Non-Alcoholic Steatohepatitis and Hepatocellular Carcinoma: Implications for Lycopene Intervention

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Mechanisms Associated with Metabolic Syndrome, Chronic Inflammation and HCC Progression
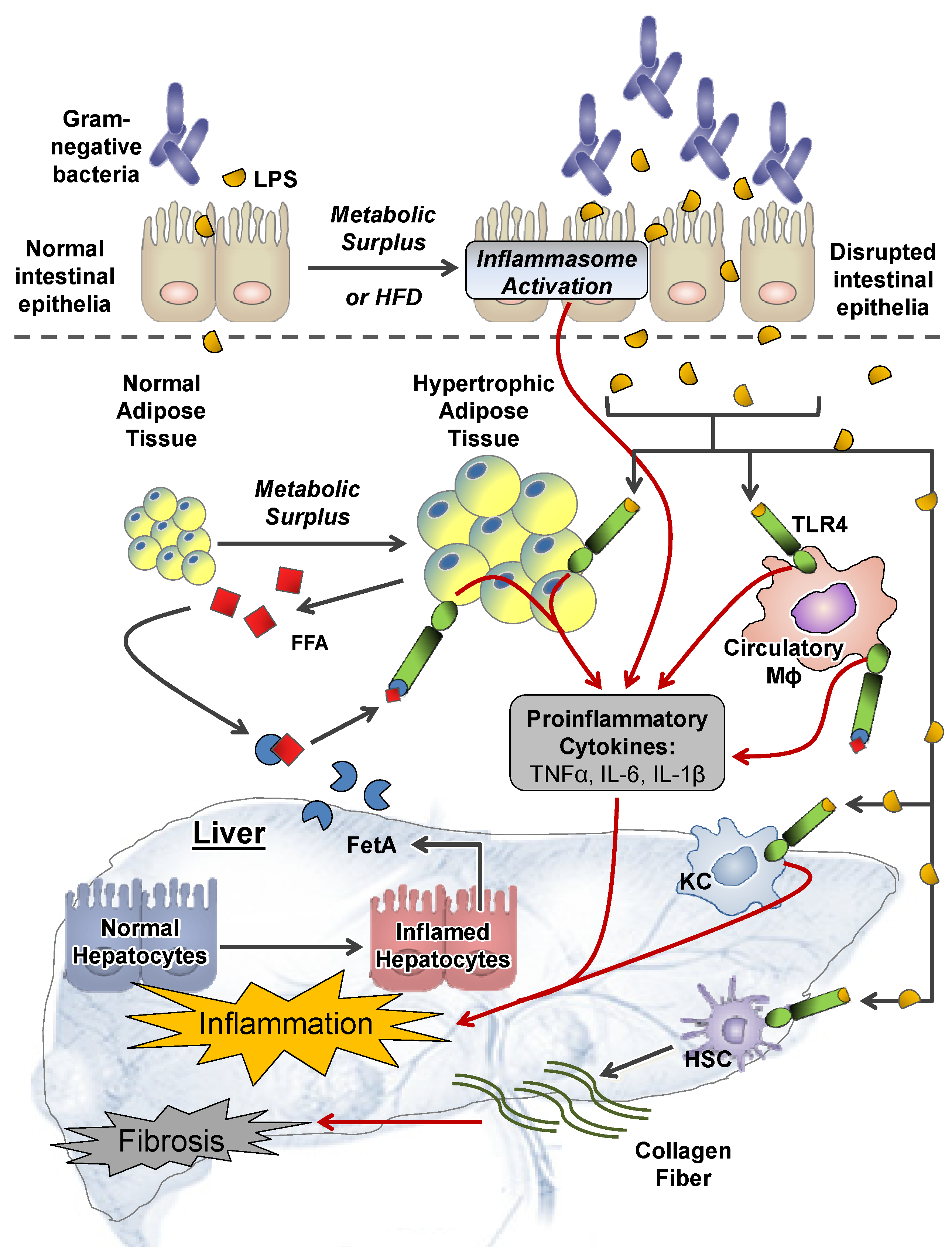
2.1. Extrahepatic Perturbations
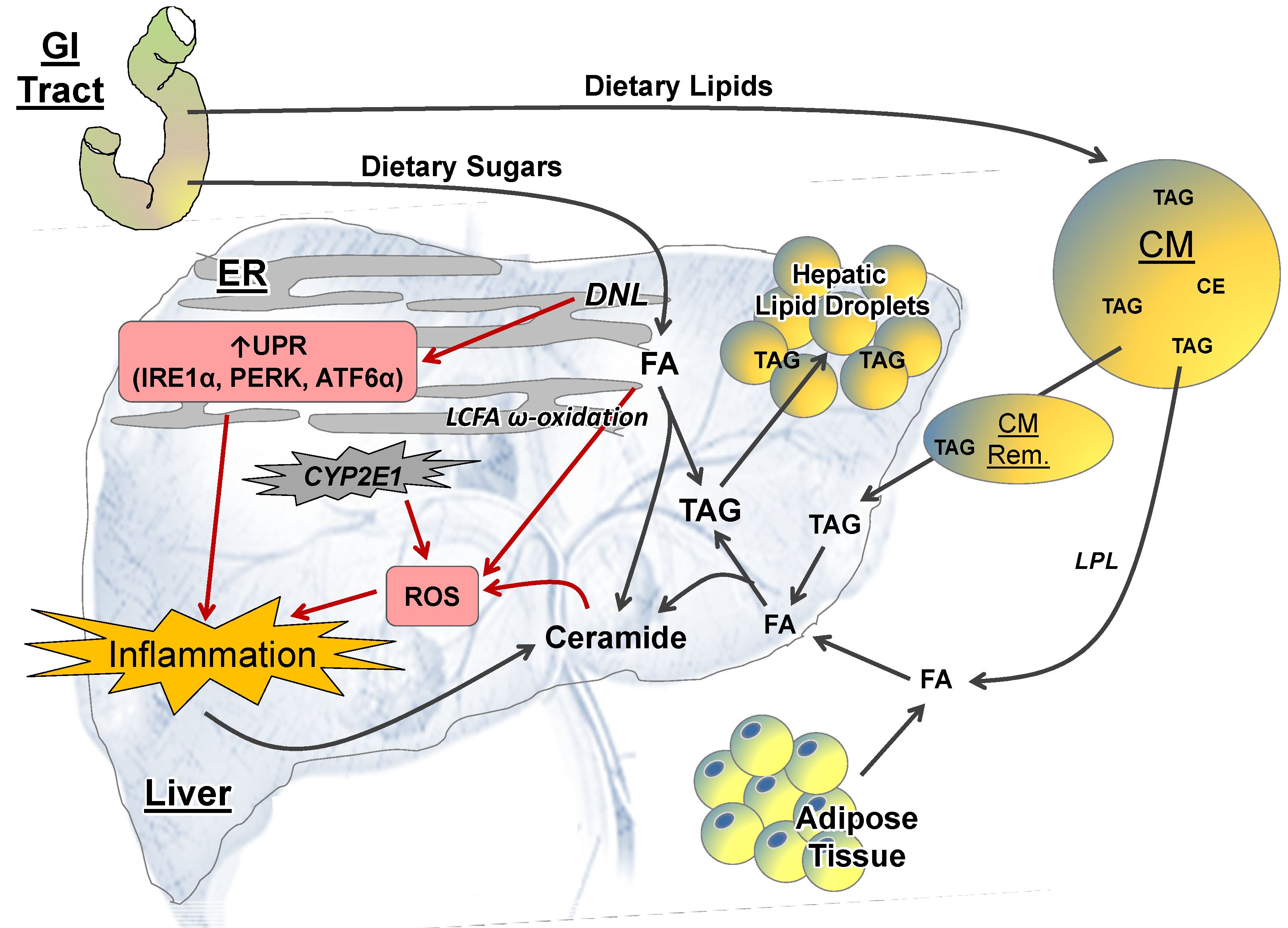
2.1.1. GI Tract
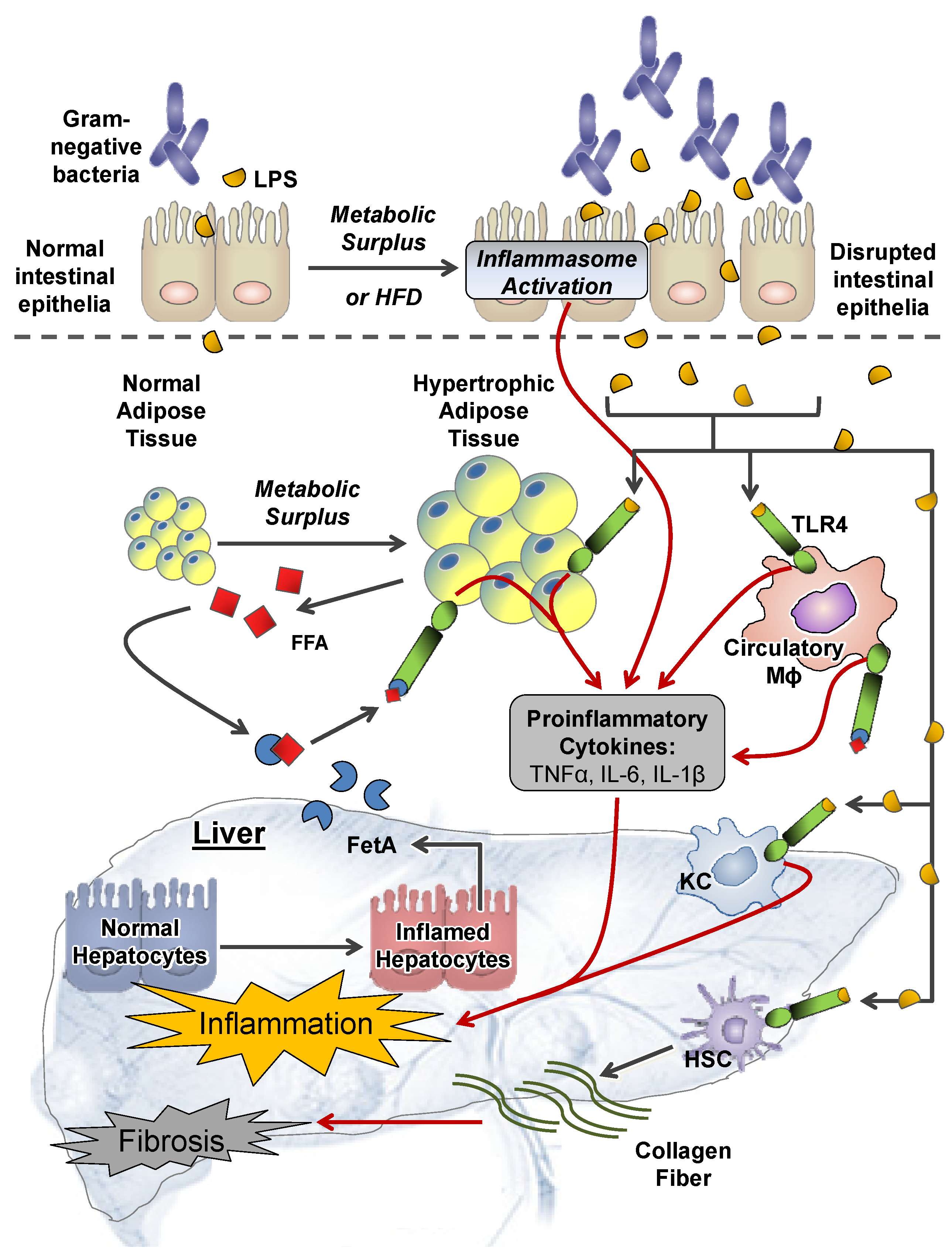
2.1.2. Adipose Tissue
2.1.3. Other Systemic Perturbations
2.2. Intrahepatic Perturbations
2.2.1. Lipid Metabolism
2.2.2. ER Stress
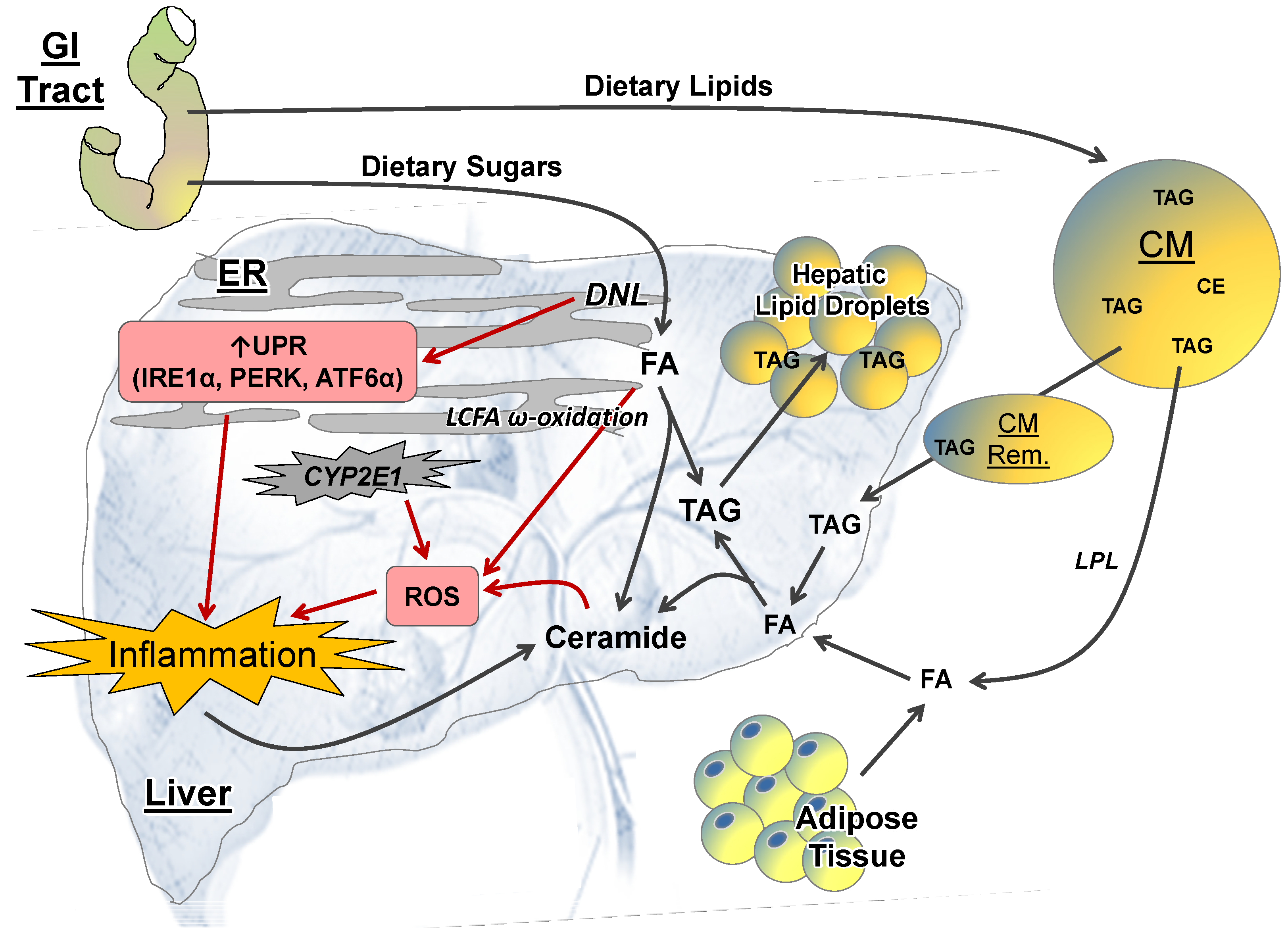
2.2.3. ROS and CYP-P450 Enzymes
2.2.4. Inflammation and TLR4
3. Effects of Tomato and Lycopene Consumption against NASH and HCC
3.1. Tomato Effects
3.2. Lycopene Effects
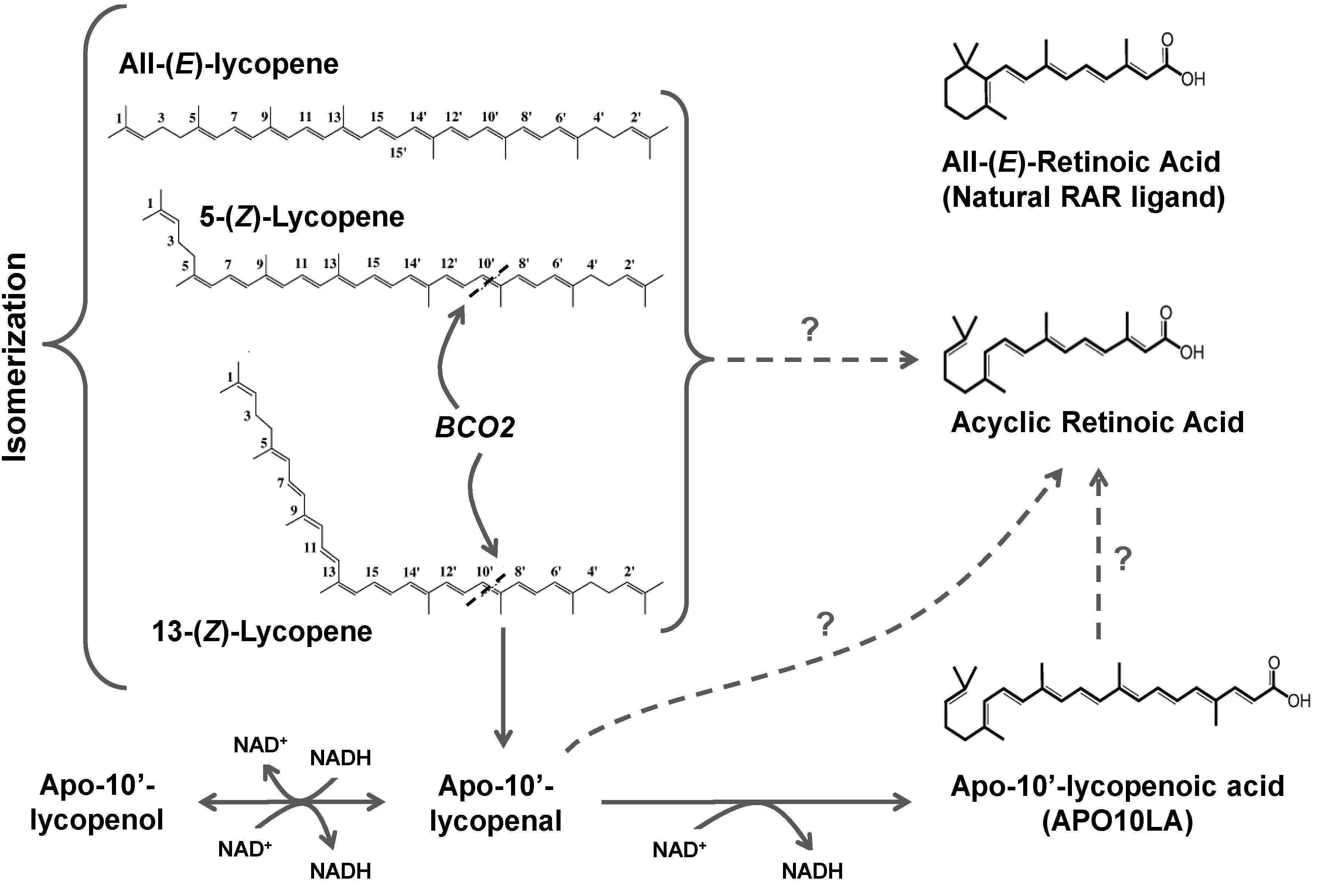
3.3. Lycopene Metabolism
3.3.1. Chemical Oxidation
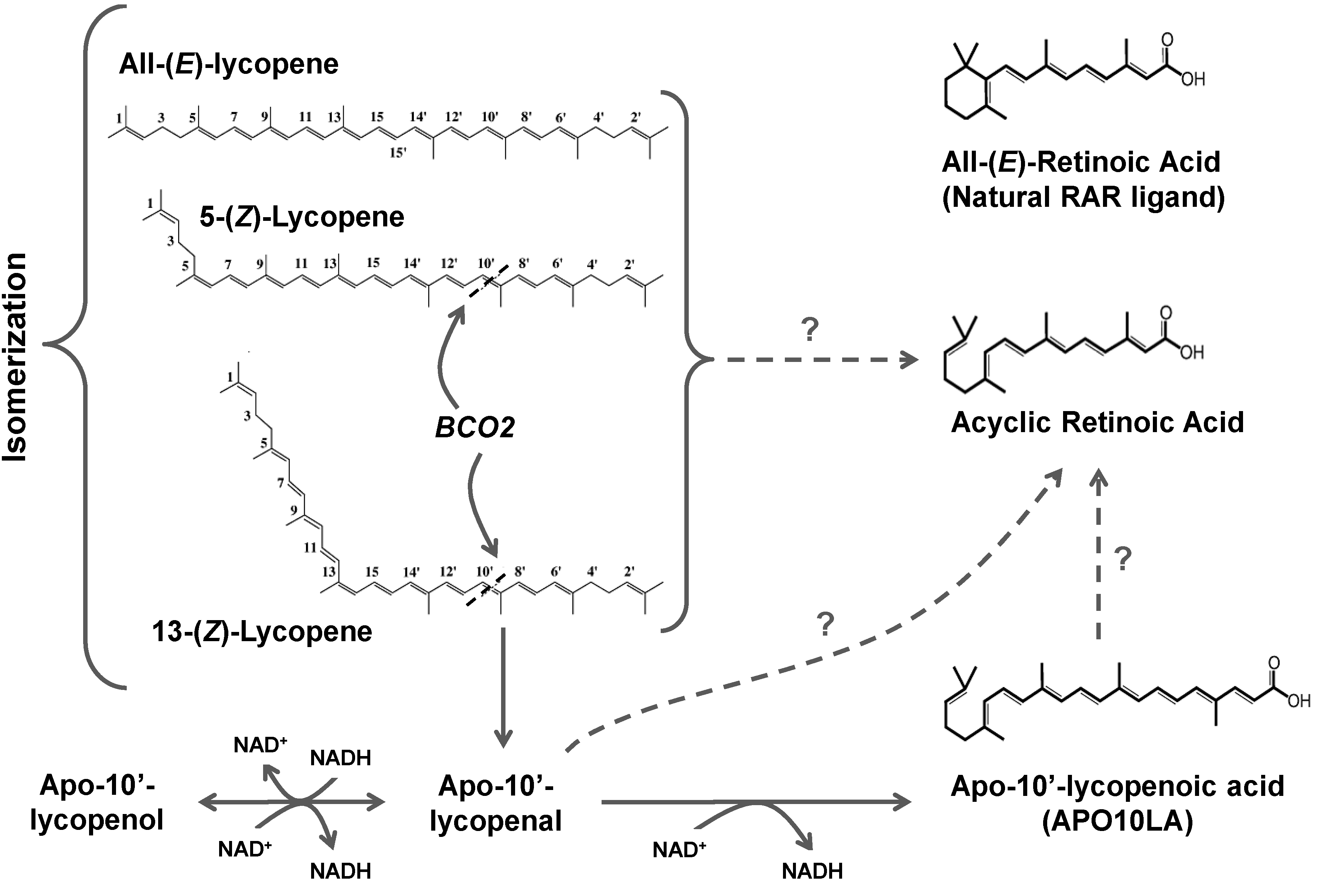
3.3.2. Oxidative Metabolism in Plants
3.3.3. Oxidative Metabolism in Mammals
3.3.3.1. BCO1
3.3.3.2. BCO2
3.3.3.3. Potential Alternative Pathways
3.3.4. Oxidation of Apolycopenoids
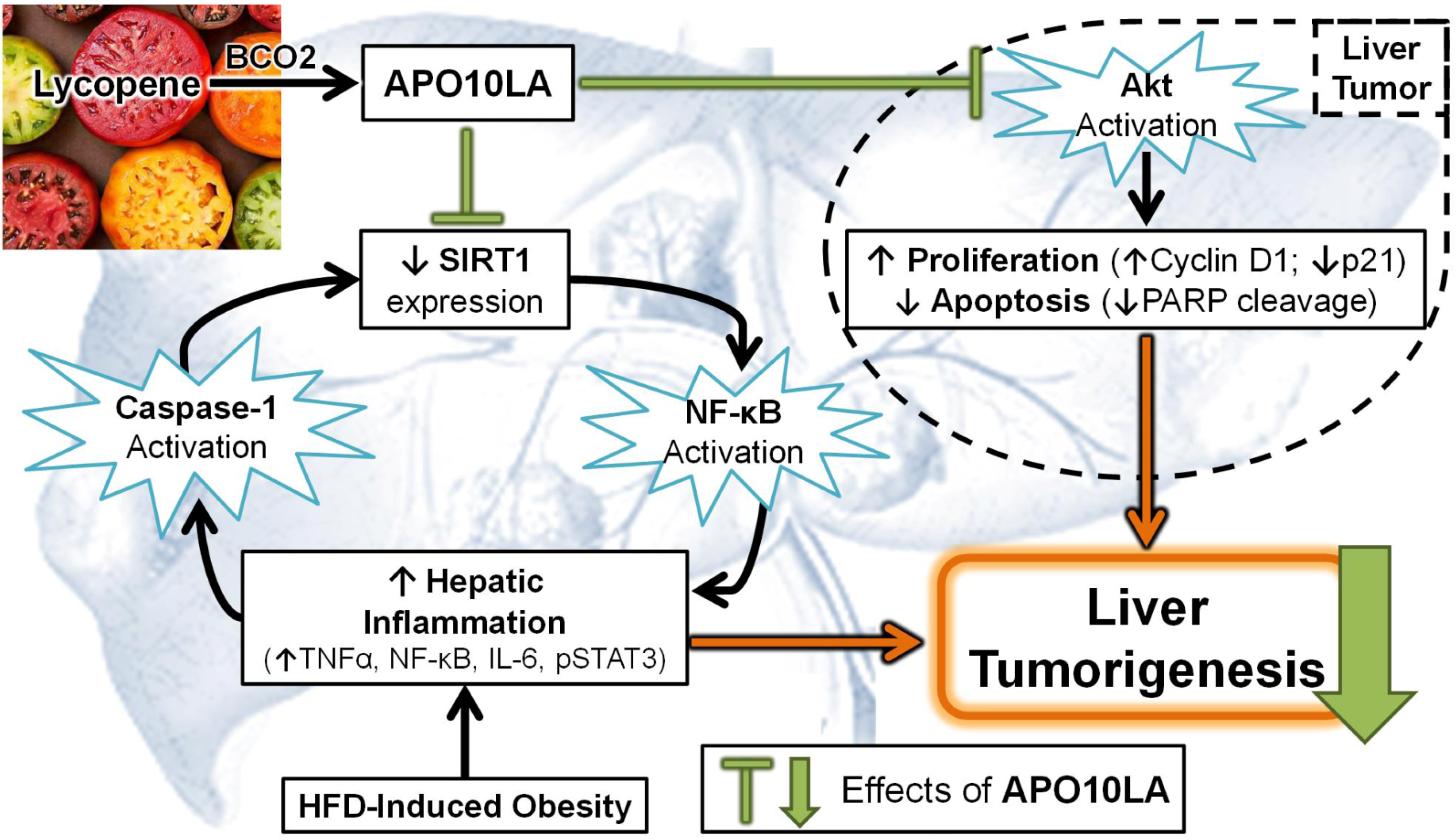
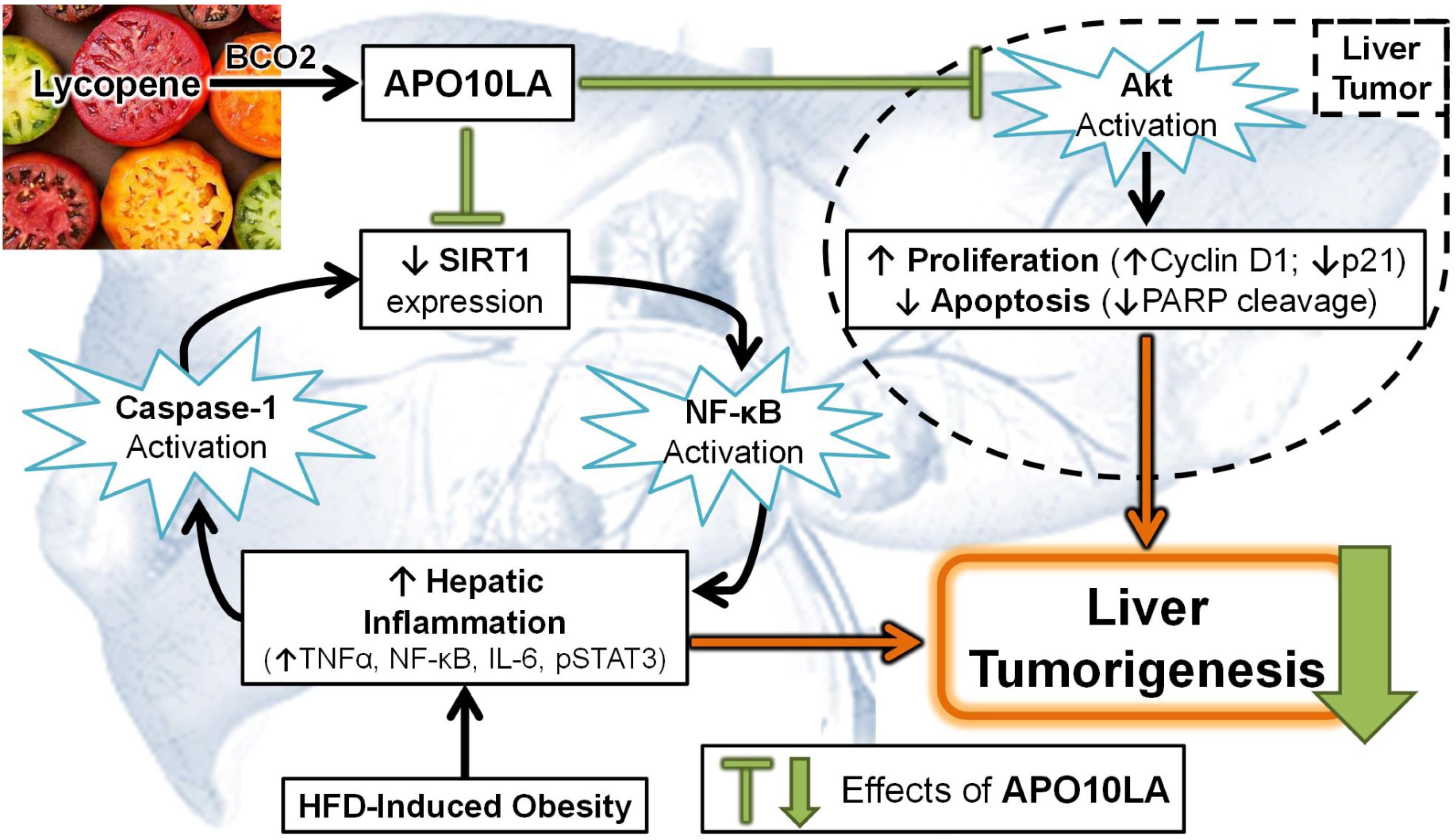
3.4. Effects of Apolycopenoids
3.5. Potential Molecular Mechanisms
3.5.1. Modulating Pro-Inflammatory Signaling and Cytokine Expression
3.5.2. Antioxidant Mechanism
3.5.3. Retinoid Receptors Interactions
3.5.4. Anti-Metastatic Effects
3.5.5. SIRT1 Up-Regulation
4. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Siegel, A.B.; Zhu, A.X. Metabolic syndrome and hepatocellular carcinoma: Two growing epidemics with a potential link. Cancer 2009, 115, 5651–5661. [Google Scholar]
- El-Serag, H.B.; Mason, A.C. Rising incidence of hepatocellular carcinoma in the United States. N. Engl. J. Med. 1999, 340, 745–750. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012, 56, 704–713. [Google Scholar] [CrossRef]
- Page, J.M.; Harrison, S.A. NASH and HCC. Clin. Liver Dis. 2009, 13, 631–647. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530.e1, quiz e60. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Nakagawa, H.; Maeda, S. Inflammation- and stress-related signaling pathways in hepatocarcinogenesis. World J. Gastroenterol. 2012, 18, 4071–4081. [Google Scholar] [CrossRef]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Sinha, R.; Fisch, G.; Teague, B.; Tamborlane, W.V.; Banyas, B.; Allen, K.; Savoye, M.; Rieger, V.; Taksali, S.; Barbetta, G.; et al. Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. N. Engl. J. Med. 2002, 346, 802–810. [Google Scholar] [CrossRef]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M. Hepatocellular cancer—The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Bianchini, F.; Kaaks, R.; Vainio, H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002, 3, 565–574. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- De Angel, R.E.; Conti, C.J.; Wheatley, K.E.; Brenner, A.J.; Otto, G.; Degraffenried, L.A.; Hursting, S.D. The enhancing effects of obesity on mammary tumor growth and Akt/mTOR pathway activation persist after weight loss and are reversed by RAD001. Mol. Carcinog. 2013, 52, 446–458. [Google Scholar] [CrossRef]
- Tilg, H.; Kaser, A. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Investig. 2011, 121, 2126–2132. [Google Scholar] [CrossRef]
- Brun, P.; Castagliuolo, I.; di Leo, V.; Buda, A.; Pinzani, M.; Palu, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Farhadi, A.; Gundlapalli, S.; Shaikh, M.; Frantzides, C.; Harrell, L.; Kwasny, M.M.; Keshavarzian, A. Susceptibility to gut leakiness: A possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008, 28, 1026–1033. [Google Scholar] [CrossRef]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Duerkop, B.A.; Vaishnava, S.; Hooper, L.V. Immune responses to the microbiota at the intestinal mucosal surface. Immunity 2009, 31, 368–376. [Google Scholar] [CrossRef]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Tandon, P.; Garcia-Tsao, G. Bacterial infections, sepsis, and multiorgan failure in cirrhosis. Semin. Liver Dis. 2008, 28, 26–42. [Google Scholar] [CrossRef]
- Cirera, I.; Bauer, T.M.; Navasa, M.; Vila, J.; Grande, L.; Taura, P.; Fuster, J.; Garcia-Valdecasas, J.C.; Lacy, A.; Suarez, M.J.; et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 2001, 34, 32–37. [Google Scholar]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferriéres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar]
- Thuy, S.; Ladurner, R.; Volynets, V.; Wagner, S.; Strahl, S.; Königsrainer, A.; Maier, K.-P.; Bischoff, S.C.; Bergheim, I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J. Nutr. 2008, 138, 1452–1455. [Google Scholar]
- Dolganiuc, A.; Norkina, O.; Kodys, K.; Catalano, D.; Bakis, G.; Marshall, C.; Mandrekar, P.; Szabo, G. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology 2007, 133, 1627–1636. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef]
- Seki, E.; de Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Wiest, R.; Garcia-Tsao, G. Bacterial translocation (BT) in cirrhosis. Hepatology 2005, 41, 422–433. [Google Scholar] [CrossRef]
- Fukui, H.; Brauner, B.; Bode, J.C.; Bode, C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: Reevaluation with an improved chromogenic assay. J. Hepatol. 1991, 12, 162–169. [Google Scholar] [CrossRef]
- Nolan, J.P.; Leibowitz, A.I. Endotoxins in liver disease. Gastroenterology 1978, 75, 765–766. [Google Scholar]
- Broitman, S.A.; Gottlieb, L.S.; Zamcheck, N. Influence of neomycin and ingested endotoxin in the pathogenesis of choline deficiency cirrhosis in the adult rat. J. Exp. Med. 1964, 119, 633–642. [Google Scholar] [CrossRef]
