Avenanthramides Prevent Osteoblast and Osteocyte Apoptosis and Induce Osteoclast Apoptosis in Vitro in an Nrf2-Independent Manner

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. AVAs Preparation and Cell Treatment
2.2. Cell Culture
2.3. RNA Extraction and Quantitative RT-PCR (qPCR)
2.4. TRAPase Staining and Osteoclast Counting
2.5. Quantification of Osteoblastic Cell Viability
2.6. Statistical Analysis
3. Results
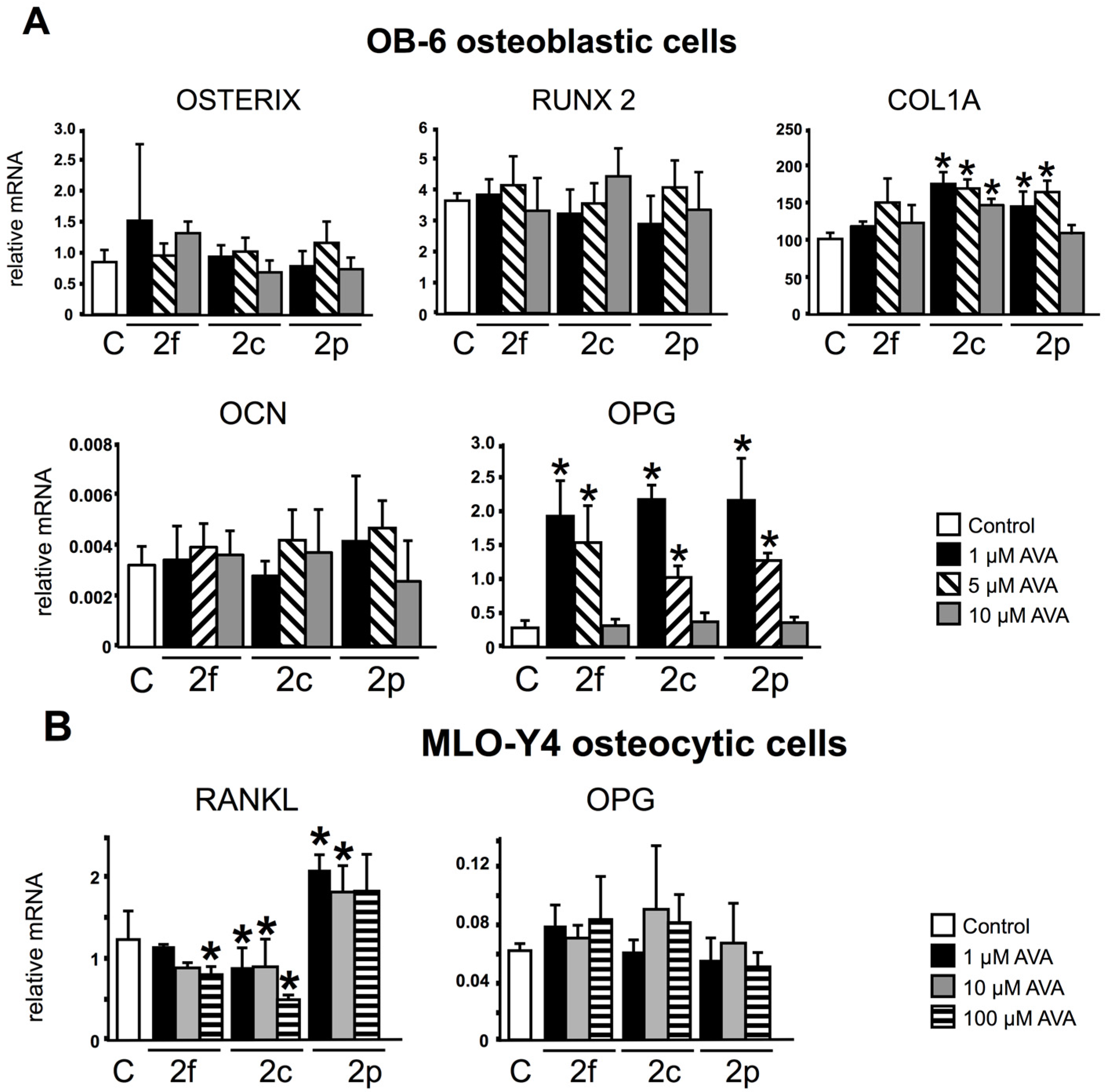
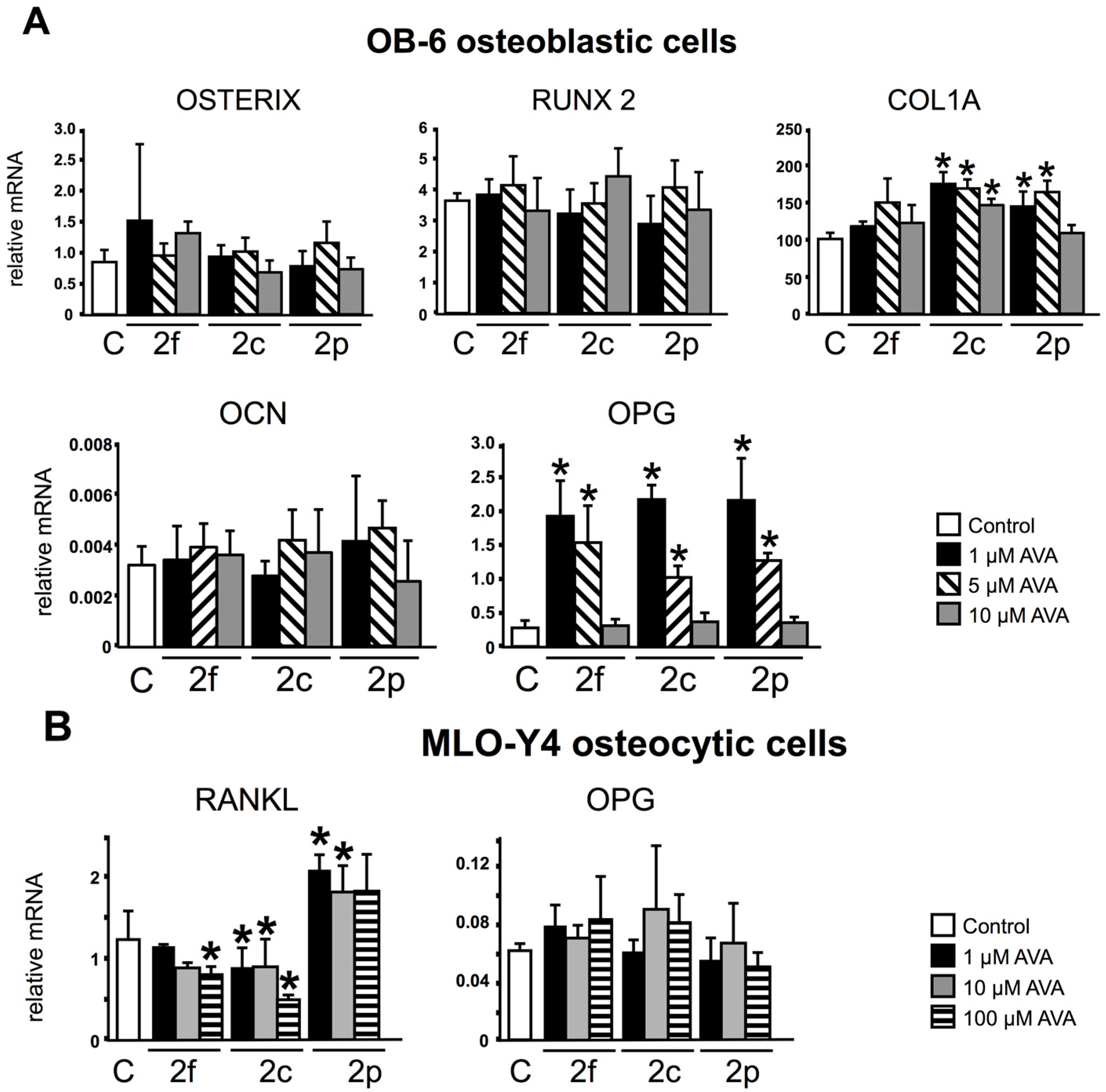
3.1. AVAs Regulate OPG and RANKL Gene Expression in OB-6 Osteoblastic and MLO-Y4 Osteocytic Cells
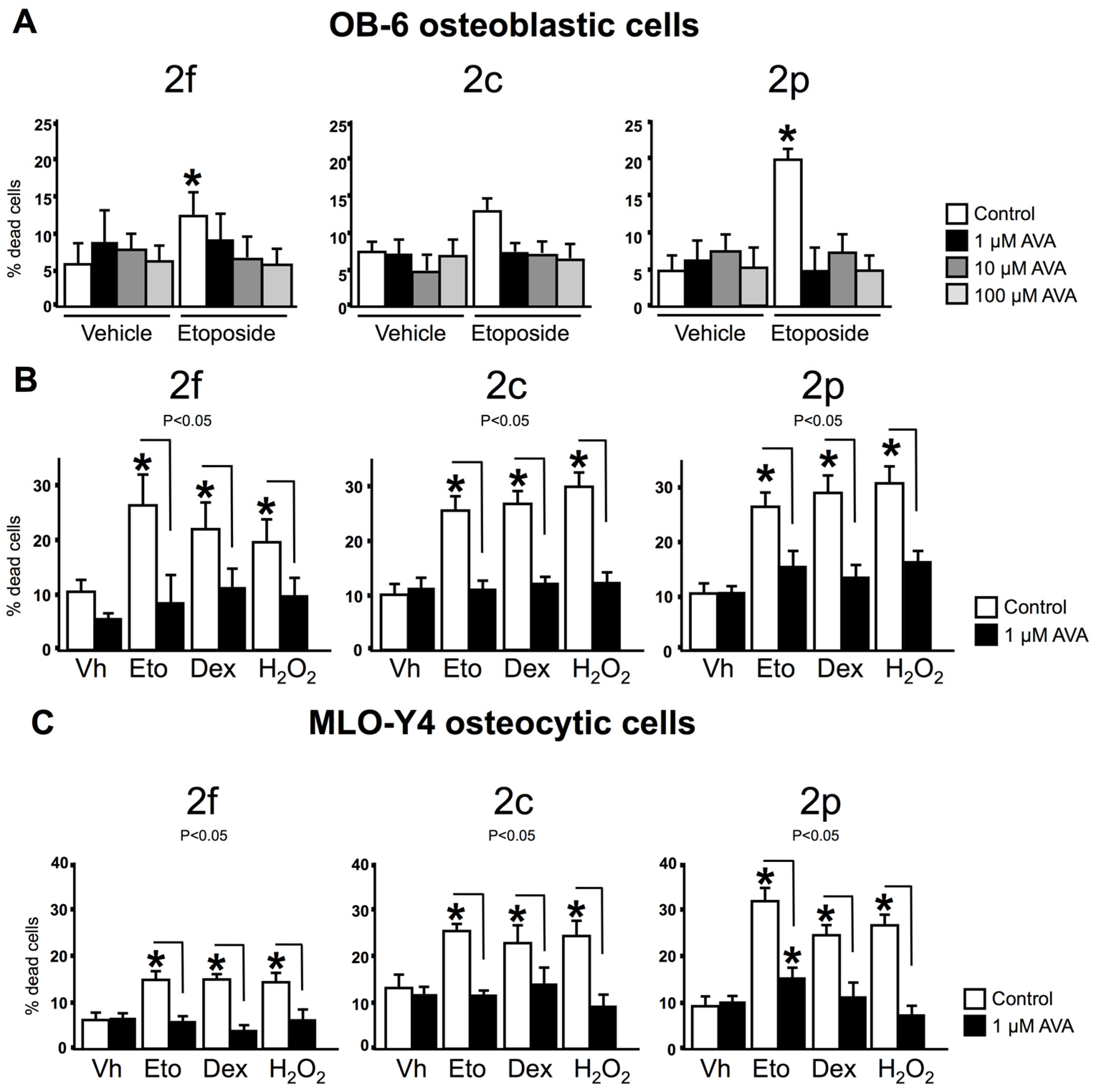
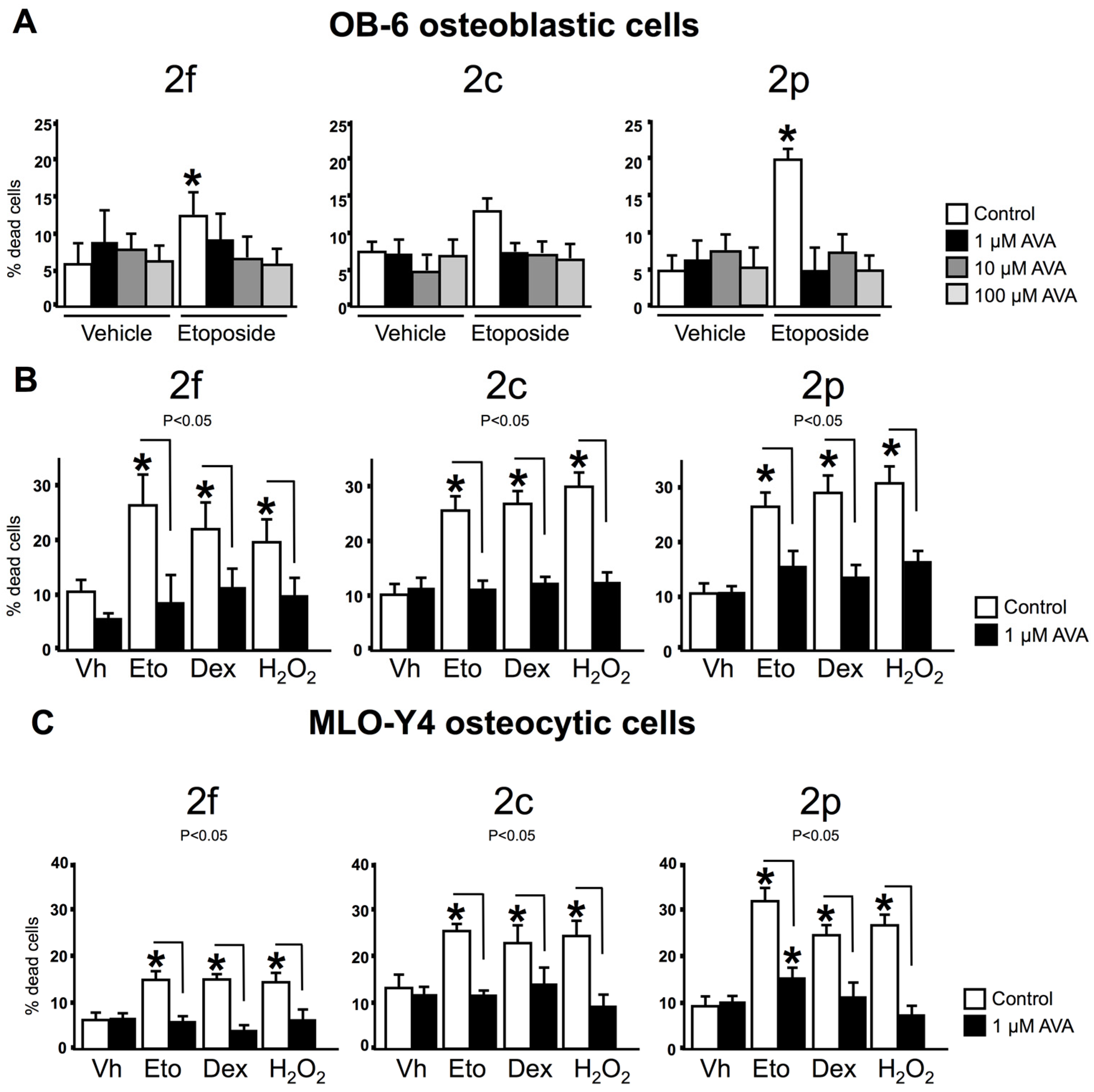
3.2. AVAs Do Not Affect Cell Death in the Absence of Pro-Apoptotic Agents but Prevent the Effect Induced by Pro-Apoptotic Agents in Ob-6 Osteoblastic and Mlo-Y4 Osteocytic Cells
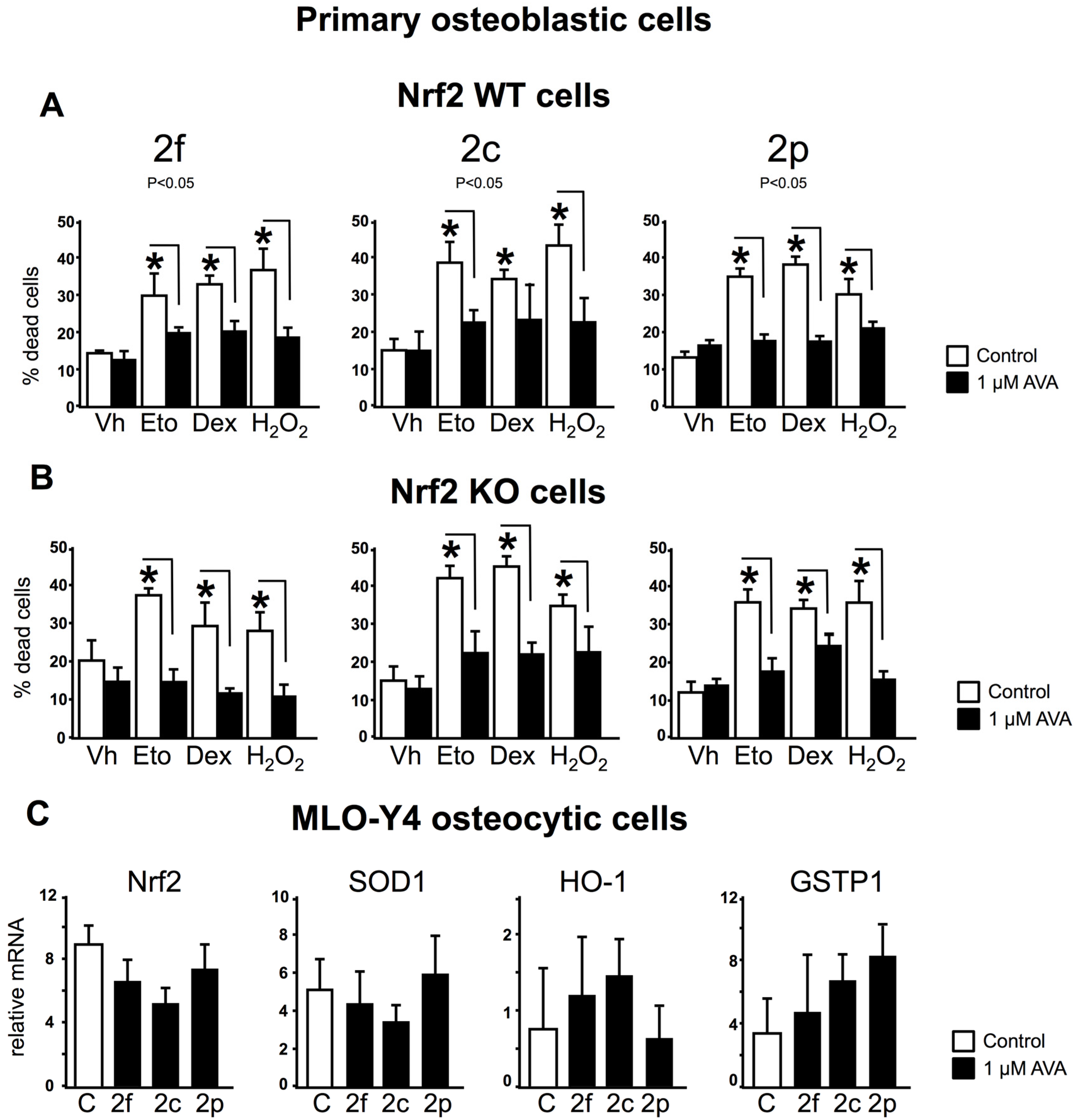
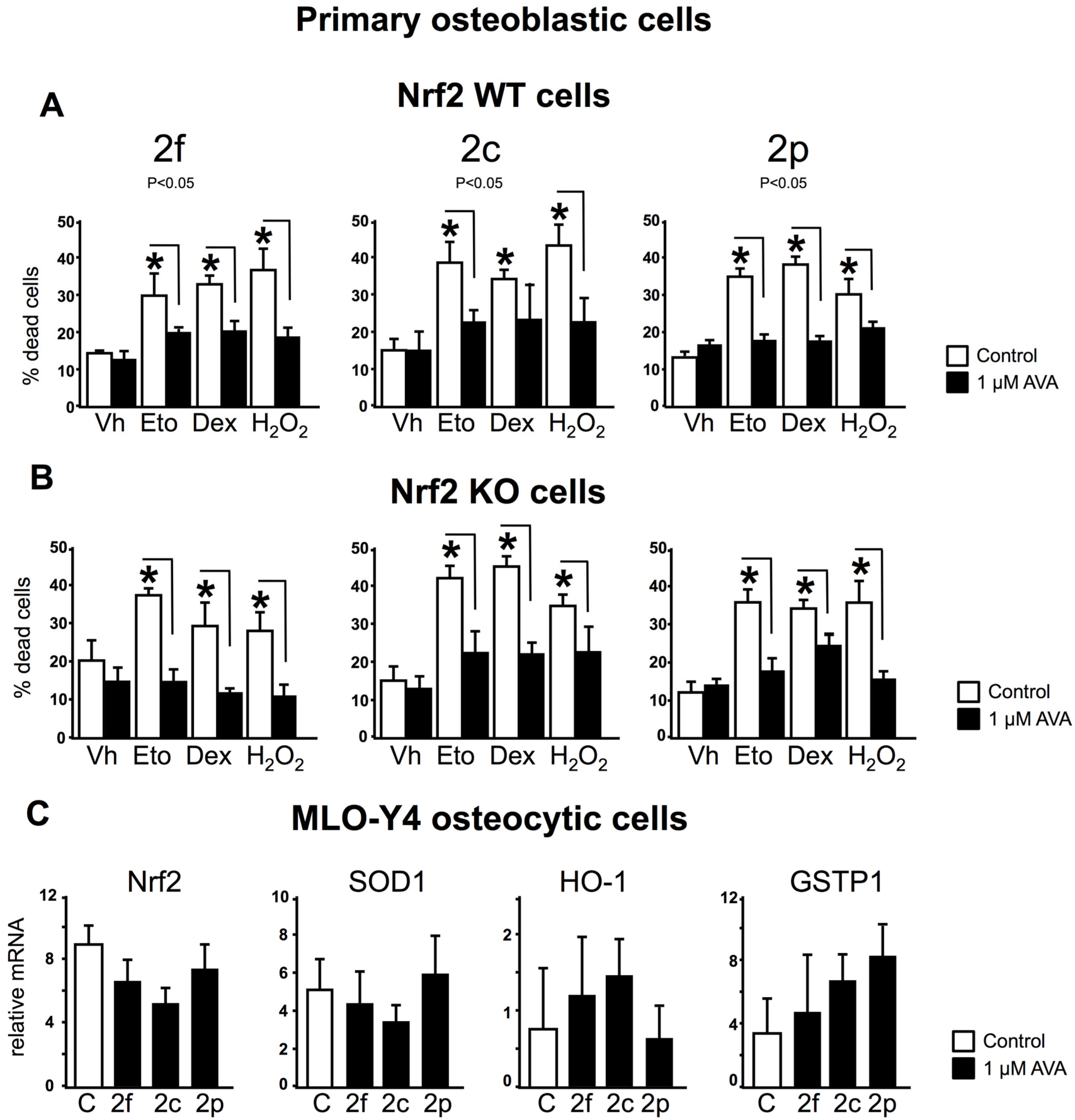
3.3. The Survival Effect of Avas in Osteoblastic Cells Does Not Require Nrf2 Expression
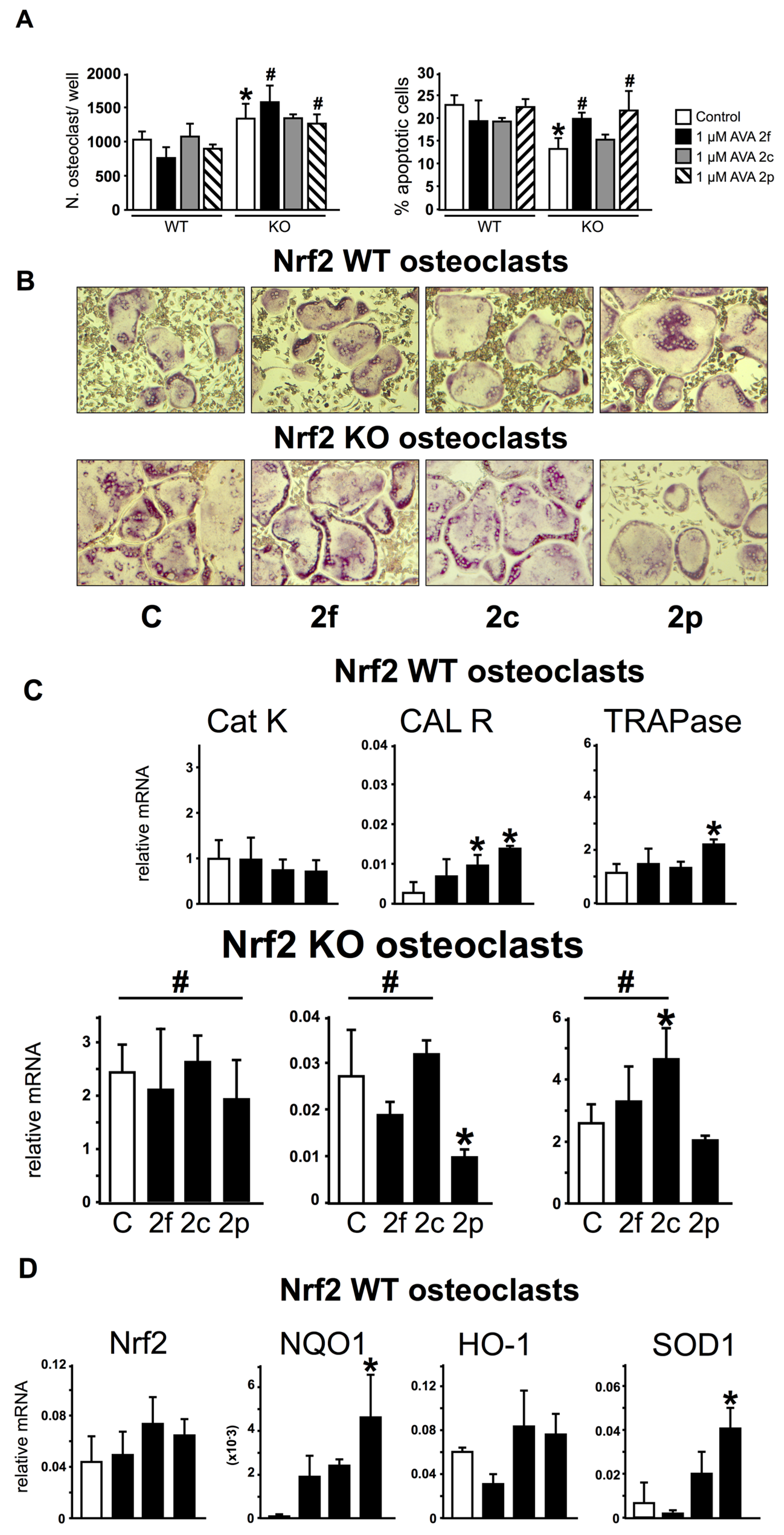
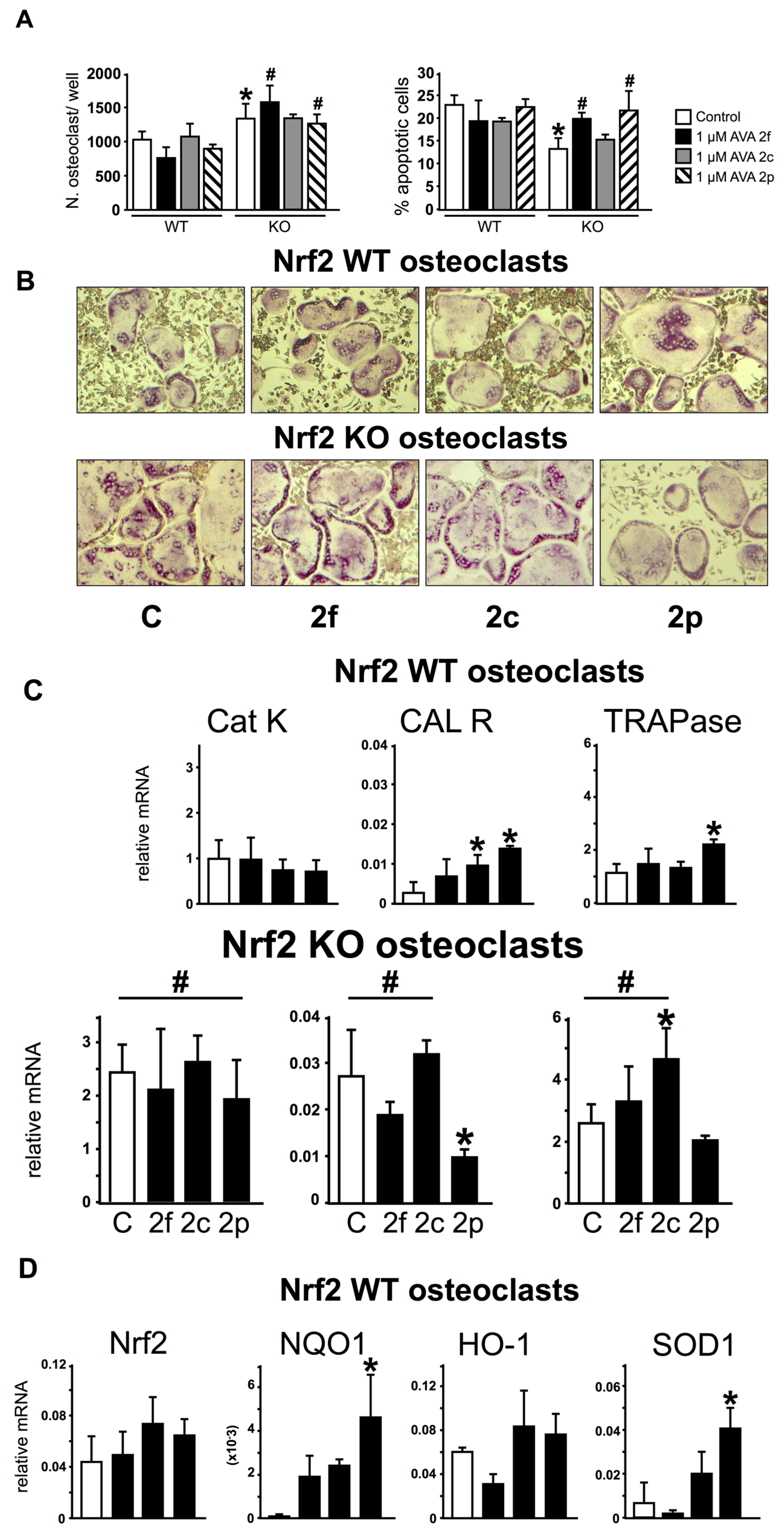
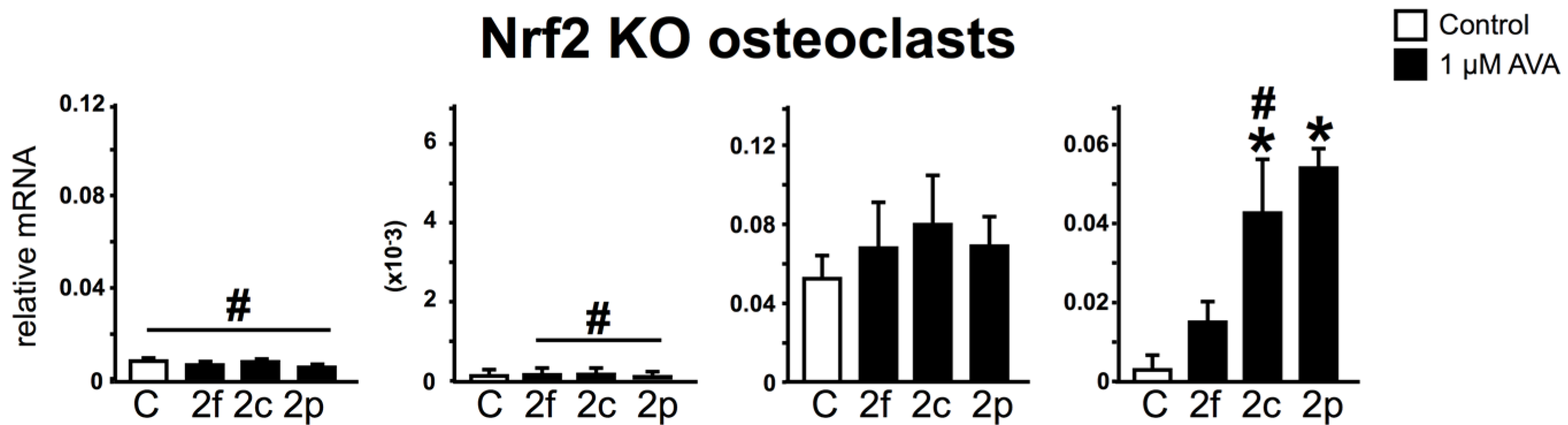
3.4. Nrf2 Is Not Required for the Regulation of Gene Expression and Survival of Osteoclasts by AVAs
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lin, H.; Gao, X.; Chen, G.; Sun, J.; Chu, J.; Jing, K.; Li, P.; Zeng, R.; Wei, B. Indole-3-carbinol as inhibitors of glucocorticoid-induced apoptosis in osteoblastic cells through blocking ROS-mediated Nrf2 pathway. Biochem. Biophys. Res. Commun. 2015, 460, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Benz, C.C.; Yau, C. Ageing, oxidative stress and cancer: Paradigms in parallax. Nat. Rev. Cancer 2008, 8, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxid. Med. Cell. Longev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M. Aging mechanisms in bone. Bonekey Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Ambrogini, E.; Weinstein, R.S.; Manolagas, S.C. Glucocorticoids and tumor necrosis factor (TNF) alpha increase oxidative stress and suppress WNT signaling in osteoblasts. J. Biol. Chem. 2011, 286, 44326–44335. [Google Scholar] [CrossRef] [PubMed]
