2.1. Production and characterization of D. septosporum dotC mutants and complemented strains
D. septosporum strains transformed with a
dotC replacement construct (pR260) were selected by hygromycin resistance. Two out of 26 hygromcyin resistant colonies (named FJT15 and FJT16) showed correct targeted replacement of the
dotC gene when tested by PCR (results not shown) and Southern blotting (
Figure 1). Other transformants had ectopic integration of the construct or vector integration only in the 3' region of
dotC. The Southern blot also confirmed that there is only one copy of
dotC in the
D. septosporum genome. The
dotC mutant FJT15 was subsequently transformed with a complementation construct (pR282) to restore
dotC function and integration of the construct in the complemented strain FJT93 was confirmed by PCR (results not shown). The copy number of
dotC in the complemented strain was estimated by real-time PCR to be 10 copies, making FJT93 a DotC-overproducing strain.
No significant difference was seen between sporulation of the dotC mutants FJT15 (1.48 ± 0.32 ± 106 spores/mL; P = 0.725) and FJT16 (1.17 ± 0.29 ± 106 spores/mL; P = 0.706) compared to the wild type NZE10 (1.32 ± 0.29 ± 106 spores/mL). Likewise the radial growth rate of the dotC mutants was similar to that of the wild type with FJT15 (0.60 ± 0.03 mm/day; P = 0.092) and FJT16 (0.58 ± 0.02 mm/day; P = 0.182) not significantly different from the wild type (0.54 ± 0.02 mm/day). The dotC-complemented strain FJT93 grew slightly slower than the wild-type but in these experiments the difference was not significant (0.50 ± 0.02 mm/day; P = 0.119).
Figure 1.
KpnI-digested genomic DNA of D. septosporum wild-type (lanes 1 & 2) or transformed with pR260 (lanes 3-8) and hybridized with a dotC coding region probe. KpnI sites flank the dotC gene, hence replacement of a 1.4 kb central portion of dotC with a 2.4 kb hygromycin resistance gene cassette increased the dotC-containing KpnI fragment size from 3.5 kb (wild-type) to 4.5 kb (dotC replacement). The dotC replacement mutants FJT16 and FJT15 are shown in lanes 3 and 8 respectively. Lane 6 contains another dotC mutant that had an additional ectopic integration of the pR260 plasmid.
Figure 1.
KpnI-digested genomic DNA of D. septosporum wild-type (lanes 1 & 2) or transformed with pR260 (lanes 3-8) and hybridized with a dotC coding region probe. KpnI sites flank the dotC gene, hence replacement of a 1.4 kb central portion of dotC with a 2.4 kb hygromycin resistance gene cassette increased the dotC-containing KpnI fragment size from 3.5 kb (wild-type) to 4.5 kb (dotC replacement). The dotC replacement mutants FJT16 and FJT15 are shown in lanes 3 and 8 respectively. Lane 6 contains another dotC mutant that had an additional ectopic integration of the pR260 plasmid.
2.2. Resistance of mutants to exogenous dothistromin
To determine if DotC confers a protective effect against exogenously supplied toxin, the ability of
dotC mutants to grow in the presence of dothistromin was investigated (
Table 1). Although there was some variability in growth rates, as often seen with
D. septosporum, none of the strains appeared to be strongly inhibited by even the highest concentration of dothistromin. Colonies on 100 μM dothistromin grew to >96% of the diameter of colonies grown without dothistromin. This concentration of dothistromin is in excess of levels usually secreted by the strains (5-11 μM for NZE10 and 20-40 μM secreted by FJT93). Hence it appears that DotC is not required for resistance to dothistromin in
D. septosporum. Similarly in
C. nicotianae CTB4 mutants showed normal levels of resistance to exogenously supplied cercosporin, although disruption of other putative toxin pump genes in
Cercospora spp. (
CFP in
C. kikuchii and
ATR1 and
CnCFP in
C. nicotianae) had some effect on cercosporin resistance [
13,
14,
15,
16,
17].
In these experiments using a total diameter calculation the complemented (DotC-overproducing) FJT93 strain did show significantly slower growth than the wild type (P < 0.01) in all concentrations of dothistromin, in contrast to the earlier study.