- Rutenburg, A.M.; Sonnenblick, E.; Koven, I.; Aprahamian, H.A.; Reiner, L.; Fine, J. The role of intestinal bacteria in the development of dietary cirrhosis in rats. J. Exp. Med. 1957, 106, 1–14. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin—A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef]
- Hennige, A.M.; Staiger, H.; Wicke, C.; Machicao, F.; Fritsche, A.; Haring, H.U.; Stefan, N. Fetuin—A induces cytokine expression and suppresses adiponectin production. PLoS One 2008, 3, e1765. [Google Scholar]
- He, G.; Karin, M. NF-κB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. 2012, 16, 180–188. [Google Scholar] [CrossRef]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschop, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef]
- Donmez, G.; Guarente, L. Aging and disease: Connections to sirtuins. Aging Cell 2010, 9, 285–290. [Google Scholar] [CrossRef]
- Herranz, D.; Munoz-Martin, M.; Canamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1, 3. [Google Scholar]
- Herranz, D.; Serrano, M. SIRT1: Recent lessons from mouse models. Nat. Rev. Cancer 2010, 10, 819–823. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef]
- Kume, S.; Haneda, M.; Kanasaki, K.; Sugimoto, T.; Araki, S.; Isono, M.; Isshiki, K.; Uzu, T.; Kashiwagi, A.; Koya, D. Silent information regulator 2 (SIRT1) attenuates oxidative stress-induced mesangial cell apoptosis via p53 deacetylation. Free Radic. Biol. Med. 2006, 40, 2175–2182. [Google Scholar] [CrossRef]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef]
- De Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Dalla Man, C.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: Potential biochemical mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Solinas, G.; Karin, M. JNK1 and IKKβ: Molecular links between obesity and metabolic dysfunction. FASEB J. 2010, 24, 2596–2611. [Google Scholar] [CrossRef]
- Nov, O.; Kohl, A.; Lewis, E.C.; Bashan, N.; Dvir, I.; Ben-Shlomo, S.; Fishman, S.; Wueest, S.; Konrad, D.; Rudich, A. Interleukin-1β may mediate insulin resistance in liver-derived cells in response to adipocyte inflammation. Endocrinology 2010, 151, 4247–4256. [Google Scholar] [CrossRef]
- Rajala, M.W.; Scherer, P.E. Minireview: The adipocyte–At the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 2003, 144, 3765–3773. [Google Scholar] [CrossRef]
- Patton, J.S.; Shepard, H.M.; Wilking, H.; Lewis, G.; Aggarwal, B.B.; Eessalu, T.E.; Gavin, L.A.; Grunfeld, C. Interferons and tumor necrosis factors have similar catabolic effects on 3T3 L1 cells. Proc. Natl. Acad. Sci. USA 1986, 83, 8313–8317. [Google Scholar] [CrossRef]
- Wanless, I.R.; Lentz, J.S. Fatty liver hepatitis (steatohepatitis) and obesity: An autopsy study with analysis of risk factors. Hepatology 1990, 12, 1106–1110. [Google Scholar] [CrossRef]
- Ix, J.H.; Shlipak, M.G.; Brandenburg, V.M.; Ali, S.; Ketteler, M.; Whooley, M.A. Association between human fetuin—A and the metabolic syndrome: Data from the heart and soul study. Circulation 2006, 113, 1760–1767. [Google Scholar] [CrossRef]
- Haukeland, J.W.; Dahl, T.B.; Yndestad, A.; Gladhaug, I.P.; Loberg, E.M.; Haaland, T.; Konopski, Z.; Wium, C.; Aasheim, E.T.; Johansen, O.E.; et al. Fetuin A in nonalcoholic fatty liver disease: In vivo and in vitro studies. Eur. J. Endocrinol. 2012, 166, 503–510. [Google Scholar] [CrossRef]
- Solinas, G.; Vilcu, C.; Neels, J.G.; Bandyopadhyay, G.K.; Luo, J.L.; Naugler, W.; Grivennikov, S.; Wynshaw-Boris, A.; Scadeng, M.; Olefsky, J.M.; et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007, 6, 386–397. [Google Scholar] [CrossRef]
- Seki, E.; Brenner, D.A.; Karin, M. A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef]
- Weinhold, B.; Ruther, U. Interleukin-6-dependent and -independent regulation of the human C-reactive protein gene. Biochem. J. 1997, 327, 425–429. [Google Scholar]
- Castell, J.V.; Gomez-Lechon, M.J.; David, M.; Fabra, R.; Trullenque, R.; Heinrich, P.C. Acute-phase response of human hepatocytes: Regulation of acute-phase protein synthesis by interleukin-6. Hepatology 1990, 12, 1179–1186. [Google Scholar] [CrossRef]
- Visser, M.; Bouter, L.M.; McQuillan, G.M.; Wener, M.H.; Harris, T.B. Elevated C-reactive protein levels in overweight and obese adults. JAMA 1999, 282, 2131–2135. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Inokuchi, S.; Aoyama, T.; Miura, K.; Osterreicher, C.H.; Kodama, Y.; Miyai, K.; Akira, S.; Brenner, D.A.; Seki, E. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 844–849. [Google Scholar] [CrossRef]
- Auer, K.L.; Contessa, J.; Brenz-Verca, S.; Pirola, L.; Rusconi, S.; Cooper, G.; Abo, A.; Wymann, M.P.; Davis, R.J.; Birrer, M. The Ras/Rac1/Cdc42/SEK/JNK/c-Jun cascade is a key pathway by which agonists stimulate DNA synthesis in primary cultures of rat hepatocytes. Mol. Biol. Cell 1998, 9, 561–573. [Google Scholar] [CrossRef]
- Shimizu, M.; Sakai, H.; Shirakami, Y.; Iwasa, J.; Yasuda, Y.; Kubota, M.; Takai, K.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. Acyclic retinoid inhibits diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BLKS/J- +Leprdb/+Leprdb mice. Cancer Prev. Res. 2011, 4, 128–136. [Google Scholar] [CrossRef]
- Moriwaki, H.; Shimizu, M.; Okuno, M.; Nishiwaki-Matsushima, R. Chemoprevention of liver carcinogenesis with retinoids: Basic and clinical aspects. Hepatol. Res. 2007, 37, S299–S302. [Google Scholar] [CrossRef]
- Matsushima-Nishiwaki, R.; Okuno, M.; Adachi, S.; Sano, T.; Akita, K.; Moriwaki, H.; Friedman, S.L.; Kojima, S. Phosphorylation of retinoid X receptor α at serine 260 impairs its metabolism and function in human hepatocellular carcinoma. Cancer Res. 2001, 61, 7675–7682. [Google Scholar]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Crowe, F.L.; Key, T.J.; Allen, N.E.; Appleby, P.N.; Overvad, K.; Grønbæk, H.; Tjønneland, A.; Halkjær, J.; Dossus, L.; Boeing, H. A cross-sectional analysis of the associations between adult height, BMI and serum concentrations of IGF-I and IGFBP-1, -2 and -3 in the European Prospective Investigation into Cancer and Nutrition (EPIC). Ann. Hum. Biol. 2011, 38, 194–202. [Google Scholar] [CrossRef]