- Callaway, D.A.; Jiang, J.X. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J. Bone Miner. Metab. 2015, 33, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; Lu, D.; Liu, A.L.; Zhang, Z.M.; Li, X.M.; Zou, Z.P.; Zeng, W.S.; Cheng, B.L.; Luo, S.Q. Reactive oxygen species stimulates receptor activator of NF-kappaB ligand expression in osteoblast. J. Biol. Chem. 2005, 280, 17497–17506. [Google Scholar] [CrossRef] [PubMed]
- Lean, J.M.; Jagger, C.J.; Kirstein, B.; Fuller, K.; Chambers, T.J. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology 2005, 146, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.S.; Wang, X.Y.; Xiao, D.M.; Hu, L.F.; Lu, M.; Wu, Z.Y.; Bian, J.S. Hydrogen sulfide protects MC3T3-E1 osteoblastic cells against H2O2-induced oxidative damage-implications for the treatment of osteoporosis. Free Radic. Biol. Med. 2011, 50, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Mody, N.; Parhami, F.; Sarafian, T.A.; Demer, L.L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic. Biol. Med. 2001, 31, 509–519. [Google Scholar] [CrossRef]
- Bai, X.; Lu, D.; Bai, J.; Zheng, H.; Ke, Z.; Li, X.; Luo, S. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem. Biophys. Res. Commun. 2004, 314, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Shibata, Y.; Pugdee, K.; Abiko, Y.; Ogata, Y. Effects of reactive oxygen species (ROS) on antioxidant system and osteoblastic differentiation in MC3T3-E1 cells. IUBMB Life 2007, 59, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Ambrogini, E.; Bartell, S.M.; Manolagas, S.C. Oxidative stress stimulates apoptosis and activates NF-κB in osteoblastic cells via a PKCβ/p66shc signaling cascade: Counter regulation by estrogens or androgens. Mol. Endocrinol. 2010, 24, 2030–2037. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, G.; Bilezikian, J.; Canalis, E.; Cocchi, D.; Giustina, A. New understanding and treatments for osteoporosis. Endocrine 2012, 41, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, L.; Ferrandiz, M.L.; Brines, R.; Guede, D.; Cuadrado, A.; Alcaraz, M.J. Effects of Nrf2 deficiency on bone microarchitecture in an experimental model of osteoporosis. Oxid. Med. Cell. Longev. 2014. [Google Scholar] [CrossRef]