Table 1.
Growth of D. septosporum in the presence of exogenous dothistromin (doth). Mean radial growth (mm) ± standard error shown for two independent experiments each with 10 replicates. *Significant differences between growth of each strain with dothistromin compared to without dothistromin (P < 0.05) are indicated by asterisks.
Table 1.
Growth of D. septosporum in the presence of exogenous dothistromin (doth). Mean radial growth (mm) ± standard error shown for two independent experiments each with 10 replicates. *Significant differences between growth of each strain with dothistromin compared to without dothistromin (P < 0.05) are indicated by asterisks.
| Strain | 0 μM doth | 20 μM doth | 40 μM doth | 100 μM doth |
|---|
| NZE10 (wildtype) | 14.7 ± 0.19 | 14.6 ± 0.27 | 14.0 ± 0.11* | 14.2 ± 0.12* |
| FJT15 (ΔdotC) | 15.5 ± 0.43 | 16.1 ± 0.30 | 16.1 ± 0.36 | 16.7 ± 0.33* |
| FJT16 (ΔdotC) | 17.3 ± 0.29 | 16.6 ± 0.23 | 16.4 ± 0.25* | 16.9 ± 0.20 |
| FJT93 (ΔdotC +dotC) | 13.4 ± 0.21 | 12.6 ± 0.14* | 13.2 ± 0.18 | 13.3 ± 0.21 |
2.3. Dothistromin production by dotC disruption mutants
Amounts of dothistromin secreted into broth, and remaining in the mycelium, were determined by ELISA following a standardised extraction procedure (
Table 2). Strikingly, the
dotC disruption mutants (FJT15 and FJT16) secreted only 4-7% of the amount of dothistromin secreted by the wild type NZE10 strain. The amounts of dothistromin in the mycelia were also lower in these mutants than the wild-type, but these differences were not significant, possibly due to large variation between replicates.
It is possible that the lower levels of secreted dothistromin in the
dotC mutants were a direct consequence of overall lower levels of dothistromin production. The amounts of dothistromin produced by the mutants (mycelium and broth combined) were only 8.5% and 4.4% of wild type levels for the FJT15 and FJT16 mutants respectively. However the right-hand column in
Table 2 suggests a small, but not significant, difference in the percentage of dothistromin secreted: in the wild type over 90% of all dothistromin was secreted whilst in the
dotC knockout mutants only 80-83% was secreted. Thus there is only weak evidence for a role in dothistromin transport for DotC.
Table 2.
Dothistromin as measured by ELISA from broth (secreted dothistromin) and mycelium (not secreted) extracted from the same flasks using the same extraction method (n = 3). For mycelium the amounts shown are dothistromin per mg dry weight. For broth the amounts of dothistromin have been calculated to show how much dothistromin was secreted into the broth by each mg of mycelium in the flask. *Significant differences from NZE10 values (P < 0.05) are indicated by an asterisk.
Table 2.
Dothistromin as measured by ELISA from broth (secreted dothistromin) and mycelium (not secreted) extracted from the same flasks using the same extraction method (n = 3). For mycelium the amounts shown are dothistromin per mg dry weight. For broth the amounts of dothistromin have been calculated to show how much dothistromin was secreted into the broth by each mg of mycelium in the flask. *Significant differences from NZE10 values (P < 0.05) are indicated by an asterisk.
| Strain | Dothistromin in broth | Dothistromin from mycelia | % Doth secreted broth/(myc + broth) |
|---|
| Dothistromin Mean ± SE (ng/mg DW) | % of WT (NZE10) | Dothistromin Mean ± SE (ng/mg DW) | % of WT (NZE10) |
| NZE10 (wildtype) | 319.8 ± 30.3 | 100 | 30.5 ± 15.3 | 100 | 91.3% |
| NZE7 (wildtype) | 249.1 ± 32.8 | 77.9 | 26.8 ± 16.6 | 88.1 | 90.3% |
| FJT15 (ΔdotC) | 23.8 ± 5.0* | 7.4 | 6.1 ± 2.2 | 20.1 | 79.5% |
| FJT16 (ΔdotC) | 12.9 ± 2.7* | 4.0 | 2.6 ± 0.6 | 8.4 | 83.4% |
| FJT93 (ΔdotC + dotC) | 1716.6 ± 507.9 | 536.8 | 1105.3 ± 348.2* | 3629.9 | 60.8%* |
The lower levels of dothistromin in the
dotC mutants strongly suggest that DotC is required for wild-type levels of dothistromin biosynthesis. DotC is a close match to MFS proteins, with no evidence for a DNA binding domain. A dramatic reduction in toxin production was also seen in CFP mutants of
Cercospora kikuchii, with less than 5% of wild type levels of cercosporin produced [
13], and also to CTB4 and ATR1 mutants of
C. nicotianae with less than 35% and 25% of wild type cercosporin respectively [
14,
17].