- Walenbergh, S.M.; Koek, G.H.; Bieghs, V.; Shiri-Sverdlov, R. Non-alcoholic steatohepatitis: The role of oxidized low-density lipoproteins. J. Hepatol. 2013, 58, 801–810. [Google Scholar] [CrossRef]
- Patel, R.; Baker, S.S.; Liu, W.; Desai, S.; Alkhouri, R.; Kozielski, R.; Mastrandrea, L.; Sarfraz, A.; Cai, W.; Vlassara, H. Effect of dietary advanced glycation end products on mouse liver. PLoS One 2012, 7, e35143. [Google Scholar] [CrossRef]
- Paolisso, G.; Gambardella, A.; Tagliamonte, M.; Saccomanno, F.; Salvatore, T.; Gualdiero, P.; DʼOnofrio, M.; Howard, B. Does free fatty acid infusion impair insulin action also through an increase in oxidative stress? J. Clin. Endocrinol. Metab. 1996, 81, 4244–4248. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger receptors class AI/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Bieghs, V.; Verheyen, F.; van Gorp, P.J.; Hendrikx, T.; Wouters, K.; Lütjohann, D.; Gijbels, M.J.; Febbraio, M.; Binder, C.J.; Hofker, M.H. Internalization of modified lipids by CD36 and SR-A leads to hepatic inflammation and lysosomal cholesterol storage in Kupffer cells. PLoS One 2012, 7, e34378. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2012, 425, 121–126. [Google Scholar] [CrossRef]
- Groeneweg, M.; Kanters, E.; Vergouwe, M.N.; Duerink, H.; Kraal, G.; Hofker, M.H.; de Winther, M.P. Lipopolysaccharide-induced gene expression in murine macrophages is enhanced by prior exposure to oxLDL. J. Lipid Res. 2006, 47, 2259–2267. [Google Scholar] [CrossRef]
- Liao, F.; Andalibi, A. Genetic control of inflammatory gene induction and NF-κB-like transcription factor activation in response to an atherogenic diet in mice. J. Clin. Investig. 1993, 91, 2572–2579. [Google Scholar] [CrossRef]
- Bieghs, V.; van Gorp, P.J.; Walenbergh, S.; Gijbels, M.J.; Verheyen, F.; Buurman, W.A.; Briles, D.E.; Hofker, M.H.; Binder, C.J.; Shiri-Sverdlov, R. Specific immunization strategies against oxidized low-density lipoprotein: A novel way to reduce nonalcoholic steatohepatitis in mice. Hepatology 2012, 56, 894–903. [Google Scholar] [CrossRef]
- Bieghs, V.; Wouters, K.; van Gorp, P.J.; Gijbels, M.J.; de Winther, M.P.; Binder, C.J.; Lütjohann, D.; Febbraio, M.; Moore, K.J.; van Bilsen, M. Role of scavenger receptor A and CD36 in diet-induced nonalcoholic steatohepatitis in hyperlipidemic mice. Gastroenterology 2010, 138, 2477–2486. e3. [Google Scholar] [CrossRef]
- Yimin, H.F.; Matsuoka, S.; Sakurai, T.; Kohanawa, M.; Zhao, S.; Kuge, Y.; Tamaki, N.; Chiba, H. A novel murine model for non-alcoholic steatohepatitis developed by combination of a high-fat diet and oxidized low-density lipoprotein. Lab. Investig. 2011, 92, 265–281. [Google Scholar]
- Scorletti, E.; Byrne, C.D. Omega-3 fatty acids, hepatic lipid metabolism, and nonalcoholic fatty liver disease. Annu. Rev. Nutr. 2013, 33, 231–248. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef]
- Shen, L.-L.; Liu, H.; Gan, L.; Lu, L.; Zhang, Q.; Li, L.; He, F.; Jiang, Y. Effects of farnesoid X receptor on the expression of the fatty acid synthetase and hepatic lipase. Mol. Biol. Rep. 2011, 38, 553–559. [Google Scholar] [CrossRef]
- Knight, B.; Hebbachi, A.; Hauton, D.; Brown, A.; Wiggins, D.; Patel, D.; Gibbons, G. A role for PPARα in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421. [Google Scholar] [CrossRef]
- Schadinger, S.E.; Bucher, N.L.; Schreiber, B.M.; Farmer, S.R. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1195–E1205. [Google Scholar] [CrossRef]
- Dentin, R.; Girard, J.; Postic, C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): Two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005, 87, 81–86. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef]
- Longato, L.; Tong, M.; Wands, J.R.; de la Monte, S.M. High fat diet induced hepatic steatosis and insulin resistance: Role of dysregulated ceramide metabolism. Hepatol. Res. 2012, 42, 412–427. [Google Scholar] [CrossRef]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef]
- Promrat, K.; Longato, L.; Wands, J.R.; de la Monte, S.M. Weight loss amelioration of non-alcoholic steatohepatitis linked to shifts in hepatic ceramide expression and serum ceramide levels. Hepatol. Res. 2011, 41, 754–762. [Google Scholar] [CrossRef]
- Chocian, G.; Chabowski, A.; Żendzian-Piotrowska, M.; Harasim, E.; Łukaszuk, B.; Górski, J. High fat diet induces ceramide and sphingomyelin formation in rat’s liver nuclei. Mol. Cell. Biochem. 2010, 340, 125–131. [Google Scholar] [CrossRef]
- Holland, W.L.; Brozinick, J.T.; Wang, L.-P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef]
- Holland, W.L.; Bikman, B.T.; Wang, L.-P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid—Induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef]
- Chang, Z.-Q.; Lee, S.-Y.; Kim, H.-J.; Kim, J.R.; Kim, S.-J.; Hong, I.-K.; Oh, B.-C.; Choi, C.-S.; Goldberg, I.J.; Park, T.-S. Endotoxin activates de novo sphingolipid biosynthesis via nuclear factor κB-mediated upregulation of Sptlc2. Prostaglandins Other Lipid Mediat. 2011, 94, 44–52. [Google Scholar] [CrossRef]
- Levy, M.; Castillo, S.S.; Goldkorn, T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem. Biophys. Res. Commun. 2006, 344, 900–905. [Google Scholar] [CrossRef]
- Colombini, M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2010, 1797, 1239–1244. [Google Scholar] [CrossRef]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef]
