Regulation of Vitamin C Homeostasis during Deficiency



Abstract
:1. Introduction
2. Vitamin C Transport
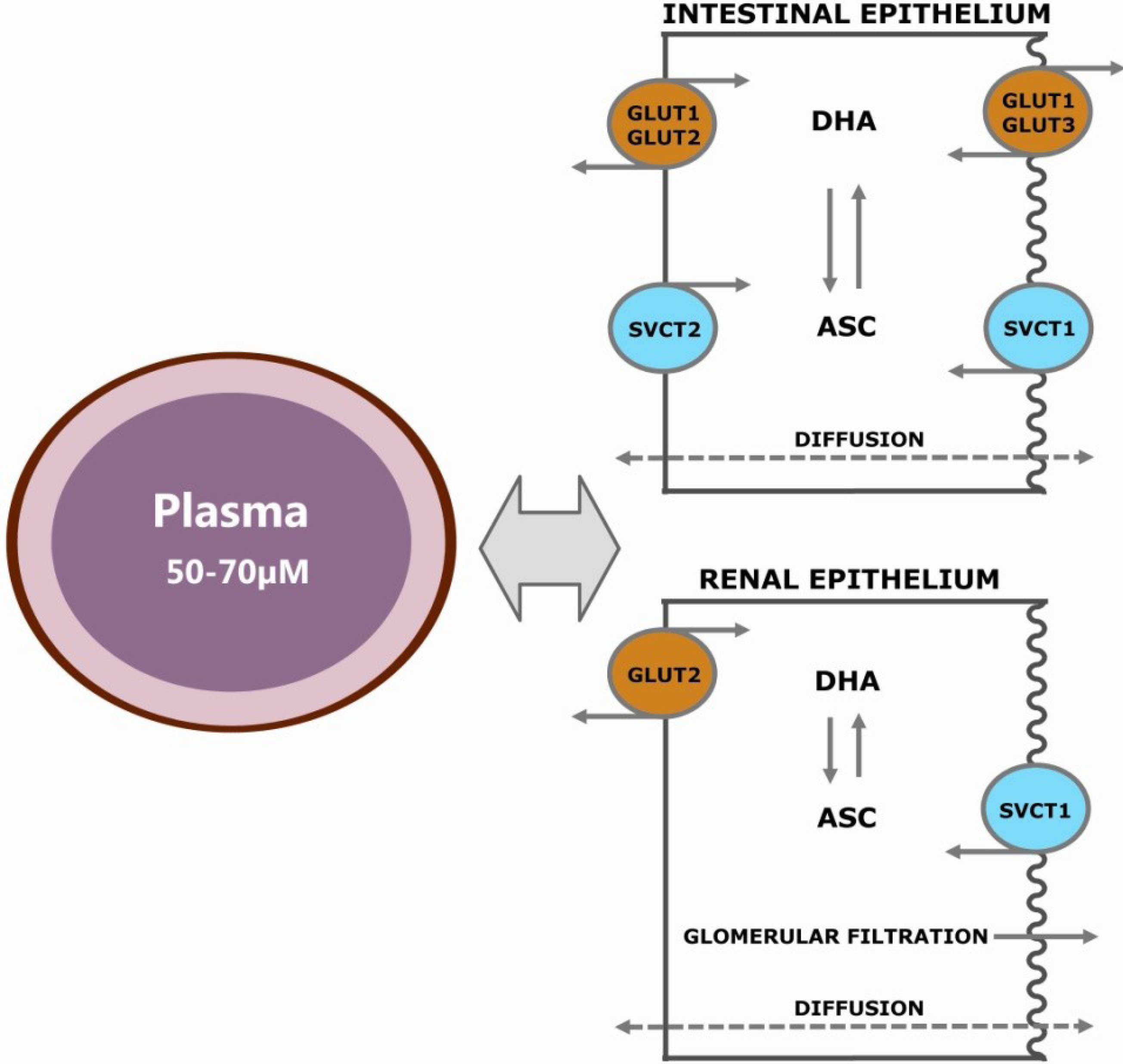
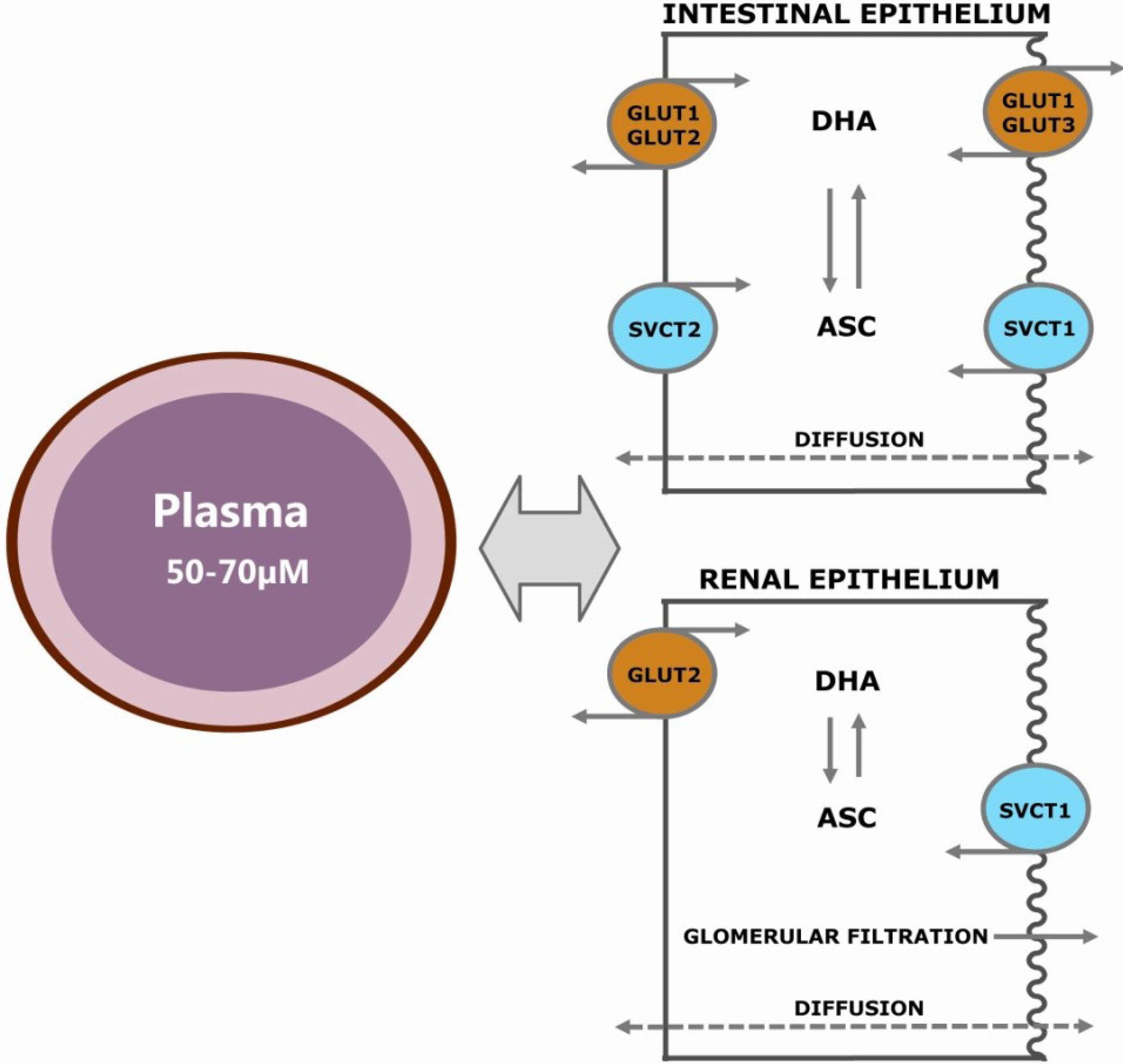
2.1. Passive Diffusion