The observation that some dothistromin was still secreted in the
dotC mutants suggests that other transporters are involved. Multiple transporters with overlapping roles have been reported in several fungi. For example in
C. nicotianae the ABC transporter ATR1 was shown to have a major role in cercosporin production and, along with another MFS transporter CnCFP, contributed to auto-resistance to cercosporin [
17].
The dotC complemented mutant (FJT93) produced and secreted very high levels of dothistromin. The secreted levels were five-fold higher than those seen with the wild-type, but the variability between replicates was high. The amount of dothistromin secreted from FJT93 was significantly higher than the combined results from wild type strains NZE7 and NZE10 (P = 0.0037) but not compared to NZE10 alone (P = 0.0516). The levels of non-secreted dothistromin remaining in the mycelium were extremely high in FJT93: more than 30-fold higher than the wild type. It appears there was a dramatic increase in dothistromin biosynthesis in FJT93 and that the cells were unable to secrete the dothistromin as quickly as it was being made: almost 40% of the total amount of dothistromin remained in the mycelium. Since FJT93 only showed a slightly slower growth rate compared to the wild type (significant in only one of two trials) the high intracellular levels of dothistromin did not appear to be toxic. These high levels of toxin further suggest that dothistromin accumulation in the cell is not a primary feedback regulator of dothistromin biosynthesis.
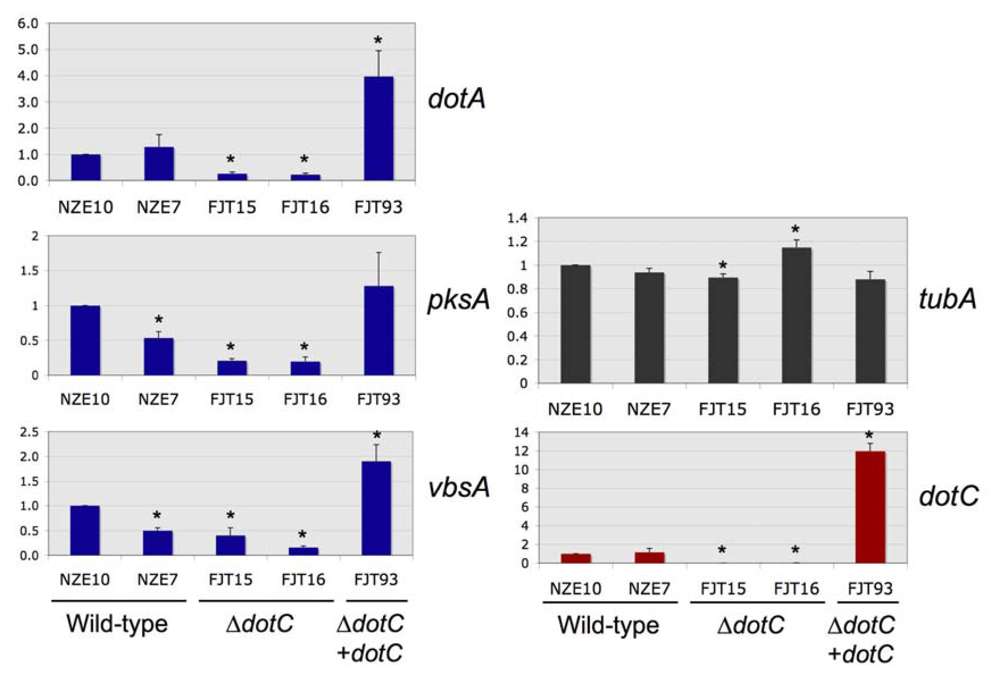
2.4. Dothistromin gene expression in dotC disruption and complemented mutants
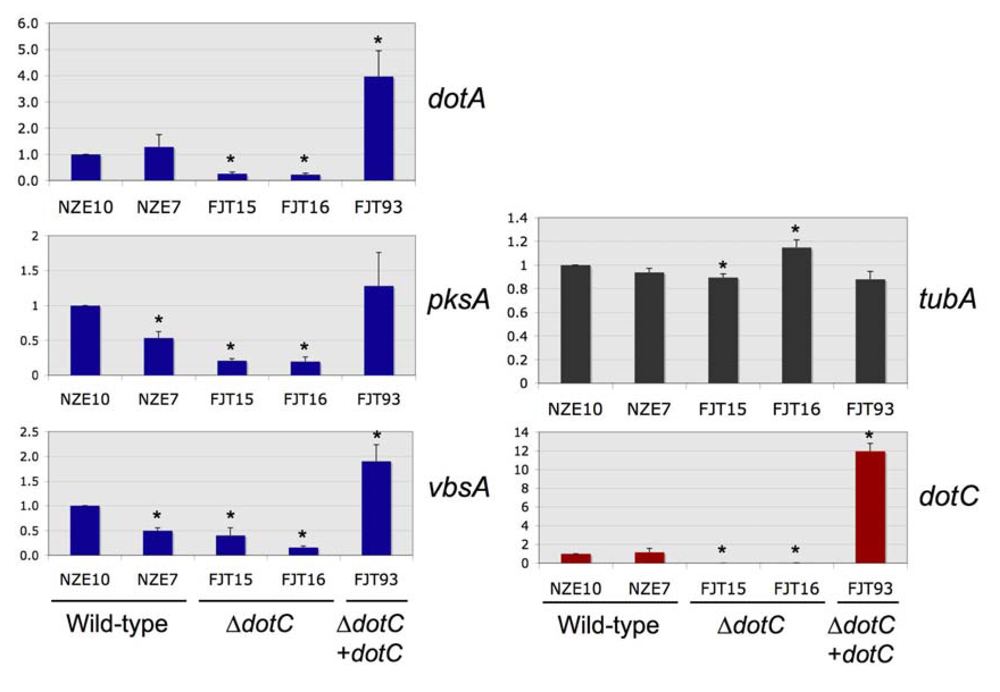
Real-time PCR was used to quantify
dotC expression in the
dotC mutants and complemented strain. As expected, the