- Stiban, J.; Caputo, L.; Colombini, M. Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J. Lipid Res. 2008, 49, 625–634. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O. Activation of the ATF6, XBP1 and GRP78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Pytel, D.; Riese, M.J.; Vaites, L.P.; Singh, N.; Koretzky, G.A.; Witze, E.S.; Diehl, J.A. PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate Akt activation, and promote adipocyte differentiation. Mol. Cell. Biol. 2012, 32, 2268–2278. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Bobrovnikova-Marjon, E.; Ji, X.; Liebhaber, S.A.; Diehl, J.A. PERK-dependent regulation of IAP translation during ER stress. Oncogene 2008, 28, 910–920. [Google Scholar]
- Kazemi, S.; Mounir, Z.; Baltzis, D.; Raven, J.F.; Wang, S.; Krishnamoorthy, J.-L.; Pluquet, O.; Pelletier, J.; Koromilas, A.E. A novel function of eIF2α kinases as inducers of the phosphoinositide-3 kinase signaling pathway. Mol. Biol. Cell 2007, 18, 3635–3644. [Google Scholar] [CrossRef]
- Kammoun, H.L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferré, P.; Foufelle, F. GRP78 expression inhibits insulin and ER stress—Induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Investig. 2009, 119, 1201–1215. [Google Scholar] [CrossRef]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system—From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar]
- Akkaoui, M.; Cohen, I.; Esnous, C.; Lenoir, V.; Sournac, M.; Girard, J.; Prip-Buus, C. Modulation of the hepatic malonyl-CoA-carnitine palmitoyltransferase 1A partnership creates a metabolic switch allowing oxidation of de novo fatty acids1. Biochem. J. 2009, 420, 429–438. [Google Scholar] [CrossRef]
- Reddy, J.K. Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal β-oxidation, PPARα, and steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1333–G1339. [Google Scholar]
- Leung, T.M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.-A.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef]
- Lieber, C.S. Cytochrome P-4502E1: Its physiological and pathological role. Physiol. Rev. 1997, 77, 517–544. [Google Scholar]
- Kathirvel, E.; Morgan, K.; French, S.W.; Morgan, T.R. Overexpression of liver-specific cytochrome P4502E1 impairs hepatic insulin signaling in a transgenic mouse model of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2009, 21, 973–983. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Farrell, G.C.; Field, J.; Bell, D.R.; Gonzalez, F.J.; Robertson, G.R. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Investig. 2000, 105, 1067–1075. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Banerjee, A.; Yoo, S.-H.; Jang, S.; Gonzalez, F.J.; Song, B.-J. Critical role of cytochrome P4502E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J. Hepatol. 2012, 57, 860–866. [Google Scholar] [CrossRef]
- Zong, H.; Armoni, M.; Harel, C.; Karnieli, E.; Pessin, J.E. Cytochrome P-450 CYP2E1 knockout mice are protected against high-fat diet-induced obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E532–E539. [Google Scholar] [CrossRef]
- Petrasek, J.; Csak, T.; Szabo, G. Toll-like receptors in liver disease. Adv. Clin. Chem. 2012, 59, 155–201. [Google Scholar] [CrossRef]
- Kesar, V.; Odin, J.A. Toll-like receptors and liver disease. Liver Int. 2013. [Google Scholar] [CrossRef]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar]
- Seki, E.; Tsutsui, H.; Tsuji, N.M.; Hayashi, N.; Adachi, K.; Nakano, H.; Futatsugi-Yumikura, S.; Takeuchi, O.; Hoshino, K.; Akira, S. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 2002, 169, 3863–3868. [Google Scholar]
- Knoll, P.; Schlaak, J.; Uhrig, A.; Kempf, P.; Zum Büschenfelde, K.-H.M.; Gerken, G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J. Hepatol. 1995, 22, 226–229. [Google Scholar] [CrossRef]
- Ford, N.; Erdman, J.W., Jr. Lycopene and Cancer. In Carotenoids and Human Health; Springer: New York, NY, USA, 2013; pp. 193–214. [Google Scholar]
- Story, E.N.; Kopec, R.E.; Schwartz, S.J.; Harris, G.K. An update on the health effects of tomato lycopene. Annu. Rev. Food Sci. Technol. 2010, 1, 189–210. [Google Scholar]
- Giovannucci, E. Tomatoes, tomato-based products, lycopene, and cancer: Review of the epidemiologic literature. J. Natl. Cancer Inst. 1999, 91, 317–331. [Google Scholar] [CrossRef]
- Kopec, R.E.; Riedl, K.M.; Harrison, E.H.; Curley, R.W., Jr.; Hruszkewycz, D.P.; Clinton, S.K.; Schwartz, S.J. Identification and quantification of apo-lycopenals in fruits, vegetables, and human plasma. J. Agric. Food Chem. 2010, 58, 3290–3296. [Google Scholar]
- Wang, Y.; Ausman, L.M.; Greenberg, A.S.; Russell, R.M.; Wang, X.D. Dietary lycopene and tomato extract supplementations inhibit nonalcoholic steatohepatitis-promoted hepatocarcinogenesis in rats. Int. J. Cancer 2010, 126, 1788–1796. [Google Scholar]
- Wang, X.D. Lycopene metabolism and its biological significance. Am. J. Clin. Nutr. 2012, 96, 1214S–1222S. [Google Scholar] [CrossRef]
- Rao, A.V.; Ray, M.R.; Rao, L.G. Lycopene. Adv. Food Nutr. Res. 2006, 51, 99–164. [Google Scholar]
- Franceschi, S.; Bidoli, E.; Vecchia, C.L.; Talamini, R.; DʼAvanzo, B.; Negri, E. Tomatoes and risk of digestive-tract cancers. Int. J. Cancer 1994, 59, 181–184. [Google Scholar] [CrossRef]
- Colditz, G.A.; Branch, L.G.; Lipnick, R.J.; Willett, W.; Rosner, B.; Posner, B.; Hennekens, C. Increased green and yellow vegetable intake and lowered cancer deaths in an elderly population. Am. J. Clin. Nutr. 1985, 41, 32–36. [Google Scholar]
- Giovannucci, E.; Rimm, E.B.; Liu, Y.; Stampfer, M.J.; Willett, W.C. A prospective study of tomato products, lycopene, and prostate cancer risk. J. Natl. Cancer Inst. 2002, 94, 391–398. [Google Scholar] [CrossRef]