- Jung, K.A.; Kwak, M.K. The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules 2010, 15, 7266–7291. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Xu, A.H.; Yang, Y.; Li, J. Role of Nrf2 in bone metabolism. J. Biomed. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Lee, Y.; Kim, K.H.; Lee, Z.H.; Joo, M.; Kim, H.H. Nrf2 is a novel regulator of bone acquisition. Bone 2014, 63, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Arts, I.C.; Hollman, P.C. Polyphenols and disease risk in epidemiologic studies. Am. J. Clin. Nutr. 2005, 81, 317S–325S. [Google Scholar] [PubMed]
- Scalbert, A.; Holvoet, S.; Mercenier, A. Dietary polyphenols and the prevention of diseases. Crit. Rev. Food Sci. Nutr. 2005, 41, 287–306. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, J.W.; Martin, B.R.; McCabe, G.P.; Ferruzzi, M.G.; Weaver, C.M. Plum and soy aglycon extracts superior at increasing bone calcium retention in ovariectomized sprague dawley rats. J. Agric. Food Chem. 2014, 62, 6108–6117. [Google Scholar] [CrossRef] [PubMed]
- Boz, H. Phenolic amides (avenanthramides) in Oats—A review. Czech J. Food Sci. 2015, 34, 399–404. [Google Scholar] [CrossRef]
- Meydani, M. Potential health benefits of avenanthramides of oats. Nutr. Rev. 2009, 67, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Emmons, C.L.; Peterson, D.M.; Paul, G.L. Antioxidant capacity of oat (Avena sativa L.) extracts. 2. In vitro antioxidant activity and contents of phenolic and tocol antioxidants. J. Agric. Food Chem. 1999, 47, 4894–4898. [Google Scholar] [CrossRef] [PubMed]
- Mourikis, P.; Sambasivan, R.; Castel, D.; Rocheteau, P.; Bizzarro, V.; Tajbakhsh, S. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 2012, 30, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Lee-Manion, A.M.; Price, R.K.; Strain, J.J.; Dimberg, L.H.; Sunnerheim, K.; Welch, R.W. In vitro antioxidant activity and antigenotoxic effects of avenanthramides and related compounds. J. Agric. Food Chem. 2009, 57, 10619–10624. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.W. Oat phenolics: Avenanthramides, novel substituted N-cinnamoylanthranilate alkaloids from oat groats and hulls. J. Agric. Food Chem. 1989, 37, 60–66. [Google Scholar] [CrossRef]
