Functional Consequences of Calcium Influx Promoted by Bacterial Pore-Forming Toxins

Université Grenoble Alpes, CNRS ERL5261, CEA BIG-BCI, INSERM UMR1036, Grenoble 38054, France
*
Author to whom correspondence should be addressed.
Toxins 2018, 10(10), 387; https://doi.org/10.3390/toxins10100387
Submission received: 3 September 2018
/
Revised: 14 September 2018
/
Accepted: 20 September 2018
/
Published: 25 September 2018
(This article belongs to the Special Issue Bacterial Pore-Forming Toxins)
Abstract
:Bacterial pore-forming toxins induce a rapid and massive increase in cytosolic Ca2+ concentration due to the formation of pores in the plasma membrane and/or activation of Ca2+-channels. As Ca2+ is an essential messenger in cellular signaling, a sustained increase in Ca2+ concentration has dramatic consequences on cellular behavior, eventually leading to cell death. However, host cells have adapted mechanisms to protect against Ca2+ intoxication, such as Ca2+ efflux and membrane repair. The final outcome depends upon the nature and concentration of the toxin and on the cell type. This review highlights the repercussions of Ca2+ overload on the induction of cell death, repair mechanisms, cellular adhesive properties, and the inflammatory response.
Keywords:
host–pathogen interaction; bacterial virulence factor; cell death; signal transduction; ion fluxKey Contribution: This review summarizes the numerous host cell alterations induced by Ca2+ overload triggered by bacterial pore-forming toxins, as well as the defense mechanisms implemented by the host to limit Ca2+ intoxication and plasma membrane perforation.
1. Introduction
Bacterial pore-forming toxins (PFTs) are the most frequently encountered virulence factors among bacterial pathogens [1,2,3,4]. They are secreted in the extracellular milieu from Gram-negative and Gram-positive bacteria by various bacterial secretion systems. It is generally accepted that PFTs need a specific cellular receptor to bind to the host cell and to form a pore, hence preventing bacterial self-toxicity. Several cellular receptors for PFTs have been identified, so far, from membrane proteins specific to one or more cell types of multicellular organisms, to lipids like cholesterol or sphingomyelin, which are frequently present in vertebrate tissues [3,5]. These two lipids are concentrated in specialized membrane domains called lipid rafts, which are also the site of localization of specific membrane proteins or receptors. As a result, several PFTs are guided toward these rafts.
Once bound to a target cell receptor, PFTs form a pore in the plasma membrane, using a complex multistep process. The first step is toxin oligomerization. Oligomerization may occur once the toxins are inserted into the membrane or otherwise bound to the cell’s surface, forming a prepore. The prepore then inserts into the plasma membrane, forming a ring-shaped pore, which alters the target cell’s integrity [3]. Pore formation may have dramatic consequences for the host cell, unless it can mount a process to eliminate the membrane domain containing the pore. Importantly, mechanisms dependent on the toxins, but independent of pore formation, have also been reported. They involve receptor activation and downstream signaling that can also alter Ca2+ concentration within host cells. The various scenarios described after PFT intoxication depend mainly on PFT identity and its local concentration, but also on the host cell type (e.g., immune vs nonimmune cells, epithelium vs. endothelium). For all these effects, Ca2+ is a central player in PFT-induced toxicity and downregulation of its cytosolic concentration is critical for cell fate.
2. How Do PFTs Increase Intracellular Ca2+?
In general, one of the immediate consequences of PFT insertion into the plasma membrane is ion exchange between the extracellular environment and the cytosol through the open pore. For intracellular bacteria, pore formation and ion flux can also occur within the phagosome [6]. Other mechanisms have been described, such as the activation of endogenous ion channels, either located in the plasma membrane or in organelles accumulating Ca2+, like the endoplasmic reticulum (ER) and lysosomes (Figure 1) [7,8,9,10,11,12,13,14,15].
Pores or ion channels can exchange a range of ion types, but most previous studies have concentrated on Ca2+ and K+ flux, because they are known to have important functional consequences. These ions passively flow through the open pores as a result of the concentration gradients between the external milieu and the cytosol: millimolar Ca2+ concentrations are reported in animal tissues and fluids, and 10,000-fold lower concentrations are found in the cytosol [16]; in contrast, K+ concentration is in the millimolar range in animal fluids, and 130 mM in the cytosol [17]. Hence, pore formation triggers passive K+ efflux and Ca2+ influx. Although passive ion flow is the rule (see Table 1), at least one PFT, the Vibrio cholerae cytolysin, creates a pore that is too narrow to allow passage of Ca2+ [18]. The related PFT, phobalysin, from Photobacterium dameselae, is large enough to allow Ca2+ flux [18]. A single mutation in V. cholerae cytolysin, rendering the channel domain similar to that of phobalysin and enlarging the pore, makes Ca2+ influx possible and modifies the host response to pore formation. Interestingly, K+ can flow out through the unmodified cytolysin, suggesting that some ion selection may exist in the pores created by PFTs.
Importantly, K+ efflux is well known to induce several host cell alterations, including the activation of the NLRP3 inflammasome and p38 MAP kinase. The functional implications of K+ efflux have been reviewed in details elsewhere [19,20,21,22]. However, it has also been reported that mitochondrial Ca2+ elevation, a secondary effect of cytosolic Ca2+ rise, can promote NLRP3 activation [23,24], hence positioning Ca2+ as another potent initiator of inflammasome activation.
Ca2+ influx through the pore is usually massive because of the very steep concentration gradient between the extracellular and cytosolic compartments. Therefore, Ca2+ entry usually displays monophasic kinetics, eventually followed by a sudden drop if the cell bursts, delivering its content in the extracellular milieu (biphasic kinetics). However, multiphasic kinetics may also be observed, either because of rapid opening/closing of the pore or because forming pores are progressively eliminated by the host cell’s repair mechanisms (see below and references in Table 1).
Ca2+ oscillations have also been described for some PFTs when Ca2+ channels are activated (Table 1). Release of Ca2+ from internal stores has been reported for several PFTs using different pathways. In addition to formation of Ca2+-permeable pores, some PFTs, like aerolysin from Aeromonas hydrophila, streptolysin O (SLO) from Streptococcus pyogenes, and Staphylococcus aureus hemolysin A (Hla) [12], induce the release of Ca2+ from the ER by two different mechanisms successively: (i) a transient Ca2+ release from inositol (1,4,5)P3-sensitive stores which involves G-proteins and phospholipase C, and (ii) a delayed and sustained release, the activation mechanisms of which remain to be determined [12].
Similarly, listeriolysin (LLO) from Listeria monocytogenes induces Ca2+ release from the ER via the G protein-phospholipase C-inositol (1,4,5)P3 pathway, as well as a second wave of Ca2+ release involving damage to intracellular stores (ER and lysosomes) [9]. The mechanism leading to organelle perforation is unknown but seems to be Ca2+–independent. Interestingly, organelle damage is reversible and does not result in cell death. This is an unconventional but efficient way to deliver Ca2+ in the cytosol, because of the high Ca2+ content of the ER.