- Giovannucci, E.; Ascherio, A.; Rimm, E.B.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C. Intake of carotenoids and retino in relation to risk of prostate cancer. J. Natl. Cancer Inst. 1995, 87, 1767–1776. [Google Scholar] [CrossRef]
- Yuan, J.-M.; Gao, Y.-T.; Ong, C.-N.; Ross, R.K.; Mimi, C.Y. Prediagnostic level of serum retinol in relation to reduced risk of hepatocellular carcinoma. J. Natl. Cancer Inst. 2006, 98, 482–490. [Google Scholar] [CrossRef]
- Yu, M.-W.; Chiu, Y.-H.; Chiang, Y.-C.; Chen, C.-H.; Lee, T.-H.; Santella, R.M.; Chern, H.-D.; Liaw, Y.-F.; Chen, C.-J. Plasma carotenoids, glutathione S-transferase M1 andT1 genetic polymorphisms, and risk of hepatocellular carcinoma: Independent and interactive effects. Am. J. Epidemiol. 1999, 149, 621–629. [Google Scholar] [CrossRef]
- Erhardt, A.; Stahl, W.; Sies, H.; Lirussi, F.; Donner, A.; Haussinger, D. Plasma levels of vitamin E and carotenoids are decreased in patients with nonalcoholic steatohepatitis (NASH). Eur. J. Med. Res. 2011, 16, 76–78. [Google Scholar] [CrossRef]
- Huang, C.S.; Liao, J.W.; Hu, M.L. Lycopene inhibits experimental metastasis of human hepatoma SK-Hep-1 cells in athymic nude mice. J. Nutr. 2008, 138, 538–543. [Google Scholar]
- Huang, C.S.; Shih, M.K.; Chuang, C.H.; Hu, M.L. Lycopene inhibits cell migration and invasion and upregulates Nm23-H1 in a highly invasive hepatocarcinoma, SK-Hep-1 cells. J. Nutr. 2005, 135, 2119–2123. [Google Scholar]
- Astorg, P.; Gradelet, S.; Berges, R.; Suschetet, M. Dietary lycopene decreases the initiation of liver preneoplastic foci by diethylnitrosamine in the rat. Nutr. Cancer 1997, 29, 60–68. [Google Scholar] [CrossRef]
- Matsushima-Nishiwaki, R.; Shidoji, Y.; Nishiwaki, S.; Yamada, T.; Moriwaki, H.; Muto, Y. Suppression by carotenoids of microtenoids of microcystin-induced morphological changes in mouse hepatocytes. Lipids 1995, 30, 1029–1034. [Google Scholar] [CrossRef]
- Toledo, L.P.; Ong, T.P.; Pinho, A.L.; Jordao, A., Jr.; Vanucchi, H.; Moreno, F.S. Inhibitory effects of lutein and lycopene on placental glutathione S-transferase-positive preneoplastic lesions and DNA strand breakage induced in Wistar rats by the resistant hepatocyte model of hepatocarcinogenesis. Nutr. Cancer 2003, 47, 62–69. [Google Scholar] [CrossRef]
- Watanabe, S.; Kitade, Y.; Masaki, T.; Nishioka, M.; Satoh, K.; Nishino, H. Effects of lycopene and Sho-saiko-to on hepatocarcinogenesis in a rat model of spontaneous liver cancer. Nutr. Cancer 2001, 39, 96–101. [Google Scholar] [CrossRef]
- Lucia dos Anjos Ferreira, A.; Yeum, K.-J.; Russell, R.M.; Krinsky, N.I.; Tang, G. Enzymatic and oxidative metabolites of lycopene. J. Nutr. Biochem. 2004, 15, 493–502. [Google Scholar] [CrossRef]
- Britton, G.; Liaaen-Jensen, S.; Pfander, H.P. Carotenoids: Handbook; Springer: Basle, Switzerland, 2004. [Google Scholar]
- Kim, S.-J.; Nara, E.; Kobayashi, H.; Terao, J.; Nagao, A. Formation of cleavage products by autoxidation of lycopene. Lipids 2001, 36, 191–200. [Google Scholar] [CrossRef]
- Caris-Veyrat, C.; Schmid, A.; Carail, M.; Böhm, V. Cleavage products of lycopene produced by in vitro oxidations: Characterization and mechanisms of formation. J. Agric. Food Chem. 2003, 51, 7318–7325. [Google Scholar] [CrossRef]
- Rodriguez, E.B.; Rodriguez-Amaya, D.B. Lycopene epoxides and apo-lycopenals formed by chemical reactions and autoxidation in model systems and processed foods. J. Food Sci. 2009, 74, C674–C682. [Google Scholar] [CrossRef]
- DellaPenna, D.; Pogson, B.J. Vitamin synthesis in plants: Tocopherols and carotenoids. Annu. Rev. Plant Biol. 2006, 57, 711–738. [Google Scholar] [CrossRef]
- Khachik, F.; Goli, M.B.; Beecher, G.R.; Holden, J.; Lusby, W.R.; Tenorio, M.D.; Barrera, M.R. Effect of food preparation on qualitative and quantitative distribution of major carotenoid constituents of tomatoes and several green vegetables. J. Agric. Food Chem. 1992, 40, 390–398. [Google Scholar] [CrossRef]
- Kamer, M.; Pfander, H. Separation carotenoids by high-performance liquid chromatography: III. 1,2-Epoxycarotenoids. J. Chromatogr. 1984, 295, 295–298. [Google Scholar]
- Britton, G.; Goodwin, T.W. Carotene epoxides from the delta tomato mutant. Phytochemistry 1975, 14, 2530–2532. [Google Scholar] [CrossRef]
- Ben-Aziz, A.; Britton, G.; Goodwin, T.W. Carotene epoxides of Lycopersicon esculentum. Phytochemistry 1973, 12, 2759–2764. [Google Scholar] [CrossRef]
- Eroglu, A.; Harrison, E.H. Carotenoid metabolism in mammals, including man: Formation, occurrence, and function of apocarotenoids thematic review series: Fat-soluble vitamins: Vitamin A. J. Lipid Res. 2013, 54, 1719–1730. [Google Scholar] [CrossRef]
- Schwartz, S.H.; Qin, X.; Zeevaart, J.A. Characterization of a novel carotenoid cleavage dioxygenase from plants. J. Biol. Chem. 2001, 276, 25208–25211. [Google Scholar] [CrossRef]
- Rachel, E.K.; Steven, J.S. Carotenoid Cleavage Dioxygenase and Presence of Apo-Carotenoids in Biological Matrices. In Carotenoid Cleavage Products; American Chemical Society: Washington, DC, USA, 2013; pp. 31–41. [Google Scholar]
- Wilfried, S.; Fong-Chin, H.; Péter, M. Carotenoid Cleavage Dioxygenase Genes from Fruit. In Carotenoid Cleavage Products; American Chemical Society: Washington, DC, USA, 2013; pp. 11–19. [Google Scholar]
- Schwartz, S.H.; Qin, X.; Loewen, M.C. The biochemical characterization of two carotenoid cleavage enzymes from Arabidopsis indicates that a carotenoid-derived compound inhibits lateral branching. J. Biol. Chem. 2004, 279, 46940–46945. [Google Scholar] [CrossRef]
- Takitani, K.; Zhu, C.-L.; Inoue, A.; Tamai, H. Molecular cloning of the rat β-carotene 15,15′-monooxygenase gene and its regulation by retinoic acid. Eur. J. Nutr. 2006, 45, 320–326. [Google Scholar] [CrossRef]
