google Scholar] [PubMed]
- Chiarella, P.; Massi, E.; De Robertis, M.; Signori, E.; Fazio, V.M. Adjuvants in vaccines and for immunisation: current trends. Expert. Opin. Biol. Ther. 2007, 7, 1551–1562. [Google Scholar] [PubMed]
- Munro, P.; Flatau, G.; Anjuère, F.; Hofman, V.; Czerkinsky, C.; Lemichez, E. The Rho GTPase activators CNF1 and DNT bacterial toxins have mucosal adjuvant properties. Vaccine 2005, 23, 2551–2556. [Google Scholar] [PubMed]
- Munro, P.; Flatau, G.; Lemichez, E. Intranasal immunization with tetanus toxoid and CNF1 as a new mucosal adjuvant protects BALB/c mice against lethal challenge. Vaccine 2007, 25, 8702–8706. [Google Scholar] [PubMed]
- Bhave, G.; Gereau, R.W. Growing pains: the cytoskeleton as a critical regulator of pain plasticity. Neuron 2003, 39, 577–579. [Google Scholar] [PubMed]
- Dina, O.A.; McCarter, G.C.; de Coupade, C.; Levine, J.D. Role of the sensory neuron cytoskeleton in second messenger signaling for inflammatory pain. Neuron 2003, 39, 613–624. [Google Scholar] [PubMed]
- Goswami, C.; Dreger, M.; Otto, H.; Schwappach, B.; Hucho, F. Rapid disassembly of dynamic microtubules upon activation of the capsaicin receptor TRPV1. J. Neurochem. 2006, 96, 254–266. [Google Scholar] [PubMed]
- Porro, C.A.; Cavazzuti, M. Spatial and temporal aspects of spinal cord and brainstem activation in the formalin pain model. Prog. Neurobiol. 1993, 41, 565–607. [Google Scholar] [PubMed]
- Pavone, F.; Luvisetto, S.; Marinelli, S.; Straface, E.; Fabbri, A.; Falzano, L.; Fiorentini, C.; Malorni, W. The Rac GTPase-activating bacterial protein toxin CNF1 induces analgesia up-regulating mu-opioid receptors. Pain 2009, 145, 219–229. [Google Scholar] [PubMed]
- Mason, P. Deconstructing endogenous pain modulations. J. Neurophysiol. 2005, 94, 1659–1663. [Google Scholar] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fabbri, A.; Travaglione, S.; Fiorentini, C. Escherichia coli Cytotoxic Necrotizing Factor 1 (CNF1): Toxin Biology, in Vivo Applications and Therapeutic Potential. Toxins 2010, 2, 283-296. https://doi.org/10.3390/toxins2020282
Fabbri A, Travaglione S, Fiorentini C. Escherichia coli Cytotoxic Necrotizing Factor 1 (CNF1): Toxin Biology, in Vivo Applications and Therapeutic Potential. Toxins. 2010; 2(2):283-296. https://doi.org/10.3390/toxins2020282
Chicago/Turabian StyleFabbri, Alessia, Sara Travaglione, and Carla Fiorentini. 2010. "Escherichia coli Cytotoxic Necrotizing Factor 1 (CNF1): Toxin Biology, in Vivo Applications and Therapeutic Potential" Toxins 2, no. 2: 283-296. https://doi.org/10.3390/toxins2020282