Finally, Pasteurella haemolytica leukotoxin (LKT) induces increased cytosolic Ca2+ by activating voltage-gated channels in the plasma membrane via a G-protein-coupled mechanism involving activation of phospholipases A2 and C [7,10],
The mechanism of G-protein activation by PFTs remains undetermined. It is tempting to speculate that PFTs interact with a G-protein coupled receptor at the cell surface, which is the common way for G-protein activation. Alternatively, the transmembrane pore formed by PFTs may interact directly with G-proteins in the cytosol, without the need of a specific receptor.
Other examples of PFTs with specific modes of action are S. aureus leukotoxins (γ-hemolysin, Hlg, and Panton–Valentine leukocidin, PVL), which increase Ca2+ levels by triggering its release from lysosomes followed by a second release from endoplasmic reticulum. This in turn stimulates the activation of store-operated Ca2+-channels in the plasma membrane, a process normally used when intracellular organelles are discharged of Ca2+ [11]. The initial signal linking leukotoxin binding to acidic stores is the activation of the ADP-ribosyl cyclase CD38 [11]. CD38 is a membrane receptor and a nicotinic acid adenine dinucleotide phosphate (NAADP) synthase required for coupling receptor activation to NAADP-mediated Ca2+ release from lysosomal stores through the two-pore Ca2+ channels [25,26].
Finally, Bordetella pertussis ACT, through its adenylate cyclase properties, activates non-voltage-dependent Ca2+ channels with L-type characteristics. This process involves ACT-induced cAMP production and subsequent protein kinase A activation [13].
Thus, although passive influx through the pore is the most widespread mode of Ca2+ entry exploited by bacterial PFTs, several toxins can also use other pathways to increase cytosolic Ca2+ concentration. It is yet unknown whether Ca2+ channel opening occurs before or simultaneously with pore formation.
In general, Ca2+ is maintained at low levels in the cytosol because it is an important second messenger activating several signaling pathways. Ca2+ can interact with and activate a number of cytoplasmic proteins [16] that are potential sensors of the presence of PFT pores. Effective activation by PFTs have been reported for calmodulin [34,48], calpains [34,37,61], protein kinase C (PKC) [43], phospholipases [50], and calcineurin [54]. Sustained activation of calpain, PKC, and calcineurin pathways leads to cell death. Therefore, several mechanisms are employed by the cell to maintain low concentrations of cytosolic Ca2+. Extrusion of cytosolic Ca2+ can be carried out by plasma membrane pumps (the plasma membrane Ca2+-ATPase; PMCA) [62], and was reported for Hla [49]. Large amounts of Ca2+ can also be efficiently sequestered in the ER thanks to the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) [63] or into mitochondria via the mitochondrial Ca2+ uniporter (MCU) [64], which eventually leads to mitochondrial intoxication. However, massive Ca2+ entry rapidly overloads the cytosol and exceeds the capacity of internal stores and the capability of Ca2+ pumps. If no membrane repair mechanism is activated, ion imbalance eventually triggers osmotic rupture of the plasma membrane or cell death by another pathway (see below).
In the following sections, we will focus on the main consequences of sustained PFT-dependent increase in cytosolic Ca2+ reported in the literature. Other major effects of pore formation by PFTs, for which the link with Ca2+ has not been established, are not dealt with here.
3. Cell Repair Mechanisms
Host cells have adapted to PFT injury by creating several mechanisms to eliminate pores that efficiently combat the dramatic effects of cell perforation by PFTs. The repair mechanism used depends upon the nature and number of pores, and on the cell type. As for the Ca2+ exporting systems, when the repair mechanisms are overwhelmed, the cellular ion imbalance reaches a point-of-no-return and cells engage in an irreversible process of cell death. Several excellent reviews have recently been published on this topic [1,2,4,65,66]; here, we will simply summarize the main mechanisms used by the cell to repair its plasma membrane.
Membrane repair mechanisms have been studied for cholesterol-binding PFTs (SLO, Streptococcus pneumoniae pneumolysin (PLY), Clostridium perfringens perfringolysin (PFO), and Streptococcus intermedius intermedilysin (ILY)) forming large pores (30 nm in diameter) that are surprisingly more efficiently eliminated by host cells than small-pore forming toxins.
The primary mechanism of pore clearance involves the externalization of microvesicles (also called ectocytosis) containing PFT pores [33,67,68,69,70]. This can be performed via two different ways involving either (i) annexins, which migrate to the injured site, avidly bind to Ca2+ and interact with the plasma membrane; because of their fusogenic activity, annexins induce the formation of membrane folds that can be expelled; or (ii) the endosomal sorting complex required for transport (ESCRT) machinery, which drives microvesicle shedding [71,72].
The other process described is the endocytic model, whereby a portion of pore-containing membrane is internalized and then targeted to the lysosome for degradation [60,73]. This process seems to be restricted to the elimination of inactivated or monomeric toxins [33].
Both processes of membrane repair (ectocytosis or endocytosis) require RAB-5 and RAB-11, two important regulators of vesicle trafficking [74].
As mentioned above, PFTs forming large pores trigger repair mechanisms much more efficiently than small-pore toxins. The reasons for this difference in triggering capacity remain elusive. One obvious hypothesis would be that small pores do not promote a Ca2+ influx sufficient for repair mechanism activation, however this hypothesis is contradicted by Ca2+ imaging data showing massive influx when cells are incubated with small-pore forming toxins [48,49,50]. As PFTs inducing efficient membrane repair interact directly with lipids and stimulate blebbing at the prepore stage, it is possible that they are cleared before membrane damage. Conversely, PFTs interacting with proteinaceous receptors may not induce the intrinsic pathway and cells may only depend upon the endocytic repair mechanism. Other collateral factors may include the capacity of PFTs to induce Ca2+ channel opening or pore stability in the plasma membrane.
Interestingly, ACT controls the path and kinetics of endocytic removal of toxin pores in a K+-dependent manner [32]. As many PFT pores were reported to trigger K+ efflux, this repair mechanism may also be true for most PFT pores.
4. Cell Death
If the repair mechanisms fail to remove PFT pores, the cells will eventually commit to Ca2+-dependent death programs (Table 2).
PFT-induced cell death is often reported as “osmotic lysis”, a type of necrosis involving cell dilation and membrane rupture due to excessive intracellular pressure [34,35,37,59]. As previously mentioned, this results from sustained ion flux causing osmotic imbalance due to the high concentration of macromolecules inside the cell [78]. However, the kinetics of PFT-mediated Ca2+ influx induces cell death that may be more related to Ca2+ toxicity than ion imbalance [30], and a number of PFTs trigger cell death by apoptosis, alone or in parallel with necrosis [29,34,59,61,79].
In some examples [29,34,59,79], low doses of PFTs can promote Ca2+-dependent apoptosis by eliciting the release of apoptosis-inducing factor (AIF) and cytochrome c, owing to Ca2+-induced opening of the mitochondrial permeability transition pore. Both proteins are known proapoptotic factors: AIF is translocated to the nucleus where it causes DNA degradation and chromatin condensation; cytochrome c activates the caspase cascade leading to DNA fragmentation [80]. Independently, calpain protease activation by Ca2+ can also trigger caspase-dependent or independent apoptosis as well as necrosis [78,81]. All three pathways (cytochrome c, AIF, and calpains) may be instrumental for PFTs to promote cell death of intoxicated cells. For example, C. perfringens enterotoxin (CPE) elicits apoptosis at low doses and necrosis at high doses, both pathways being caspase- and calpain-dependent [34]. In addition, calmodulin, a cytosolic protein with high affinity for Ca2+, is also involved in CPE-dependent apoptosis and necrosis processes [34]. The mechanism of calmodulin-induced cell death was not determined in this context, but recent work in breast cancer cells demonstrated that calmodulin can bind to death receptor-5 (DR5) in a Ca2+-dependent manner, which triggers apoptotic signaling [82]. This mechanism may also occur when cells are intoxicated with CPE.