- Von Lintig, J.; Vogt, K. Filling the gap in vitamin A research. Molecular identification of an enzyme cleaving β-carotene to retinal. J. Biol. Chem. 2000, 275, 11915–11920. [Google Scholar]
- Wyss, A.; Wirtz, G.; Woggon, W.-D.; Brugger, R.; Wyss, M.; Friedlein, A.; Bachmann, H.; Hunziker, W. Cloning and expression of β,β-carotene 15,15′-dioxygenase. Biochem. Biophys. Res. Commun. 2000, 271, 334–336. [Google Scholar] [CrossRef]
- Lindqvist, A.; Andersson, S. Biochemical properties of purified recombinant human β-carotene 15,15′-monooxygenase. J. Biol. Chem. 2002, 277, 23942–23948. [Google Scholar] [CrossRef]
- Redmond, T.M.; Gentleman, S.; Duncan, T.; Yu, S.; Wiggert, B.; Gantt, E.; Cunningham, F.X. Identification, expression, and substrate specificity of a mammalian β-carotene 15,15′-dioxygenase. J. Biol. Chem. 2001, 276, 6560–6565. [Google Scholar]
- Dela Sena, C.; Narayanasamy, S.; Riedl, K.M.; Curley, R.W.; Schwartz, S.J.; Harrison, E.H. Substrate specificity of purified recombinant human β-carotene 15,15′-oxygenase (BCO1). J. Biol. Chem. 2013. [Google Scholar] [CrossRef]
- Yan, W.; Jang, G.F.; Haeseleer, F.; Esumi, N.; Chang, J.; Kerrigan, M.; Campochiaro, M.; Campochiaro, P.; Palczewski, K.; Zack, D.J. Cloning and characterization of a human β,β-carotene-15,15′-dioxygenase that is highly expressed in the retinal pigment epithelium. Genomics 2001, 72, 193–202. [Google Scholar] [CrossRef]
- Nagao, A.; Olson, J.A. Enzymatic formation of 9-cis, 13-cis, and all-trans retinals from isomers of β-carotene. FASEB J. 1994, 8, 968–973. [Google Scholar]
- Ross, A.B.; Ruckle, J.; Synal, H.A.; Schulze-König, T.; Wertz, K.; Rümbeli, R.; Liberman, R.G.; Skipper, P.L.; Tannenbaum, S.R.; Bourgeois, A. Lycopene bioavailability and metabolism in humans: An accelerator mass spectrometry study. Am. J. Clin. Nutr. 2011, 93, 1263–1273. [Google Scholar] [CrossRef]
- Kiefer, C.; Hessel, S.; Lampert, J.M.; Vogt, K.; Lederer, M.O.; Breithaupt, D.E.; von Lintig, J. Identification and characterization of a mammalian enzyme catalyzing the asymmetric oxidative cleavage of provitamin A. J. Biol. Chem. 2001, 276, 14110–14116. [Google Scholar]
- Hu, K.Q.; Liu, C.; Ernst, H.; Krinsky, N.I.; Russell, R.M.; Wang, X.D. The biochemical characterization of ferret carotene-9′,10′-monooxygenase catalyzing cleavage of carotenoids in vitro and in vivo. J. Biol. Chem. 2006, 281, 19327–19338. [Google Scholar]
- Amengual, J.; Lobo, G.P.; Golczak, M.; Li, H.N.M.; Klimova, T.; Hoppel, C.L.; Wyss, A.; Palczewski, K.; von Lintig, J. A mitochondrial enzyme degrades carotenoids and protects against oxidative stress. FASEB J. 2011, 25, 948–959. [Google Scholar] [CrossRef]
- Ford, N.A.; Clinton, S.K.; von Lintig, J.; Wyss, A.; Erdman, J.W. Loss of carotene-9′,10′-monooxygenase expression increases serum and tissue lycopene concentrations in lycopene-fed mice. J. Nutr. 2010, 140, 2134–2138. [Google Scholar] [CrossRef]
- Lindqvist, A.; He, Y.G.; Andersson, S. Cell type-specific expression of β-carotene 9′,10′-monooxygenase in human tissues. J. Histochem. Cytochem. 2005, 53, 1403–1412. [Google Scholar] [CrossRef]
- Lietz, G.; Oxley, A.; Boesch-Saadatmandi, C.; Kobayashi, D. Importance of β-carotene 15,15′-monooxygenase 1 (BCMO1) and β-carotene 9′,10′-dioxygenase 2 (BCDO2) in nutrition and health. Mol. Nutr. Food Res. 2012, 56, 241–250. [Google Scholar] [CrossRef]
- He, M.; Cornelis, M.C.; Kraft, P.; van Dam, R.M.; Sun, Q.; Laurie, C.C.; Mirel, D.B.; Chasman, D.I.; Ridker, P.M.; Hunter, D.J. Genome-wide association study identifies variants at the IL18-BCO2 locus associated with interleukin-18 levels. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 885–890. [Google Scholar] [CrossRef]
- Tourniaire, F.; Minihane, A.M.; Hesketh, J.; Lietz, G. Do single nucleotide polymorphisms in β-carotene dioxygenase-2 (BCDO2) gene affect the postprandial response? Proc. Nutr. Soc. 2008, 67, E187. [Google Scholar]
- Tian, R.; Pitchford, W.S.; Morris, C.A.; Cullen, N.G.; Bottema, C.D. Genetic variation in the β,β-carotene-9′,10′-dioxygenase gene and association with fat colour in bovine adipose tissue and milk. Anim. Genet. 2010, 41, 253–259. [Google Scholar] [CrossRef]
- Berry, S.D.; Davis, S.R.; Beattie, E.M.; Thomas, N.L.; Burrett, A.K.; Ward, H.E.; Stanfield, A.M.; Biswas, M.; Ankersmit-Udy, A.E.; Oxley, P.E.; et al. Mutation in bovine β-carotene oxygenase 2 affects milk color. Genetics 2009, 182, 923–926. [Google Scholar] [CrossRef]
- Vage, D.I.; Boman, I.A. A nonsense mutation in the β-carotene oxygenase 2 (BCO2) gene is tightly associated with accumulation of carotenoids in adipose tissue in sheep (Ovis aries). BMC Genet. 2010, 11, 10. [Google Scholar] [CrossRef]
- Eriksson, J.; Larson, G.; Gunnarsson, U.; Bed’hom, B.; Tixier-Boichard, M.; Stromstedt, L.; Wright, D.; Jungerius, A.; Vereijken, A.; Randi, E.; et al. Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. PLoS Genet. 2008, 4, e1000010. [Google Scholar] [CrossRef]
- Giuliano, G.; Al-Babili, S.; von Lintig, J. Carotenoid oxygenases: Cleave it or leave it. Trends Plant Sci. 2003, 8, 145–149. [Google Scholar] [CrossRef]
- Gajic, M.; Zaripheh, S.; Sun, F.; Erdman, J.W., Jr. Apo-8′-lycopenal and apo-12′-lycopenal are metabolic products of lycopene in rat liver. J. Nutr. 2006, 136, 1552–1557. [Google Scholar]
- Gouranton, E.; Aydemir, G.; Reynaud, E.; Marcotorchino, J.; Malezet, C.; Caris-Veyrat, C.; Blomhoff, R.; Landrier, J.F.; Ruhl, R. Apo-10′-lycopenoic acid impacts adipose tissue biology via the retinoic acid receptors. Biochim. Biophys. Acta 2011, 1811, 1105–1114. [Google Scholar] [CrossRef]
- Amengual, J.; Widjaja-Adhi, M.A.K.; Rodriguez-Santiago, S.; Hessel, S.; Golczak, M.; Palczewski, K.; von Lintig, J. Two carotenoid oxygenases contribute to mammalian provitamin A metabolism. J. Biol. Chem. 2013, 288, 34081–34096. [Google Scholar] [CrossRef]