dotC disruption mutants FJT15 & 16 showed no
dotC expression above background, as they lacked this single-copy gene. Expression of the
dotA dothistromin biosynthetic gene was significantly reduced in the
dotC mutants, to around 18-26% of levels in wild type strains (
Figure 2 and
Supplementary Table S3). It therefore seems likely that the lower levels of dothistromin made by the mutants are at least partly due to lower levels of biosynthetic enzymes. Both FJT15 and FJT16 also showed different
tubA (constitutive control) expression levels compared to the wild type, but one had a higher and one a lower level of
tubA so no consistent effects were noted. The
dotC mutants showed lower levels of expression of the dothistromin biosynthetic genes
pksA and
vbsA, but the NZE7 wild type strain also had reduced levels of these transcripts compared to the NZE10 wild type. NZE7 and NZE10, like all New Zealand isolates of
D. septosporum, are considered clonal but strain degeneration has sometimes been noted over time in axenic culture. The NZE7 strain was isolated from infected pine needles three years before NZE10 and the differences in gene expression may reflect this. Inter-strain differences were also noted between the independent
dotC mutants FJT15 and FJT16, making the
pksA and
vbsA results less reliable than those for
dotA.
Figure 2.
Expression of dotC, dothistromin genes dotA, pksA and vbsA, and beta tubulin (tubA, constitutive control) in wild type, dotC knockout (∆dotC) and complemented (∆dotC + dotC) strains. 18S ribosomal RNA was used as a reference gene for standardisation. Values are normalised expression ratios relative to the NZE10 wild type, shown as mean ± standard error (n = 6). *Significant differences from NZE10 (P < 0.05) are shown by an asterisk.