Similarly, C. perfringens epsilon toxin (ET) stimulates the release of cytochrome c and mitochondrial–nuclear translocation of AIF, leading to chromatin condensation and nuclear shrinkage. However, ET fails to induce DNA fragmentation and thus to achieve apoptosis; cell death being then executed by necrosis [35]. A possible explanation is that the energy-dependent process of apoptosis is dissipated as a result of the loss of ATP through the pore, whereas necrosis can be completed as it requires no energy [35].
In the case of α-toxin from C. septicum, Ca2+ influx induces a mechanism of programmed necrosis involving calpain activation, release of cathepsins from lysosomes and increased reactive oxygen species (ROS) levels produced by deregulation of mitochondrial activity [37]. Thus, PFT-dependent necrosis may not just result from oncosis, but from damage of cellular organelles together with activation of cytosolic Ca2+-sensor proteins.
Finally, two PFTs, ShlA and PLY, can trigger Ca2+-dependent necroptosis in pneumocytes [83]. Necroptosis is a regulated cell death program leading to cell membrane rupture, and as such is considered as a form of necrosis [84]. In general, necroptosis is engaged by membrane receptors, tumor necrosis factor receptors (TNFRs) or Toll-like receptors (TLRs), and is followed by a cascade of signaling that occurs only when caspases are inactivated [84]. Signaling proteins include the receptor interacting proteins (RIP) 1 and 3, and the mixed-lineage kinase domain-like protein (MLKL), the executer of necroptosis. Gonzales-Juarbe et al. [83] showed that Serratia marcescens hemolysin (ShlA)- and PLY-induced necroptosis were independent of TNFR or TLR activation, but required RIP1, RIP3, and MLKL. This was confirmed in vivo in mice deficient either in RIP3 or in MLKL that exhibited increased survival when challenged with S. marcescens.
Taken together, PFTs can act on several pathways, probably in combination, to provoke cell death. It is noteworthy to point out the central role of mitochondria in this context. Still, much work has to be done to fully elucidate this critical effect of PFTs in order to identify the missing links in these pathways. Most of these findings result from investigations in one cell type, while most PFTs intoxicate several. As death programs are cell type-dependent, it would be important to extend these investigations to other cells and tissues. [29,34,55,59]
5. Intercellular Junction Disruption
Although not precisely demonstrated, PFTs are probably present at sublytic concentrations in vivo [19]. However, bacterial PFTs trigger additional toxic mechanisms modifying cell behavior.
Cadherins, located in adherens junctions and required for intercellular adhesion, were recently shown to be targeted by PFTs. PFT-triggered cadherin cleavage was first described by Inoshima et al. after incubation of cells with Hla [51]. The mechanism involves the subversion of ADAM10, a transmembrane protease, whose major substrates are adhesive receptors, including E- and VE-cadherins located at epithelial and endothelial intercellular junctions, respectively. In normal settings, when activated by outside-in signals, ADAM10 cleaves cadherin extracellular domains close to the transmembrane region to remove the homophilic adhesive domain (Figure 1). This proteolytic cleavage considerably modifies the cell’s adhesive properties and induces their extrusion from tissues.
ADAM10 is the cellular receptor for Hla. Extracellular Ca2+ is required for Hla-dependent cadherin cleavage by ADAM10, suggesting that the Hla-ADAM10 interaction alone cannot activate ADAM10; indeed, Ca2+ influx generated by pore formation is involved in this process. A similar effect was confirmed for PLY, which binds cholesterol rather than ADAM10 [51,85,86].
More recently, two PFTs secreted by Pseudomonas aeruginosa (ExlA) and S. marcescens (ShlA) were also shown to induce rapid E- and VE-cadherin cleavage through ADAM10 activation, even though ADAM10 is not a cellular receptor for these PFTs [48]. In resting conditions, intracellular pro-ADAM10 is bound to calmodulin, preventing its cleavage and activation by furin [87,88]. Both ExlA and ShlA promote a sustained elevation of cytosolic concentration of Ca2+ [48], which interacts with high affinity with calmodulin. Once bound to Ca2+, calmodulin releases pro-ADAM10, which is in turn activated by furin and transported to the plasma membrane where it induces cadherin shedding (Figure 1). Thus, these PFTs subvert a tightly regulated host pathway, which controls cellular adhesive properties within tissues. Although this effect has only been demonstrated for four PFTs so far [48,51,85,86], it is likely that most PFTs promoting Ca2+ influx would induce cadherin cleavage via ADAM10 activation, eventually permitting bacterial transmigration across tissue barriers after complete destruction of intercellular junctions.
ADAM10 substrates also include kinase receptors and matrix proteins (see past reviews [89,90]), which are additional potential targets of PFTs. If confirmed, PFTs might also manipulate signal transduction pathways and the extracellular environment through the same initial mechanism.
Interestingly, tight junctions, which control barrier permeability, are also disrupted by A. hydrophila aerolysin via Ca2+- and myosin light chain kinase (MLCK)-dependent pathways [28] (Figure 1). This feature further supports the hypothesis that intercellular junctions are one of the main targets of PFTs.
6. Other PFT-Mediated Effects
A number of Ca2+-dependent effects have been reported in immune cells, including granulocyte chemotaxis [12,40], reactive oxygen species (ROS), cytokine and leukotriene B4 production by granulocytes [40,46,56], cytokine production by macrophages [10], and degranulation and cytokine synthesis in mast cells [9]. Cytokine production is also elicited by PFTs in epithelial cells [39]. All these effects are pro-inflammatory and are expected to promote elimination of bacteria.
In general, erythrocytes are rapidly hemolyzed by PFTs because they have no means of resisting pore formation. Platelets may be activated by pore-induced Ca2+ flux, hence providing an explanation for the prothrombotic action of some pathogens [57].
Importantly, pore formation may also facilitate bacterial internalization by triggering a Ca2+-dependent protein kinase C-Rac1-Arp2/3 signaling pathway acting on F-actin [43].
In addition to the mechanisms presented above, several intracellular signaling molecules are stimulated by PFTs, such as phospholipase A2 (PLA2), whose activity is enhanced by Ca2+ binding [50], and early growth response protein 1 (EGR-1) transcription factor via the calcineurin-nuclear factor of activated T cells (NFAT) pathway [54]. The final impact of these modifications has yet to be determined.
7. Concluding Remarks
Because Ca2+ is a very important communicator in cell signaling and drives important cellular functions, its manipulation by bacterial PFTs has profound consequences on cell behavior and homeostasis.
Increased cytosolic Ca2+ concentrations can have dramatic consequences, including tissue destruction and bacterial dissemination, or more subtle effects, such as bacterial internalization or thrombosis. However, PFT-induced cell rupture and cytokine production are also alarming signals engaging a strong immune response that counteract the infection.