- Hébuterne, X.; Wang, X.-D.; Smith, D.; Tang, G.; Russell, R.M. In vivo biosynthesis of retinoic acid from β-carotene involves and excentric cleavage pathway in ferret intestine. J. Lipid Res. 1996, 37, 482–492. [Google Scholar]
- Shimizu, M.; Shirakami, Y.; Imai, K.; Takai, K.; Moriwaki, H. Acyclic retinoid in chemoprevention of hepatocellular carcinoma: Targeting phosphorylated retinoid X receptor-α for prevention of liver carcinogenesis. J. Carcinog. 2012, 11, 11. [Google Scholar] [CrossRef]
- Lian, F.; Smith, D.E.; Ernst, H.; Russell, R.M.; Wang, X.D. Apo-10′-lycopenoic acid inhibits lung cancer cell growth in vitro, and suppresses lung tumorigenesis in the A/J mouse model in vivo. Carcinogenesis 2007, 28, 1567–1574. [Google Scholar] [CrossRef]
- Yang, C.M.; Hu, T.Y.; Hu, M.L. Antimetastatic effects and mechanisms of apo-8′-lycopenal, an enzymatic metabolite of lycopene, against human hepatocarcinoma SK-Hep-1 cells. Nutr. Cancer 2012, 64, 274–285. [Google Scholar] [CrossRef]
- Chung, J.; Koo, K.; Lian, F.; Hu, K.Q.; Ernst, H.; Wang, X.D. Apo-10′-lycopenoic acid, a lycopene metabolite, increases sirtuin 1 mRNA and protein levels and decreases hepatic fat accumulation in ob/ob mice. J. Nutr. 2012, 142, 405–410. [Google Scholar] [CrossRef]
- Borel, P. Genetic variations involved in interindividual variability in carotenoid status. Mol. Nutr. Food Res. 2012, 56, 228–240. [Google Scholar] [CrossRef]
- Sakabe, T.; Tsuchiya, H.; Endo, M.; Tomita, A.; Ishii, K.; Gonda, K.; Murai, R.; Takubo, K.; Hoshikawa, Y.; Kurimasa, A. An antioxidant effect by acyclic retinoid suppresses liver tumor in mice. Biochem. Pharmacol. 2007, 73, 1405–1411. [Google Scholar] [CrossRef]
- Sano, T.; Kagawa, M.; Okuno, M.; Ishibashi, N.; Hashimoto, M.; Yamamoto, M.; Suzuki, R.; Kohno, H.; Matsushima-Nishiwaki, R.; Takano, Y.; et al. Prevention of rat hepatocarcinogenesis by acyclic retinoid is accompanied by reduction in emergence of both TGF-α-expressing oval-like cells and activated hepatic stellate cells. Nutr. Cancer 2005, 51, 197–206. [Google Scholar] [CrossRef]
- Kagawa, M.; Sano, T.; Ishibashi, N.; Hashimoto, M.; Okuno, M.; Moriwaki, H.; Suzuki, R.; Kohno, H.; Tanaka, T. An acyclic retinoid, NIK-333, inhibits N-diethylnitrosamine-induced rat hepatocarcinogenesis through suppression of TGF-α expression and cell proliferation. Carcinogenesis 2004, 25, 979–985. [Google Scholar] [CrossRef]
- Tatsukawa, H.; Sano, T.; Fukaya, Y.; Ishibashi, N.; Watanabe, M.; Okuno, M.; Moriwaki, H.; Kojima, S. Dual induction of caspase 3- and transglutaminase-dependent apoptosis by acyclic retinoid in hepatocellular carcinoma cells. Mol. Cancer 2011, 10, 4. [Google Scholar] [CrossRef]
- Suzui, M.; Shimizu, M.; Masuda, M.; Lim, J.T.; Yoshimi, N.; Weinstein, I.B. Acyclic retinoid activates retinoic acid receptor β and induces transcriptional activation of p21CIP1 in HepG2 human hepatoma cells. Mol. Cancer Ther. 2004, 3, 309–316. [Google Scholar]
- Suzui, M.; Masuda, M.; Lim, J.T.; Albanese, C.; Pestell, R.G.; Weinstein, I.B. Growth inhibition of human hepatoma cells by acyclic retinoid is associated with induction of p21(CIP1) and inhibition of expression of cyclin D1. Cancer Res. 2002, 62, 3997–4006. [Google Scholar]
- Nakamura, N.; Shidoji, Y.; Yamada, Y.; Hatakeyama, H.; Moriwaki, H.; Muto, Y. Induction of apoptosis by acyclic retinoid in the human hepatoma-derived cell line, HuH-7. Biochem. Biophys. Res. Commun. 1995, 207, 382–388. [Google Scholar] [CrossRef]
- Komi, Y.; Sogabe, Y.; Ishibashi, N.; Sato, Y.; Moriwaki, H.; Shimokado, K.; Kojima, S. Acyclic retinoid inhibits angiogenesis by suppressing the MAPK pathway. Lab. Investig. 2010, 90, 52–60. [Google Scholar] [CrossRef]
- Muto, Y.; Moriwaki, H.; Saito, A. Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. N. Engl. J. Med. 1999, 340, 1046–1047. [Google Scholar] [CrossRef]
- Muto, Y.; Moriwaki, H.; Ninomiya, M.; Adachi, S.; Saito, A.; Takasaki, K.T.; Tanaka, T.; Tsurumi, K.; Okuno, M.; Tomita, E.; et al. Prevention of second primary tumors by an acyclic retinoid, polyprenoic acid, in patients with hepatocellular carcinoma. Hepatoma Prevention Study Group. N. Engl. J. Med. 1996, 334, 1561–1567. [Google Scholar] [CrossRef]
- Okita, K.; Matsui, O.; Kumada, H.; Tanaka, K.; Kaneko, S.; Moriwaki, H.; Izumi, N.; Okusaka, T.; Ohashi, Y.; Makuuchi, M. Effect of peretinoin on recurrence of hepatocellular carcinoma (HCC): Results of a phase II/III randomized placebo-controlled trial. J. Clin. Oncol. 2010, 28, 4024. [Google Scholar]
- Hadad, N.; Levy, R. The synergistic anti-inflammatory effect of lycopene, lutein, β-carotene and carnosic acid combinations via redox-based inhibition of NF-κB signaling. Free Radic. Biol. Med. 2012, 53, 1381–1391. [Google Scholar] [CrossRef]
- Simone, R.E.; Russo, M.; Catalano, A.; Monego, G.; Froehlich, K.; Boehm, V.; Palozza, P. Lycopene inhibits NF-κB-mediated IL-8 expression and changes redox and PPARγ signalling in cigarette smoke—Stimulated macrophages. PLoS One 2011, 6, e19652. [Google Scholar]
- Gouranton, E.; Thabuis, C.; Riollet, C.; Malezet-Desmoulins, C.; El Yazidi, C.; Amiot, M.J.; Borel, P.; Landrier, J.F. Lycopene inhibits proinflammatory cytokine and chemokine expression in adipose tissue. J. Nutr. Biochem. 2011, 22, 642–648. [Google Scholar] [CrossRef]
- Feng, D.; Ling, W.-H.; Duan, R.-D. Lycopene suppresses LPS-induced NO and IL-6 production by inhibiting the activation of ERK, p38MAPK, and NF-κB in macrophages. Inflamm. Res. 2010, 59, 115–121. [Google Scholar] [CrossRef]