- Bryngelsson, S.; Dimberg, L.H.; Kamal-Eldin, A. Effects of commercial processing on levels of antioxidants in oats (Avena sativa L.). J. Agric. Food Chem. 2002, 50, 1890–1896. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Milbury, P.E.; Kwak, H.K.; Collins, F.W.; Samuel, P.; Blumberg, J.B. Avenanthramides and phenolic acids from oats are bioavailable and act synergistically with vitamin C to enhance hamster and human LDL resistance to oxidation. J. Nutr. 2004, 134, 1459–1466. [Google Scholar] [PubMed]
- Wang, P.; Chen, H.; Zhu, Y.; McBride, J.; Fu, J.; Sang, S. Oat avenanthramide-C (2c) is biotransformed by mice and the human microbiota into bioactive metabolites. J. Nutr. 2015, 145, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zubik, L.; Collins, F.W.; Marko, M.; Meydani, M. The antiatherogenic potential of oat phenolic compounds. Atherosclerosis 2004, 175, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Koenig, R.; Dickman, J.R.; Kang, C.; Zhang, T.; Chu, Y.F.; Ji, L.L. Avenanthramide supplementation attenuates exercise-induced inflammation in postmenopausal women. Nutr. J. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Yang, X.; Niu, X.; Liu, S.; Ren, G. Chemical characterization of the avenanthramide-rich extract from oat and its effect on d-galactose-induced oxidative stress in mice. J. Agric. Food Chem. 2011, 59, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhu, Y.; Yerke, A.; Wise, M.L.; Johnson, J.; Chu, Y.; Sang, S. Oat avenanthramides induce heme oxygenase-1 expression via Nrf2-mediated signaling in HK-2 cells. Mol. Nutr. Food Res. 2015, 59, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Wise, M.; Peterson, D.; Meydani, M. Mechanism by which avenanthramide-c, a polyphenol of oats, blocks cell cycle progression in vascular smooth muscle cells. Free Radic. Biol. Med. 2006, 41, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Lezcano, V.; Bellido, T.; Plotkin, L.I.; Boland, R.; Morelli, S. Role of connexin 43 in the mechanism of action of alendronate: Dissociation of anti-apoptotic and proliferative signaling pathways. Arch. Biochem. Biophys. 2012, 518, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Lecanda, F.; Warlow, P.M.; Sheikh, S.; Furlan, F.; Steinberg, T.H.; Civitelli, R. Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J. Cell Biol. 2000, 151, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Bellido, T.; Ali, A.A.; Plotkin, L.I.; Fu, Q.; Gubrij, I.; Roberson, P.K.; Weinstein, R.S.; O’Brien, C.A.; Manolagas, S.C.; Jilka, R.L. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J. Biol. Chem. 2003, 278, 50259–50272. [Google Scholar] [CrossRef] [PubMed]
- Lecka-Czernik, B.; Gubrij, I.; Moerman, E.A.; Kajkenova, O.; Lipschitz, D.A.; Manolagas, S.C.; Jilka, R.L. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPAR-gamma 2. J. Cell. Biochem. 1999, 74, 357–371. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Weinstein, R.S.; Parfitt, A.M.; Roberson, P.K.; Manolagas, S.C.; Bellido, T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J. Clin. Investig. 1999, 104, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Windle, J.J.; Koop, B.A.; Mundy, G.R.; Bonewald, L.F. Establishment of an osteocyte-like cell line, MLO-Y4. J. Bone Miner. Res. 1997, 12, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Costa, R.; Hassan, I.; Reginato, R.D.; Davis, H.M.; Bruzzaniti, A.; Allen, M.R.; Plotkin, L.I. High bone mass in mice lacking Cx37 due to defective osteoclast differentiation. J. Biol. Chem. 2014, 289, 8508–8520. [Google Scholar] [CrossRef] [PubMed]
- Ben-Awadh, A.; Delgado-Calle, J.; Tu, X.; Kuhlenschmidt, K.; Allen, M.R.; Plotkin, L.I.; Bellido, T. Parathyroid hormone receptor signaling induces bone resorption in the adult skeleton by directly regulating the RANKL gene in osteocytes. Endocrinology 2014, 155, 2797–2809. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Plotkin, L.I.; Galli, C.; Goellner, J.; Gortazar, A.R.; Allen, M.R.; Robling, A.G.; Bouxsein, M.; Schipani, E.; Turner, C.H.; et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 2008, 3, e2942. [Google Scholar]
- Bellido, T.; Plotkin, L.I.; Bruzzaniti, A. Bone cells. In Basic and Applied Bone Biology, 1st ed.; Burr, D., Allen, M., Eds.; Elsevier: Oxford, UK, 2014; pp. 27–45. [Google Scholar]
- Bellido, T.; Plotkin, L.I. Detection of apoptosis of bone cells in vitro. In Osteoporosis; Westendorf, J.J., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 51–75. [Google Scholar]