As indicated above, PFTs from various bacteria may induce diverse—sometimes opposing—effects, however the mechanisms activated by these toxins have not been systematically investigated for all PFTs and much work remains to be done to obtain a general view of the action of PFTs in various infection scenarios.
Author Contributions
All authors wrote the review.
Funding
The work of the team is supported by grants from Laboratoire of Excellence “GRAL” (ANR-10-LABX-49-01), Agence Nationale de la Recherche (ANR-15-CE11-0018-01), and Fondation pour la Recherche Médicale “Equipe FRM 2017” (DEQ20170336705). We further acknowledge financial support from CNRS, INSERM, CEA and University Grenoble-Alpes.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bischofberger, M.; Gonzalez, M.R.; van der Goot, F.G. Membrane injury by pore-forming proteins. Curr. Opin. Cell Biol. 2009, 21, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.K.; O’Riordan, M.X. More than a pore: The cellular response to cholesterol-dependent cytolysins. Toxins 2013, 5, 618–636. [Google Scholar] [CrossRef] [PubMed]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Los, F.C.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [PubMed]
- DuMont, A.L.; Torres, V.J. Cell targeting by the staphylococcus aureus pore-forming toxins: It’s not just about lipids. Trends Microbiol. 2014, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, L.M.; Hoppe, A.D.; Christensen, K.A.; Swanson, J.A. Membrane perforations inhibit lysosome fusion by altering ph and calcium in listeria monocytogenes vacuoles. Cell. Microbiol. 2006, 8, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Cudd, L.; Clarke, C.; Clinkenbeard, K. Contribution of intracellular calcium stores to an increase in cytosolic calcium concentration induced by mannheimia haemolytica leukotoxin. FEMS Microbiol. Lett. 2003, 225, 23–27. [Google Scholar] [CrossRef]
- Gekara, N.O.; Groebe, L.; Viegas, N.; Weiss, S. Listeria monocytogenes desensitizes immune cells to subsequent Ca2+ signaling via listeriolysin o-induced depletion of intracellular Ca2+ stores. Infect. Immun. 2008, 76, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Gekara, N.O.; Westphal, K.; Ma, B.; Rohde, M.; Groebe, L.; Weiss, S. The multiple mechanisms of Ca2+ signalling by listeriolysin o, the cholesterol-dependent cytolysin of listeria monocytogenes. Cell. Microbiol. 2007, 9, 2008–2021. [Google Scholar] [CrossRef] [PubMed]
- Hsuan, S.L.; Kannan, M.S.; Jeyaseelan, S.; Prakash, Y.S.; Sieck, G.C.; Maheswaran, S.K. Pasteurella haemolytica a1-derived leukotoxin and endotoxin induce intracellular calcium elevation in bovine alveolar macrophages by different signaling pathways. Infect. Immun. 1998, 66, 2836–2844. [Google Scholar] [PubMed]
- Jover, E.; Tawk, M.Y.; Laventie, B.J.; Poulain, B.; Prevost, G. Staphylococcal leukotoxins trigger free intracellular Ca2+ rise in neurones, signalling through acidic stores and activation of store-operated channels. Cell. Microbiol. 2013, 15, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.H.; Fivaz, M.; Monod, A.; van der Goot, F.G. Aerolysin induces g-protein activation and Ca2+ release from intracellular stores in human granulocytes. J. Biol. Chem. 1998, 273, 18122–18129. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Gomez-Bilbao, G.; Ostolaza, H. Bordetella adenylate cyclase toxin promotes calcium entry into both cd11b+ and cd11b- cells through camp-dependent l-type-like calcium channels. J. Biol. Chem. 2010, 285, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Staali, L.; Monteil, H.; Colin, D.A. The staphylococcal pore-forming leukotoxins open Ca2+ channels in the membrane of human polymorphonuclear neutrophils. J. Membr. Biol. 1998, 162, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Tanoue, N.; Nakano, M.; Hamamoto, A.; Okamoto, K.; Fujii, Y.; Harada, N.; Nakaya, Y. A pore-forming toxin produced by aeromonas sobria activates Ca2+ dependent cl- secretion. Microb. Pathog. 2005, 38, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Krebs, J. Why calcium? How calcium became the best communicator. J. Biol. Chem. 2016, 291, 20849–20857. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Matsudaira, P. Molecular Cell Biology; Freeman: New York, NY, USA, 2000. [Google Scholar]
- Von Hoven, G.; Rivas, A.J.; Neukirch, C.; Meyenburg, M.; Qin, Q.; Parekh, S.; Hellmann, N.; Husmann, M. Repair of a bacterial small beta-barrel toxin pore depends on channel width. mBio 2017, 8, e02083-16. [Google Scholar] [CrossRef] [PubMed]
- Aroian, R.; van der Goot, F.G. Pore-forming toxins and cellular non-immune defenses (CNIDs). Curr. Opin. Microbiol. 2007, 10, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kroemer, G. Necrosis: Linking the inflammasome to inflammation. Cell Rep. 2015, 11, 1501–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, D.; Ahn, D.; Cohen, T.; Prince, A. Innate immune signaling activated by mdr bacteria in the airway. Physiol. Rev. 2016, 96, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Bezzerri, V.; Patergnani, S.; Marchi, S.; Cabrini, G.; Pinton, P. Mitochondrial Ca2+-dependent nlrp3 activation exacerbates the pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 2015, 6, 6201. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Cosker, F.; Cheviron, N.; Yamasaki, M.; Menteyne, A.; Lund, F.E.; Moutin, M.J.; Galione, A.; Cancela, J.M. The ecto-enzyme CD38 is a nicotinic acid adenine dinucleotide phosphate (NAADP) synthase that couples receptor activation to Ca2+ mobilization from lysosomes in pancreatic acinar cells. J. Biol. Chem. 2010, 285, 38251–38259. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.X.; Ma, J.; Parrington, J.; Galione, A.; Evans, A.M. Tpcs: Endolysosomal channels for Ca2+ mobilization from acidic organelles triggered by NAADP. FEBS Lett. 2010, 584, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- Iwase, M.; Korchak, H.M.; Lally, E.T.; Berthold, P.; Taichman, N.S. Lytic effects of actinobacillus actinomycetemcomitans leukotoxin on human neutrophil cytoplasts. J. Leukoc. Biol. 1992, 52, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Bucker, R.; Krug, S.M.; Rosenthal, R.; Gunzel, D.; Fromm, A.; Zeitz, M.; Chakraborty, T.; Fromm, M.; Epple, H.J.; Schulzke, J.D. Aerolysin from aeromonas hydrophila perturbs tight junction integrity and cell lesion repair in intestinal epithelial ht-29/b6 cells. J. Infect. Dis. 2011, 204, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.L.; Brodsky, R.A.; Buckley, J.T. Channels formed by subnanomolar concentrations of the toxin aerolysin trigger apoptosis of t lymphomas. Cell. Microbiol. 1999, 1, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Tschodrich-Rotter, M.; Kubitscheck, U.; Ugochukwu, G.; Buckley, J.T.; Peters, R. Optical single-channel analysis of the aerolysin pore in erythrocyte membranes. Biophys. J. 1996, 70, 723–732. [Google Scholar] [CrossRef] [Green Version]