Figure 2.
Expression of dotC, dothistromin genes dotA, pksA and vbsA, and beta tubulin (tubA, constitutive control) in wild type, dotC knockout (∆dotC) and complemented (∆dotC + dotC) strains. 18S ribosomal RNA was used as a reference gene for standardisation. Values are normalised expression ratios relative to the NZE10 wild type, shown as mean ± standard error (n = 6). *Significant differences from NZE10 (P < 0.05) are shown by an asterisk.
In
C. nicotianae, although disruption of the
CTB4 MFS gene led to significantly lower levels of cercosporin biosynthesis compared to the wild type, expression of the four cercosporin biosynthetic genes tested was not noticeably reduced in northern blots [
14]. Likewise disruption of the
aflT gene in
A. parasiticus had no effect on the expression of aflatoxin biosynthetic genes, as well as no effect on aflatoxin production [
16].
In contrast to the dotC mutants, the complemented FJT93 strain that contained multiple copies of dotC produced 12-fold more dotC transcript than the wild type, suggesting over-expression of DotC protein. It is possible that the dotC complementation construct may have integrated into a site that affects expression of dothistromin genes. However the increased gene expression is consistent with a role for DotC in regulating dothistromin biosynthesis and with the decline in dothistromin production seen in the dotC disruption mutants. Further to this, high intracellular concentrations of dothistromin in a strain that appears to make an excess of DotC adds support to the hypothesis that DotC is not the major efflux pump for dothistromin and suggests that there are other factors besides DotC limiting secretion.
The FJT93 complemented strain also over-expressed two of the dothistromin biosynthetic genes (
dotA and
vbsA), although levels were only 2-4 fold higher than the wild type. The fact that
pksA gene expression was not significantly increased in the DotC and dothistromin over-expressing mutant is interesting because
pksA encodes a polyketide synthase required for an early step of dothistromin biosynthesis. Since the