- Kim, G.Y.; Kim, J.H.; Ahn, S.C.; Lee, H.J.; Moon, D.O.; Lee, C.M.; Park, Y.M. Lycopene suppresses the lipopolysaccharide-induced phenotypic and functional maturation of murine dendritic cells through inhibition of mitogen-activated protein kinases and nuclear factor-κB. Immunology 2004, 113, 203–211. [Google Scholar] [CrossRef]
- Marcotorchino, J.; Romier, B.; Gouranton, E.; Riollet, C.; Gleize, B.; Malezet-Desmoulins, C.; Landrier, J.F. Lycopene attenuates LPS-induced TNF-α secretion in macrophages and inflammatory markers in adipocytes exposed to macrophage-conditioned media. Mol. Nutr. Food Res. 2012, 56, 725–732. [Google Scholar] [CrossRef]
- De Azevedo Melo Luvizotto, R.; Nascimento, A.F.; Imaizumi, E.; Pierine, D.T.; Conde, S.J.; Correa, C.R.; Yeum, K.-J.; Ferreira, A.L.A. Lycopene supplementation modulates plasma concentrations and epididymal adipose tissue mRNA of leptin, resistin and IL-6 in diet-induced obese rats. Br. J. Nutr. 2013, 110, 1803–1809. [Google Scholar] [CrossRef]
- Huang, C.-S.; Fan, Y.-E.; Lin, C.-Y.; Hu, M.-L. Lycopene inhibits matrix metalloproteinase-9 expression and down-regulates the binding activity of nuclear factor-κB and stimulatory protein-1. J. Nutr. Biochem. 2007, 18, 449–456. [Google Scholar] [CrossRef]
- Catalano, A.; Simone, R.E.; Cittadini, A.; Reynaud, E.; Caris-Veyrat, C.; Palozza, P. Comparative antioxidant effects of lycopene, apo-10′-lycopenoic acid and apo-14′-lycopenoic acid in human macrophages exposed to H2O2 and cigarette smoke extract. Food Chem. Toxicol. 2013, 51, 71–79. [Google Scholar] [CrossRef]
- Clinton, S.K. Lycopene: Chemistry, biology, and implications for human health and disease. Nutr. Rev. 1998, 56, 35–51. [Google Scholar] [CrossRef]
- Lian, F.; Wang, X.D. Enzymatic metabolites of lycopene induce Nrf2-mediated expression of phase II detoxifying/antioxidant enzymes in human bronchial epithelial cells. Int. J. Cancer 2008, 123, 1262–1268. [Google Scholar] [CrossRef]
- Liu, C.; Lian, F.; Smith, D.E.; Russell, R.M.; Wang, X.-D. Lycopene supplementation inhibits lung squamous metaplasia and induces apoptosis via up-regulating insulin-like growth factor-binding protein 3 in cigarette smoke-exposed ferrets. Cancer Res. 2003, 63, 3138–3144. [Google Scholar]
- Ip, B.C.; Hu, K.Q.; Liu, C.; Smith, D.E.; Obin, M.S.; Ausman, L.M.; Wang, X.D. Lycopene metabolite, apo-10′-lycopenoic acid, inhibits diethylnitrosamine-initiated, high fat diet-promoted hepatic inflammation and tumorigenesis in mice. Cancer Prev. Res. 2013, 6, 1304–1316. [Google Scholar] [CrossRef]
- Agarwal, S.; Rao, A.V. Tomato lycopene and its role in human health and chronic diseases. Can. Med. Assoc. J. 2000, 163, 739–744. [Google Scholar]
- Agarwal, S.; Rao, A.V. Tomato lycopene and low density lipoprotein oxidation: A human dietary intervention study. Lipids 1998, 33, 981–984. [Google Scholar] [CrossRef]
- Sies, H.; Stahl, W. Vitamins E and C, β-carotene, and other carotenoids as antioxidants. Am. J. Clin. Nutr. 1995, 62, 1315S–1321S. [Google Scholar]
- Palozza, P.; Parrone, N.; Catalano, A.; Simone, R. Tomato lycopene and inflammatory cascade: Basic interactions and clinical implications. Curr. Med. Chem. 2010, 17, 2547–2563. [Google Scholar] [CrossRef]
- Mein, J.R.; Lian, F.; Wang, X.D. Biological activity of lycopene metabolites: Implications for cancer prevention. Nutr. Rev. 2008, 66, 667–683. [Google Scholar] [CrossRef]
- Yang, C.M.; Huang, S.M.; Liu, C.L.; Hu, M.L. Apo-8′-lycopenal induces expression of HO-1 and NQO-1 via the ERK/p38-Nrf2-ARE pathway in human HepG2 cells. J. Agric. Food Chem. 2012, 60, 1576–1585. [Google Scholar] [CrossRef]
- Ip, B.C.; Wang, X.D. ; Tufts University, Boston, MA, USA. Unpublished work. 2013.
- Ando, N.; Shimizu, M.; Okuno, M.; Matsushima-Nishiwaki, R.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. Expression of retinoid X receptor α is decreased in 3′-methyl-4-dimethylaminoazobenzene-induced hepatocellular carcinoma in rats. Oncol. Rep. 2007, 18, 879–884. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Mafune, K.; Mori, M.; Shiraishi, T.; Imamura, H.; Takayama, T.; Makuuchi, M. Overexpression of MMP-9 correlates with growth of small hepatocellular carcinoma. Int. J. Oncol. 2000, 17, 237–243. [Google Scholar]
- Miao, B.; Wang, X.D.; Tufts University, Boston, MA, USA. Unpublished work. 2012.
- Gao, Z.; Zhang, J.; Kheterpal, I.; Kennedy, N.; Davis, R.J.; Ye, J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J. Biol. Chem. 2011, 286, 22227–22234. [Google Scholar] [CrossRef]
- Ford, J.; Ahmed, S.; Allison, S.; Jiang, M.; Milner, J. JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle 2008, 7, 3091–3097. [Google Scholar] [CrossRef]
- Yamamoto, M.; Iguchi, G.; Fukuoka, H.; Suda, K.; Bando, H.; Takahashi, M.; Nishizawa, H.; Seino, S.; Takahashi, Y. SIRT1 regulates adaptive response of the growth hormone—Insulin-like growth factor-I axis under fasting conditions in liver. Proc. Natl. Acad. Sci. USA 2013, 110, 14948–14953. [Google Scholar]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Baur, J.A.; Ungvari, Z.; Minor, R.K.; Le Couteur, D.G.; de Cabo, R. Are sirtuins viable targets for improving healthspan and lifespan? Nat. Rev. Drug Discov. 2012, 11, 443–461. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ip, B.C.; Wang, X.-D. Non-Alcoholic Steatohepatitis and Hepatocellular Carcinoma: Implications for Lycopene Intervention. Nutrients 2014, 6, 124-162. https://doi.org/10.3390/nu6010124
Ip BC, Wang X-D. Non-Alcoholic Steatohepatitis and Hepatocellular Carcinoma: Implications for Lycopene Intervention. Nutrients. 2014; 6(1):124-162. https://doi.org/10.3390/nu6010124
Chicago/Turabian StyleIp, Blanche C., and Xiang-Dong Wang. 2014. "Non-Alcoholic Steatohepatitis and Hepatocellular Carcinoma: Implications for Lycopene Intervention" Nutrients 6, no. 1: 124-162. https://doi.org/10.3390/nu6010124