- Fiser, R.; Masin, J.; Basler, M.; Krusek, J.; Spulakova, V.; Konopasek, I.; Sebo, P. Third activity of bordetella adenylate cyclase (ac) toxin-hemolysin. Membrane translocation of ac domain polypeptide promotes calcium influx into cd11b+ monocytes independently of the catalytic and hemolytic activities. J. Biol. Chem. 2007, 282, 2808–2820. [Google Scholar] [CrossRef] [PubMed]
- Fiser, R.; Masin, J.; Bumba, L.; Pospisilova, E.; Fayolle, C.; Basler, M.; Sadilkova, L.; Adkins, I.; Kamanova, J.; Cerny, J.; et al. Calcium influx rescues adenylate cyclase-hemolysin from rapid cell membrane removal and enables phagocyte permeabilization by toxin pores. PLoS Pathog. 2012, 8, e1002580. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, G.; McClane, B.A. The importance of calcium influx, calpain and calmodulin for the activation of caco-2 cell death pathways by clostridium perfringens enterotoxin. Cell. Microbiol. 2005, 7, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Chassin, C.; Bens, M.; de Barry, J.; Courjaret, R.; Bossu, J.L.; Cluzeaud, F.; Ben Mkaddem, S.; Gibert, M.; Poulain, B.; Popoff, M.R.; et al. Pore-forming epsilon toxin causes membrane permeabilization and rapid atp depletion-mediated cell death in renal collecting duct cells. Am. J. Physiol. Ren. Physiol. 2007, 293, F927–F937. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Maier, E.; Gibert, M.; Popoff, M.R.; Benz, R. Clostridium perfringens epsilon toxin induces a rapid change of cell membrane permeability to ions and forms channels in artificial lipid bilayers. J. Biol. Chem. 2001, 276, 15736–15740. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.L.; Smith, D.J.; Lyras, D.; Chakravorty, A.; Rood, J.I. Programmed cellular necrosis mediated by the pore-forming alpha-toxin from clostridium septicum. PLoS Pathog. 2009, 5, e1000516. [Google Scholar] [CrossRef] [PubMed]
- Koschinski, A.; Repp, H.; Unver, B.; Dreyer, F.; Brockmeier, D.; Valeva, A.; Bhakdi, S.; Walev, I. Why escherichia coli alpha-hemolysin induces calcium oscillations in mammalian cells—The pore is on its own. FASEB J. 2006, 20, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, P.; Laestadius, A.; Jahnukainen, T.; Soderblom, T.; Backhed, F.; Celsi, G.; Brismar, H.; Normark, S.; Aperia, A.; Richter-Dahlfors, A. Alpha-haemolysin of uropathogenic e. Coli induces Ca2+ oscillations in renal epithelial cells. Nature 2000, 405, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Walev, I.; Kemmer, H.; Weis, S.; Siegel, I.; Boukhallouk, F.; Wassenaar, T.M.; Chavakis, T.; Bhakdi, S. Binding of escherichia coli hemolysin and activation of the target cells is not receptor-dependent. J. Biol. Chem. 2005, 280, 36657–36663. [Google Scholar] [CrossRef] [PubMed]
- Soderblom, T.; Oxhamre, C.; Wai, S.N.; Uhlen, P.; Aperia, A.; Uhlin, B.E.; Richter-Dahlfors, A. Effects of the escherichia coli toxin cytolysin a on mucosal immunostimulation via epithelial Ca2+ signalling and toll-like receptor 4. Cell. Microbiol. 2005, 7, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Dramsi, S.; Cossart, P. Listeriolysin o-mediated calcium influx potentiates entry of listeria monocytogenes into the human hep-2 epithelial cell line. Infect. Immun. 2003, 71, 3614–3618. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.G.T.; Vadia, S.; Pathak-Sharma, S.; McLaughlin, E.; Zhang, X.; Swanson, J.; Seveau, S. Host cell perforation by listeriolysin o (llo) activates a Ca2+-dependent cpkc/rac1/arp2/3 signaling pathway that promotes listeria monocytogenes internalization independently of membrane resealing. Mol. Biol. Cell 2018, 29, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Repp, H.; Pamukci, Z.; Koschinski, A.; Domann, E.; Darji, A.; Birringer, J.; Brockmeier, D.; Chakraborty, T.; Dreyer, F. Listeriolysin of listeria monocytogenes forms Ca2+-permeable pores leading to intracellular Ca2+ oscillations. Cell. Microbiol. 2002, 4, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Cudd, L.; Clarke, C.; Clinkenbeard, K. Mannheimia haemolytica leukotoxin-induced increase in leukotriene b4 production by bovine neutrophils is mediated by a sustained and excessive increase in intracellular calcium concentration. FEMS Microbiol. Lett. 2003, 224, 85–90. [Google Scholar] [CrossRef]
- Cudd, L.; Clarke, C.; Clinkenbeard, K.; Shelton, M.; Clinkenbeard, P.; Murphy, G. Role of intracellular calcium in pasteurella haemolytica leukotoxin-induced bovine neutrophil leukotriene b4 production and plasma membrane damage. FEMS Microbiol. Lett. 1999, 172, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Carranza, O.; Czuprynski, C.J. Activation of bovine neutrophils by pasteurella haemolytica leukotoxin is calcium dependent. J. Leukoc. Biol. 1992, 52, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Reboud, E.; Bouillot, S.; Patot, S.; Beganton, B.; Attree, I.; Huber, P. Pseudomonas aeruginosa ExlA and serratia marcescens ShlA trigger cadherin cleavage by promoting calcium influx and ADAM10 activation. PLoS Pathog. 2017, 13, e1006579. [Google Scholar] [CrossRef] [PubMed]