- Plotkin, L.I.; Mathov, I.; Aguirre, J.I.; Parfitt, A.M.; Manolagas, S.C.; Bellido, T. Mechanical stimulation prevents osteocyte apoptosis: Requirement of integrins, Src kinases and ERKs. Am. J. Physiol. Cell Physiol. 2005, 289, C633–C643. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wise, M.L.; Collins, F.W.; Meydani, M. Avenanthramides, polyphenols from oats, inhibit IL-1beta-induced NF-kappaB activation in endothelial cells. Free Radic. Biol. Med. 2008, 44, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Lv, N.; Song, M.Y.; Lee, Y.R.; Choi, H.N.; Kwon, K.B.; Park, J.W.; Park, B.H. Dihydroavenanthramide D protects pancreatic beta-cells from cytokine and streptozotocin toxicity. Biochem. Biophys. Res. Commun. 2009, 387, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Tang, X.X.; He, H.Y. Gene expression during induced differentiation of sheep bone marrow mesenchymal stem cells into osteoblasts. Genet. Mol. Res. 2013, 12, 6527–6534. [Google Scholar] [CrossRef] [PubMed]
- Balcerzak, M.; Hamade, E.; Zhang, L.; Pikula, S.; Azzar, G.; Radisson, J.; Bandorowicz-Pikula, J.; Buchet, R. The roles of annexins and alkaline phosphatase in mineralization process. Acta Biochim. Pol. 2003, 50, 1019–1038. [Google Scholar] [PubMed]
- Janssens, K.; Ten, D.P.; Janssens, S.; Van, H.W. Transforming growth factor-beta1 to the bone. Endocr. Rev. 2005, 26, 743–774. [Google Scholar] [CrossRef] [PubMed]
- Osyczka, A.M.; Leboy, P.S. Bone morphogenetic protein regulation of early osteoblast genes in human marrow stromal cells is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase signaling. Endocrinology 2005, 146, 3428–3437. [Google Scholar] [CrossRef] [PubMed]
- Jimi, E.; Nakamura, I.; Amano, H.; Taguchi, Y.; Tsurkai, T.; Tamura, M.; Takahasi, N.; Suda, T. Osteoclast function is activated by osteoblastic cells through a mechanism involving cell-to-cell contact. Endocrinology 1996, 137, 2187–2190. [Google Scholar] [PubMed]
- Udagawa, N.; Takahashi, N.; Jimi, E.; Matsuzaki, K.; Tsurukai, T.; Itoh, K.; Nakagawa, N.; Yasuda, H.; Goto, M.; Tsuda, E.; et al. Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: Receptor activator of NF-kappa B ligand. Bone 1999, 25, 517–523. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Kramer, I.; Halleux, C.; Keller, H.; Pegurri, M.; Gooi, J.H.; Weber, P.B.; Feng, J.Q.; Bonewald, L.F.; Kneissel, M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol. Cell Biol. 2010, 30, 3071–3085. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.K.; Harris, S.; Ahuja, S.S.; Bonewald, L.F. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J. Bone Miner. Res. 2002, 17, 2068–2079. [Google Scholar] [CrossRef] [PubMed]
- Theoleyre, S.; Wittrant, Y.; Tat, S.K.; Fortun, Y.; Redini, F.; Heymann, D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004, 15, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Jilka, R.L.; Bellido, T.; Almeida, M.; Plotkin, L.I.; O’Brien, C.A.; Weinstein, R.S.; Manolagas, S.C. Apoptosis in bone cells. In Principles of Bone Biology, 3rd ed.; Bilezikian, J.P., Raisz, L.G., Martin, T.J., Eds.; Academic Press: San Diego/San Francisco, CA, USA; New York, NY, USA; London, UK; Sydney, Australia; Tokyo, Japan, 2008; pp. 237–261. [Google Scholar]
- Bilezikian, J.P.; Raisz, L.G.; Rodan, G.A. Principles of Bone Biology; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Bellido, T.; Plotkin, L.I. Novel actions of bisphosphonates in bone: Preservation of osteoblast and osteocyte viability. Bone 2011, 49, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, R.S.; Jilka, R.L.; Parfitt, A.M.; Manolagas, S.C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: Potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998, 102, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Jilka, R.L.; Weinstein, R.S.; Bellido, T.; Roberson, P.; Parfitt, A.M.; Manolagas, S.C. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Investig. 1999, 104, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I. Apoptotic osteocytes and the control of targeted bone resorption. Curr. Osteoporos. Rep. 2014, 12, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.I.; Plotkin, L.I.; Stewart, S.A.; Weinstein, R.S.; Parfitt, A.M.; Manolagas, S.C.; Bellido, T. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Miner. Res. 2006, 21, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Verborgt, O.; Gibson, G.; Schaffler, M.B. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J. Bone Miner. Res. 2000, 15, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I.; Bivi, N.; Bellido, T. A bisphosphonate that does not affect osteoclasts prevents osteoblast and osteocyte apoptosis and the loss of bone strength induced by glucocorticoids in mice. Bone 2011, 49, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wise, M.L.; Li, F.; Dey, M. Phytochemicals attenuating aberrant activation of beta-catenin in cancer cells. PLoS ONE 2012, 7, e50508. [Google Scholar]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Li, L.; Corry, K.A.; Zhang, P.; Yang, Y.; Himes, E.; Mihuti, C.L.; Nelson, C.; Dai, G.; Li, J. Deletion of Nrf2 reduces skeletal mechanical properties and decreases load-driven bone formation. Bone 2015, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, S.; Lee, H.; Yang, Y.; Jeong, W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic. Biol. Med. 2013, 65, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, H.; Shinohara, F.; Kajiya, M.; Kodama, T. The Keap1/Nrf2 protein axis plays a role in osteoclast differentiation by regulating intracellular reactive oxygen species signaling. J. Biol. Chem. 2013, 288, 23009–23020. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrini, G.G.; Morales, C.C.; Wallace, T.C.; Plotkin, L.I.; Bellido, T. Avenanthramides Prevent Osteoblast and Osteocyte Apoptosis and Induce Osteoclast Apoptosis in Vitro in an Nrf2-Independent Manner. Nutrients 2016, 8, 423. https://doi.org/10.3390/nu8070423
Pellegrini GG, Morales CC, Wallace TC, Plotkin LI, Bellido T. Avenanthramides Prevent Osteoblast and Osteocyte Apoptosis and Induce Osteoclast Apoptosis in Vitro in an Nrf2-Independent Manner. Nutrients. 2016; 8(7):423. https://doi.org/10.3390/nu8070423
Chicago/Turabian StylePellegrini, Gretel G., Cynthya C. Morales, Taylor C. Wallace, Lilian I. Plotkin, and Teresita Bellido. 2016. "Avenanthramides Prevent Osteoblast and Osteocyte Apoptosis and Induce Osteoclast Apoptosis in Vitro in an Nrf2-Independent Manner" Nutrients 8, no. 7: 423. https://doi.org/10.3390/nu8070423
APA StylePellegrini, G. G., Morales, C. C., Wallace, T. C., Plotkin, L. I., & Bellido, T. (2016). Avenanthramides Prevent Osteoblast and Osteocyte Apoptosis and Induce Osteoclast Apoptosis in Vitro in an Nrf2-Independent Manner. Nutrients, 8(7), 423. https://doi.org/10.3390/nu8070423