pksA gene is essential for dothistromin production [
20] this result suggests that PksA is not a major rate-limiting enzyme in dothistromin biosynthesis.
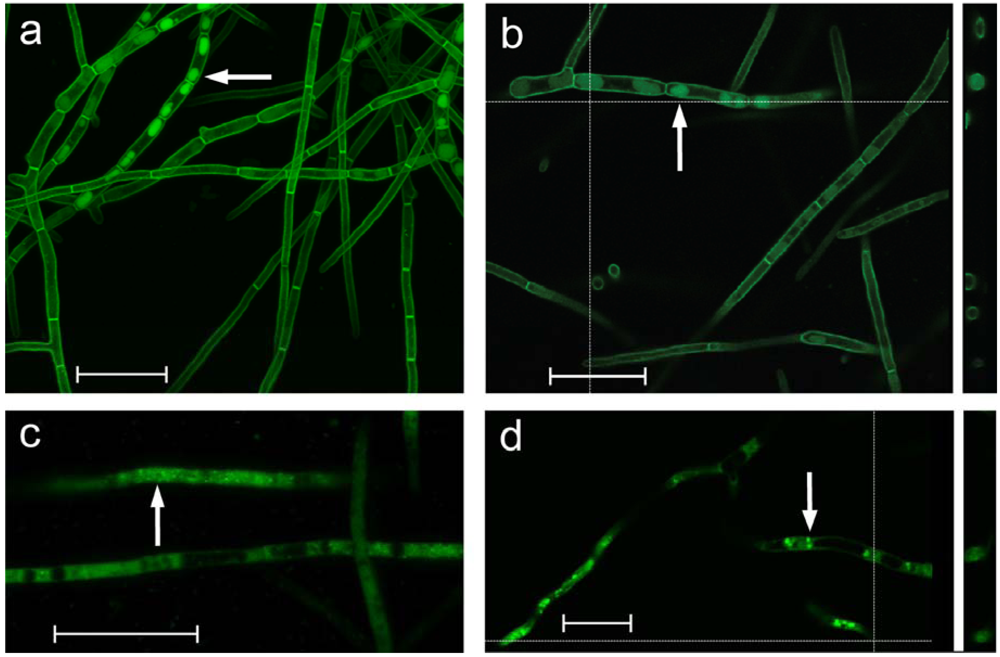
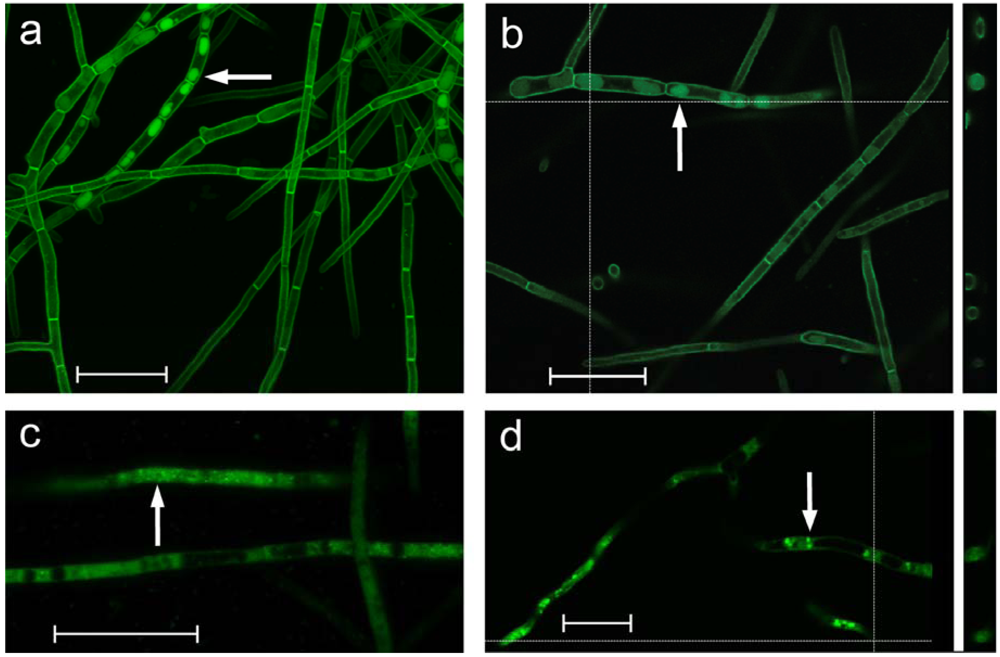
2.5. Cellular localization of DotC-GFP and DotA-GFP fusion proteins
Confocal microscopy showed that the DotC-GFP fusion protein was located around the periphery of the cells (
Figure 3), as would be expected for an MFS transporter. This is consistent with a role in secretion of dothistromin. However the images also revealed DotC-GFP localization in some intracellular regions. Cross-sections through these suggested that DotC-GFP was not confined to the perimeter but seen throughout. These GFP-containing regions varied in intensity and size but were reminiscent of vacuoles containing the aflatoxin pathway enzymes Ver-1 or Nor-1 fused to GFP [
21,
22]. Under aflatoxin-inducing conditions in
A. parasiticus a large number of vesicles accumulated that were heterogeneous in both density and size, and a functional link of vesicles (defined as vacuolar-type structures with diameters < 2.5 μm) and vacuoles with aflatoxin biosynthesis was proposed [
23].
Cellular localisation of the DotA dothistromin biosynthetic enzyme was also studied in a
D. septosporum strain expressing a DotA-GFP fusion. The DotA-GFP fusion protein was predominantly cytoplasmic or located in small (
Figure 3c) or larger (
Figure 3d) vesicles, and showed similarities to the localization of GFP fusion genes of
A. parasiticus aflatoxin genes, including the DotA homolog, Ver-1 [
21,
22]. The Ver-1 fusion protein was predominantly cytoplasmic at early stages of growth under aflatoxin-inducing conditions and found in the lumen of up to 80% of vesicle-like structures and vacuoles at later stages [
21]. Studies with a Nor-1 fusion showed a similar trend [
22]. In the present study
D. septosporum cultures were observed after 4-5 days of growth (early exponential phase), so it is possible that DotA-GFP was in the early stages of moving to vacuoles.
Figure 3.