- Eichstaedt, S.; Gabler, K.; Below, S.; Muller, C.; Kohler, C.; Engelmann, S.; Hildebrandt, P.; Volker, U.; Hecker, M.; Hildebrandt, J.P. Effects of staphylococcus aureus-hemolysin a on calcium signalling in immortalized human airway epithelial cells. Cell Calcium 2009, 45, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Contreras, M.L.; Lelkes, P.I.; Lazarovici, P. Staphylococcus aureus alpha-toxin activates phospholipases and induces a Ca2+ influx in PC12 cells. Cell. Signal. 1989, 1, 387–393. [Google Scholar] [CrossRef]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Bubeck Wardenburg, J. A staphylococcus aureus pore-forming toxin subverts the activity of adam10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.K.; Vikstrom, E.; Magnusson, K.E.; Vecsey-Semjen, B.; Colque-Navarro, P.; Mollby, R. The staphylococcus aureus alpha-toxin perturbs the barrier function in caco-2 epithelial cell monolayers by altering junctional integrity. Infect. Immun. 2012, 80, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Von Hoven, G.; Rivas, A.J.; Neukirch, C.; Klein, S.; Hamm, C.; Qin, Q.; Meyenburg, M.; Fuser, S.; Saftig, P.; Hellmann, N.; et al. Dissecting the role of adam10 as a mediator of staphylococcus aureus alpha-toxin action. Biochem. J. 2016, 473, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Susilowati, H.; Okamura, H.; Hirota, K.; Shono, M.; Yoshida, K.; Murakami, K.; Tabata, A.; Nagamune, H.; Haneji, T.; Miyake, Y. Intermedilysin induces EGR-1 expression through calcineurin/NFAT pathway in human cholangiocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2011, 404, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.S.; Sublett, J.E.; Freyer, D.; Mitchell, T.J.; Cleveland, J.L.; Tuomanen, E.I.; Weber, J.R. Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J. Clin. Investig. 2002, 109, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Fickl, H.; Cockeran, R.; Steel, H.C.; Feldman, C.; Cowan, G.; Mitchell, T.J.; Anderson, R. Pneumolysin-mediated activation of NFkappaB in human neutrophils is antagonized by docosahexaenoic acid. Clin. Exp. Immunol. 2005, 140, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nel, J.G.; Durandt, C.; Mitchell, T.J.; Feldman, C.; Anderson, R.; Tintinger, G.R. Pneumolysin mediates platelet activation in vitro. Lung 2016, 194, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Wolfmeier, H.; Schoenauer, R.; Atanassoff, A.P.; Neill, D.R.; Kadioglu, A.; Draeger, A.; Babiychuk, E.B. Ca(2)(+)-dependent repair of pneumolysin pores: A new paradigm for host cellular defense against bacterial pore-forming toxins. Biochim. Biophys. Acta 2015, 1853, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Cywes Bentley, C.; Hakansson, A.; Christianson, J.; Wessels, M.R. Extracellular group a streptococcus induces keratinocyte apoptosis by dysregulating calcium signalling. Cell. Microbiol. 2005, 7, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Fong, K.P.; Pacheco, C.M.; Otis, L.L.; Baranwal, S.; Kieba, I.R.; Harrison, G.; Hersh, E.V.; Boesze-Battaglia, K.; Lally, E.T. Actinobacillus actinomycetemcomitans leukotoxin requires lipid microdomains for target cell cytotoxicity. Cell. Microbiol. 2006, 8, 1753–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carafoli, E. Calcium pump of the plasma membrane. Physiol. Rev. 1991, 71, 129–153. [Google Scholar] [CrossRef] [PubMed]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. Serca control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Pinton, P. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Draeger, A. Defying death: Cellular survival strategies following plasmalemmal injury by bacterial toxins. Semin. Cell Dev. Biol. 2015, 45, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; McNeil, P.L. Membrane repair: Mechanisms and pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Intracellular Ca2+ operates a switch between repair and lysis of streptolysin o-perforated cells. Cell Death Differ. 2009, 16, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2011, 18, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin o clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Wolfmeier, H.; Radecke, J.; Schoenauer, R.; Koeffel, R.; Babiychuk, V.S.; Drucker, P.; Hathaway, L.J.; Mitchell, T.J.; Zuber, B.; Draeger, A.; et al. Active release of pneumolysin prepores and pores by mammalian cells undergoing a streptococcus pneumoniae attack. Biochim. Biophy. Acta 2016, 1860, 2498–2509. [Google Scholar] [CrossRef] [PubMed]
- Draeger, A.; Monastyrskaya, K.; Babiychuk, E.B. Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem. Pharmacol. 2011, 81, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. Escrt machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef] [PubMed]
- Corrotte, M.; Fernandes, M.C.; Tam, C.; Andrews, N.W. Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic 2012, 13, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Los, F.C.; Kao, C.Y.; Smitham, J.; McDonald, K.L.; Ha, C.; Peixoto, C.A.; Aroian, R.V. Rab-5- and rab-11-dependent vesicle-trafficking pathways are required for plasma membrane repair after attack by bacterial pore-forming toxin. Cell Host Microbe 2011, 9, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Dersch, K.; Bobkiewicz, W.; Beckmann, E.; Veerachato, G.; Bhakdi, S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. Aureus alpha-toxin or streptolysin O. Biochem. Biophys. Res. Commun. 2006, 344, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Los, F.C.; Huffman, D.L.; Wachi, S.; Kloft, N.; Husmann, M.; Karabrahimi, V.; Schwartz, J.L.; Bellier, A.; Ha, C.; et al. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog. 2011, 7, e1001314. [Google Scholar] [CrossRef] [PubMed]
- Bischof, L.J.; Kao, C.Y.; Los, F.C.; Gonzalez, M.R.; Shen, Z.; Briggs, S.P.; van der Goot, F.G.; Aroian, R.V. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Hail, N., Jr.; Carter, B.Z.; Konopleva, M.; Andreeff, M. Apoptosis effector mechanisms: A requiem performed in different keys. Apoptosis 2006, 11, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Braun, P.; de Groot, A.; Bitter, W.; Tommassen, J. Secretion of elastinolytic enzymes and their propeptides by pseudomonas aeruginosa. J. Bacteriol. 1998, 180, 3467–3469. [Google Scholar] [PubMed]
- Joza, N.; Kroemer, G.; Penninger, J.M. Genetic analysis of the mammalian cell death machinery. Trends Genet. 2002, 18, 142–149. [Google Scholar] [CrossRef]
- McCall, K. Genetic control of necrosis—Another type of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Fancy, R.M.; Wang, L.; Schmid, T.; Zeng, Q.; Wang, H.; Zhou, T.; Buchsbaum, D.J.; Song, Y. Characterization of the interactions between calmodulin and death receptor 5 in triple-negative and estrogen receptor-positive breast cancer cells: An integrated experimental and computational study. J. Biol. Chem. 2016, 291, 12862–12870. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Juarbe, N.; Bradley, K.M.; Shenoy, A.T.; Gilley, R.P.; Reyes, L.F.; Hinojosa, C.A.; Restrepo, M.I.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 2017, 24, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Kepp, O.; Krautwald, S.; Kroemer, G.; Linkermann, A. Molecular mechanisms of regulated necrosis. Semin. Cell Dev. Biol. 2014, 35, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.E.; Kim, H.K.; Wang, Y.; Bubeck Wardenburg, J. Adam10 mediates vascular injury induced by staphylococcus aureus alpha-hemolysin. J. Infect. Dis. 2012, 206, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.A.; Bubeck Wardenburg, J. Role of a disintegrin and metalloprotease 10 in staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef] [PubMed]
- Nagano, O.; Murakami, D.; Hartmann, D.; De Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell-matrix interaction via cd44 is independently regulated by different metalloproteinases activated in response to extracellular Ca2+ influx and pkc activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Ponnuchamy, B.; Khalil, R.A. Role of ADAMs in endothelial cell permeability: Cadherin shedding and leukocyte rolling. Circ. Res. 2008, 102, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Dreymueller, D.; Pruessmeyer, J.; Groth, E.; Ludwig, A. The role of ADAM-mediated shedding in vascular biology. Eur. J. Cell Biol. 2012, 91, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Pruessmeyer, J.; Ludwig, A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Biol. 2009, 20, 164–174. [Google Scholar] [CrossRef] [PubMed]
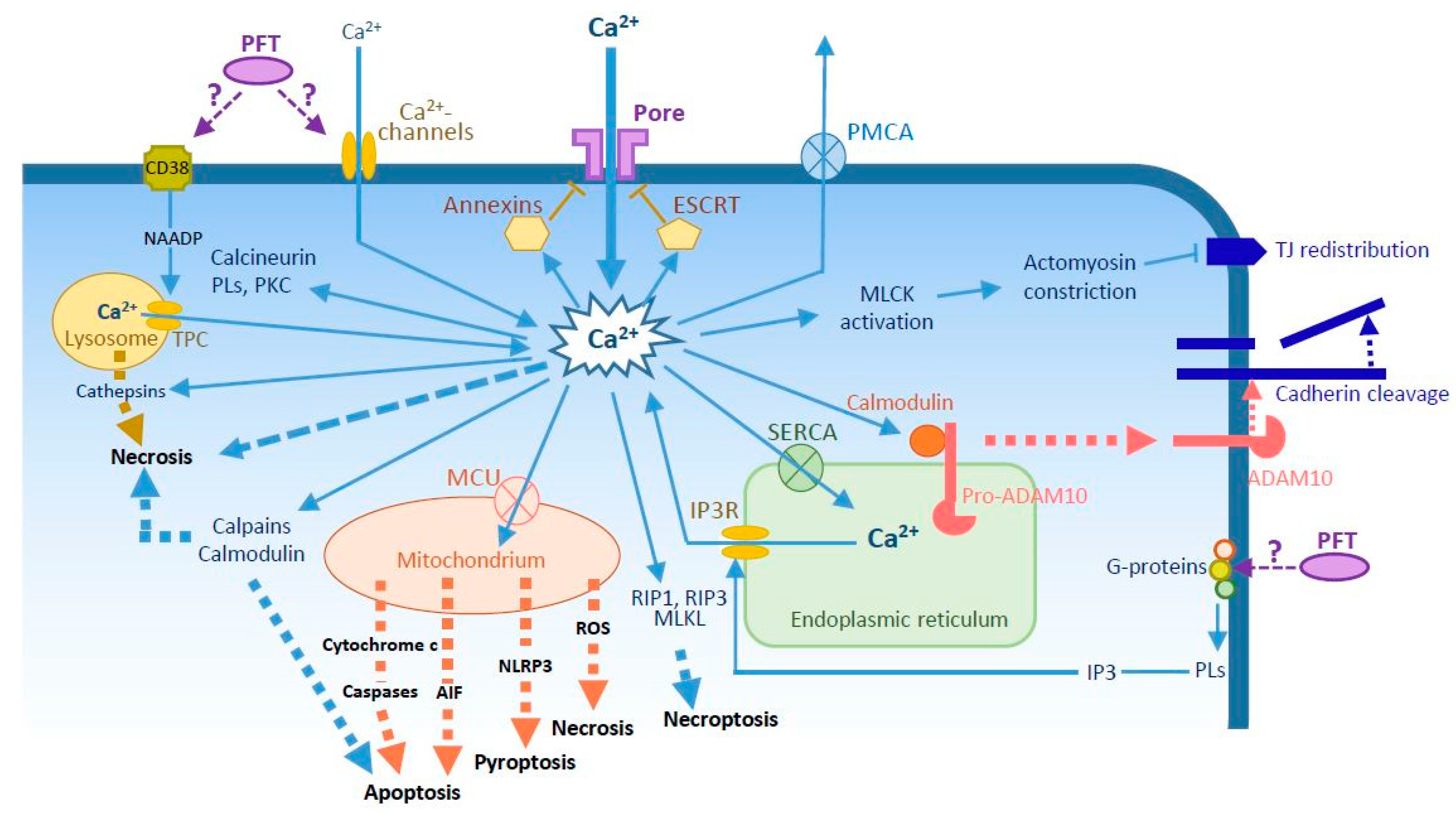
Figure 1.
Potential Ca2+ circuitry induced by pore-forming toxins (PFTs) and main reported effects of sustained Ca2+ elevation. Increased cytosolic Ca2+ concentrations can be induced by passive flow through the pore and/or activation of Ca2+-channels either in the plasma membrane, in the endoplasmic reticulum (ER) (IP3R, via G-proteins-PLs-IP3 pathway), or in the lysosome (TPC, via CD38-NAADP pathway). Ca2+ pumps in the plasma membrane (PMCA), the ER (SERCA) and the mitochondria (MCU) are employed to maintain low levels of cytosolic Ca2+. Ca2+ binds and activates members of annexin family to promote pore endocytosis, or the ESCRT complex for microvesicle secretion. Ca2+ interacts also with calmodulin, which detaches from pro-ADAM10, allowing its maturation and export to the plasma membrane where it cleaves cadherins. Ca2+ activates a number of proteins, including MLCK, which promotes actomyosin constriction and TJ protein redistribution from the junction. Ca2+ intoxication activates several cell death pathways: (i) necrosis can be induced by osmotic lysis, by activated calmodulin and calpains, by release of cathepsins from lysosomes or ROS from mitochondria; (ii) apoptosis by release of AIF and cytochrome c from mitochondria or by activated calpains and calmodulin; (iii) NLRP3-dependent pyroptosis from mitochondrial signals; and (iv) necroptosis, by activation of RIP1, RIP3, and MLKL. Abbreviations: AIF, apoptosis-inducing factor; ESCRT, endosomal sorting complex required for transport; IP3R, inositol triphosphate receptor; MLCK, myosin light chain-kinase; MCU, mitochondrial Ca2+ uniporter; MLKL, mixed-lineage kinase domain-like protein; NAADP, nicotinic acid adenine dinucleotide phosphate; PKC, protein kinase C; PL, phospholipase; PMCA, plasma membrane Ca2+-ATPase; RIP, receptor interacting protein; ROS, reactive oxygen species; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; TJ, tight junction; TPC, two-pore channel.
Figure 1.
Potential Ca2+ circuitry induced by pore-forming toxins (PFTs) and main reported effects of sustained Ca2+ elevation. Increased cytosolic Ca2+ concentrations can be induced by passive flow through the pore and/or activation of Ca2+-channels either in the plasma membrane, in the endoplasmic reticulum (ER) (IP3R, via G-proteins-PLs-IP3 pathway), or in the lysosome (TPC, via CD38-NAADP pathway). Ca2+ pumps in the plasma membrane (PMCA), the ER (SERCA) and the mitochondria (MCU) are employed to maintain low levels of cytosolic Ca2+. Ca2+ binds and activates members of annexin family to promote pore endocytosis, or the ESCRT complex for microvesicle secretion. Ca2+ interacts also with calmodulin, which detaches from pro-ADAM10, allowing its maturation and export to the plasma membrane where it cleaves cadherins. Ca2+ activates a number of proteins, including MLCK, which promotes actomyosin constriction and TJ protein redistribution from the junction. Ca2+ intoxication activates several cell death pathways: (i) necrosis can be induced by osmotic lysis, by activated calmodulin and calpains, by release of cathepsins from lysosomes or ROS from mitochondria; (ii) apoptosis by release of AIF and cytochrome c from mitochondria or by activated calpains and calmodulin; (iii) NLRP3-dependent pyroptosis from mitochondrial signals; and (iv) necroptosis, by activation of RIP1, RIP3, and MLKL. Abbreviations: AIF, apoptosis-inducing factor; ESCRT, endosomal sorting complex required for transport; IP3R, inositol triphosphate receptor; MLCK, myosin light chain-kinase; MCU, mitochondrial Ca2+ uniporter; MLKL, mixed-lineage kinase domain-like protein; NAADP, nicotinic acid adenine dinucleotide phosphate; PKC, protein kinase C; PL, phospholipase; PMCA, plasma membrane Ca2+-ATPase; RIP, receptor interacting protein; ROS, reactive oxygen species; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; TJ, tight junction; TPC, two-pore channel.