GFP fluorescence of D. septosporum transformed with dotC-gfp (a, b) or dotA-gfp (c, d). Confocal Laser Scanning Microscopy (CLSM) images on the left (a and c) are combined 0.2 μm Z-stacks. Images on the right (b and d) are single-layer snapshots, with vertical cross-section profiles illustrated on the right. White arrows indicate intracellular vesicles/vacuoles showing GFP-fluorescence. Vesicles in (c) are very small. Scale bar = 20 μm.
Figure 3.
GFP fluorescence of D. septosporum transformed with dotC-gfp (a, b) or dotA-gfp (c, d). Confocal Laser Scanning Microscopy (CLSM) images on the left (a and c) are combined 0.2 μm Z-stacks. Images on the right (b and d) are single-layer snapshots, with vertical cross-section profiles illustrated on the right. White arrows indicate intracellular vesicles/vacuoles showing GFP-fluorescence. Vesicles in (c) are very small. Scale bar = 20 μm.
Although most of the DotC-GFP protein was localized in the cell membrane and therefore could be involved in efflux from the cell, its localisation in some intracellular organelles might suggest an additional role. We propose the hypothesis that mid to late stages of dothistromin biosynthesis occur in vacuoles as proposed for aflatoxin biosynthesis [
21] and that this enables
D. septosporum to tolerate high intracellular toxin levels as seen in the over-expressing strain FJT93. We further propose the hypothesis that one function of DotC may be to transport early-stage dothistromin biosynthetic intermediates from the cytoplasm into vacuoles, thereby affecting the rate of dothistromin production. This could explain the lower levels of dothistromin produced by
dotC mutants and higher levels in the DotC over-producing strain. The CnCFP transporter in
C. nicotianae was also speculated to have a role in transport between organelles and/or transport of early pathway components within the cell [
17], although in this case no cellular localization studies were reported.
There are some anomalies in our data that make interpretation difficult. Cross-sections of vacuoles showed DotC-GFP throughout some of the vacuoles, rather than just at the periphery as would be expected if DotC has a role in intracellular transport. Furthermore the dotA-egfp gene did not function as expected: it was transformed into a dotA- dothistromin-deficient mutant strain (FJT2) and did not re-establish dothistromin production by complementation (results not shown). Therefore the observations of DotA-GFP expression might be artifacts, although the similarity to the aflatoxin biosynthesis proteins in A. parasiticus is intriguing. Further detailed studies of intracellular localization are required to determine the locations of dothistromin and the dothistromin biosynthetic enzymes, as well as the role of DotC in dothistromin biosynthesis.
According to Amnuaykanjanasin [
17], fungal transporters can be classified into three groups based on whether they have a major role in toxin efflux (group 3), self-protection (group 2) or both (group 1). In
C. nicotianae the cercosporin transporters ATR1 and CnCFP were classified as group 1 whilst CTB4, the transporter located in the cercosporin gene cluster, was group 3. On the basis of our results the
D. septosporum DotC that is encoded within the dothistromin gene cluster appears to be closest to a group 3 transporter as it may be involved in dothistromin efflux but has little or no effect on protection against exogenous dothistromin. If dothistromin is compartmentalized within the cell as hypothesized above, this may contribute to self-protection, providing one possible explanation for how the DotC-overproducing strain tolerated extremely high intracellular levels of dothistromin without any severe effects.
An additional role that has now been noted with several fungal transporters is that of regulating toxin biosynthesis [
17]. Our results clearly showed a dramatic decrease in dothistromin biosynthesis in DotC-deficient mutants and increase in a DotC-overexpressing strain. The most obvious way in which transporters might affect biosynthesis would be by a feedback mechanism due to intracellular toxin levels which would increase in mutants with deficient efflux systems. However our results suggest that a more complex type of regulation may occur. In the DotC-overproducing strain extremely high levels of dothistromin were accompanied by increased expression of dothistromin biosynthetic genes, rather than decreased expression as would be predicted from a simple dothistromin feedback regulation model. Compartmentalisation of toxin biosynthesis within the cell, as shown for
A. parasiticus [
21,
22,
23], and supported by our studies with
D. septosporum, may prove to be an important factor in regulation of toxin biosynthesis.


{kind=link}
{kind=link}
{kind=link}