{kind=link}
Table 1.
PFTs reported to promote increases in intracellular Ca2+ concentrations.
| Species | Toxin Name 1 | Pore Size 2 | Ca2+ Origin 3 | Ca2+ Kinetics | Reported Effects of PFT-Induced Ca2+ Influx | Refs |
|---|---|---|---|---|---|---|
| Actinobacillus actinomycetemcomitans | Ltx | n. d. | EC | Monophasic | ∙ Neutrophil lysis | [27] |
| Aeromonas hydrophila | Aerolysin | Small | EC + IC | Multiphasic | ∙ Granulocyte chemotaxis ∙ T cell apoptosis ∙ Actomyosin contraction and tight junction disruption | [12,28,29,30] |
| Aeromonas sobria | ASH | Small | EC + IC | Biphasic | [15] | |
| Bordetella pertussis | ACT = CyaA | Small | EC | Multiphasic via non-voltage dependent channels with L-type properties | ∙ Prevents ACT endocytosis and degradation | [31,32] |
| Clostridium perfringens | PFO | Large | EC | Unknown | ∙ Activates/enhances repair mechanism | [33] |
| CPE | Small | Unknown | Biphasic | ∙ Apoptosis and necrosis through calpain and calmodulin-dependent processes | [34] | |
| ET | Small | EC | Monophasic | [35,36] | ||
| Clostridium septicum | α-toxin | Small | EC | Biphasic | ∙ Necrosis induced by multiple pathways | [37] |
| Escherichia coli | HlyA | Small | EC | Oscillations due to Ca2+ channel activation or to rapid formation/closure of the pore | ∙ ROS production by granulocytes ∙ IL-6 and IL-8 production by epithelal cells | [38,39,40] |
| ClyA = HlyE | Small | IC | Oscillations | [41] | ||
| Listeria monocytogenes | LLO | Large | EC IC via G-protein activation-IP3 production | Oscillation due to rapid formation/closure of the pore and release from IC stores | ∙ Bacterial internalization ∙ Mast cell degranulation and cytokine synthesis ∙ Immune cell desensitization | [8,9,42,43,44] |
| Pasteurella hemolytica | LKT | n. d. | EC through voltage-gated Ca2+ channels | Monophasic | ∙ ROS and leukotriene production by neutrophils ∙ Cytokine release from macrophages | [7,10,45,46,47] |
| Pseudomonas aeruginosa | ExlA | Small | EC | Biphasic | ∙ Cadherin cleavage via ADAM10 activation ∙ Necrosis | [48] |
| Photobacterium damselae | PhlyP | Small | Monophasic | ∙ Lysosomal exocytosis | [18] | |
| Serratia marcescens | ShlA | Small | EC | Monophasic | ∙ Cadherin cleavage via ADAM10 activation ∙ Necrosis | [48] |
| Staphylococcus aureus | Hla = α-toxin | Small | EC | Monophasic | ∙ PLA2 activation ∙ Cadherin cleavage through ADAM10 activation | [49,50,51,52,53] |
| Hlg | Small | IC from lysosomes and endoplasmic reticulum EC from store-operated channels | Mono/biphasic | [11,14] | ||
| PVL | Small | As for Hlg | Mono/biphasic | [11,14] | ||
| Streptococcus intermedius | ILY | Large | Unknown | Unknown | ∙ NFAT activation and EGR-1 expression via Ca2+/calcineurin pathway ∙ Activation/enhancement of repair mechanism | [33,54] |
| Streptococcus pneumoniae | PLY | Large | EC | Multiphasic | ∙ Apoptosis ∙ IL-8 production via NFκB activation ∙ Cadherin cleavage through ADAM10 activation ∙ Activation/enhancement of repair mechanism ∙ Platelet activation ∙ NFκB-dependent IL-8 synthesis | [51,55,56,57,58] |
| Streptococcus pyogenes | SLO | Large | EC + IC | Monophasic | ∙ Granulocyte chemotaxis ∙ Keratinocyte apoptosis and ER vacuolation ∙ Membrane repair | [12,33,59,60] |
1 Ltx, leukotoxin; ASH, A. sobria hemolysin; ACT (or CyaA), adenylate cyclase toxin-hemolysin; PFO, perfringolysin O; CPE, C. perfringens enterotoxin; ET, epsilon toxin; HlyA, hemolysin-α; ClyA (or HlyE), cytolysin A; LLO, lysteriolysin O; LKT, leukotoxin A; ExlA, exolysin A; PhlyP, phobalysin; ShlA, Serratia hemolysin A; HlA, hemolysin-α; Hlg, hemolysin-γ; PVL, Panton–Valentine leukocidin; ILY, intermedilysin; PLY, pneumolysin; SLO, streptolysin O. 2 Internal pore diameter. Small: 1–2 nm; Large: up to 30 nm. n. d., not detrmined. 3 EC, from the extracellular milieu; IC, from intracellular stores; “Ca2+ channels” indicates the activation of cellular Ca2+ channels without or in addition to Ca2+ influx through the PFT.
Table 2.
Cellular death programs triggered by PFT-induced Ca2+ concentration rise.
| Pore-Forming Toxins 1 (Species) | Apoptosis | Necrosis | Necroptosis | Ref. |
|---|---|---|---|---|
| Ltx (A. actinomycetemcomitans) | In T cells. Possibly calpain-dependent | [61] | ||
| Aerolysin (A. hydophila) | At low dose in T cells | [29] | ||
| CPE (C. perfringens) | At low dose in enterocytes | At high dose in enterocytes | [34] | |
| ET (C. perfringens) | In renal collecting duct cells | [35] | ||
| α-toxin (C. septicum) | In myoblasts | [37] | ||
| PLY (S. pneumoniae) | In microglial cells | In pneumocytes | [55,83] | |
| SLO (S. pyogenes) | At low dose in keratinocytes | At high dose in keratinocytes | [59] | |
| ShlA (S. marcescens) | In pneumocytes | [83] |
1 Abbreviations as in Table 1.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bouillot, S.; Reboud, E.; Huber, P. Functional Consequences of Calcium Influx Promoted by Bacterial Pore-Forming Toxins. Toxins 2018, 10, 387. https://doi.org/10.3390/toxins10100387
AMA Style
Bouillot S, Reboud E, Huber P. Functional Consequences of Calcium Influx Promoted by Bacterial Pore-Forming Toxins. Toxins. 2018; 10(10):387. https://doi.org/10.3390/toxins10100387
Chicago/Turabian StyleBouillot, Stéphanie, Emeline Reboud, and Philippe Huber. 2018. "Functional Consequences of Calcium Influx Promoted by Bacterial Pore-Forming Toxins" Toxins 10, no. 10: 387. https://doi.org/10.3390/toxins10100387
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.