2.2. Facilitated Diffusion
2.3. Recycling
2.4. Active Transport of ASC
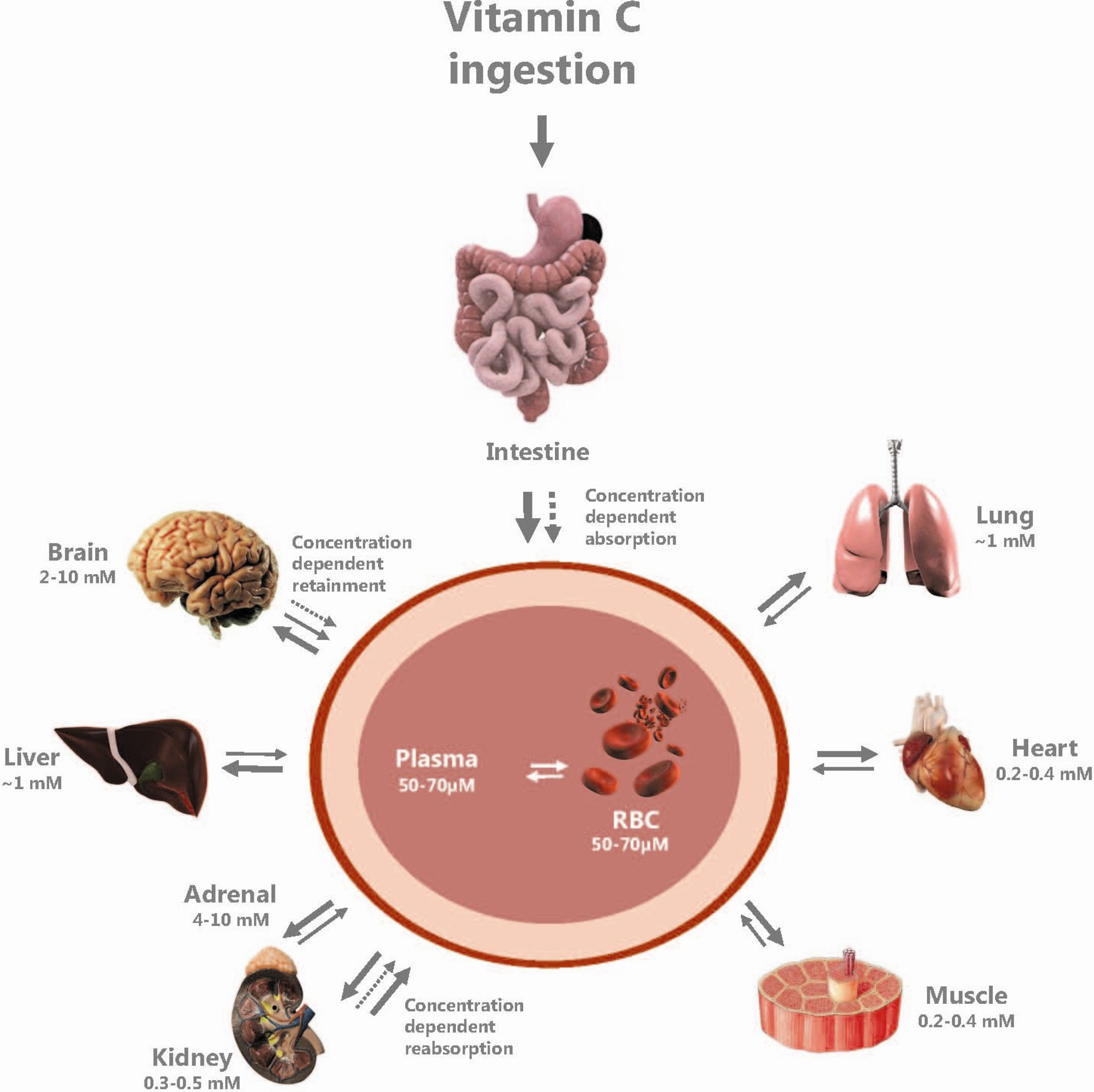
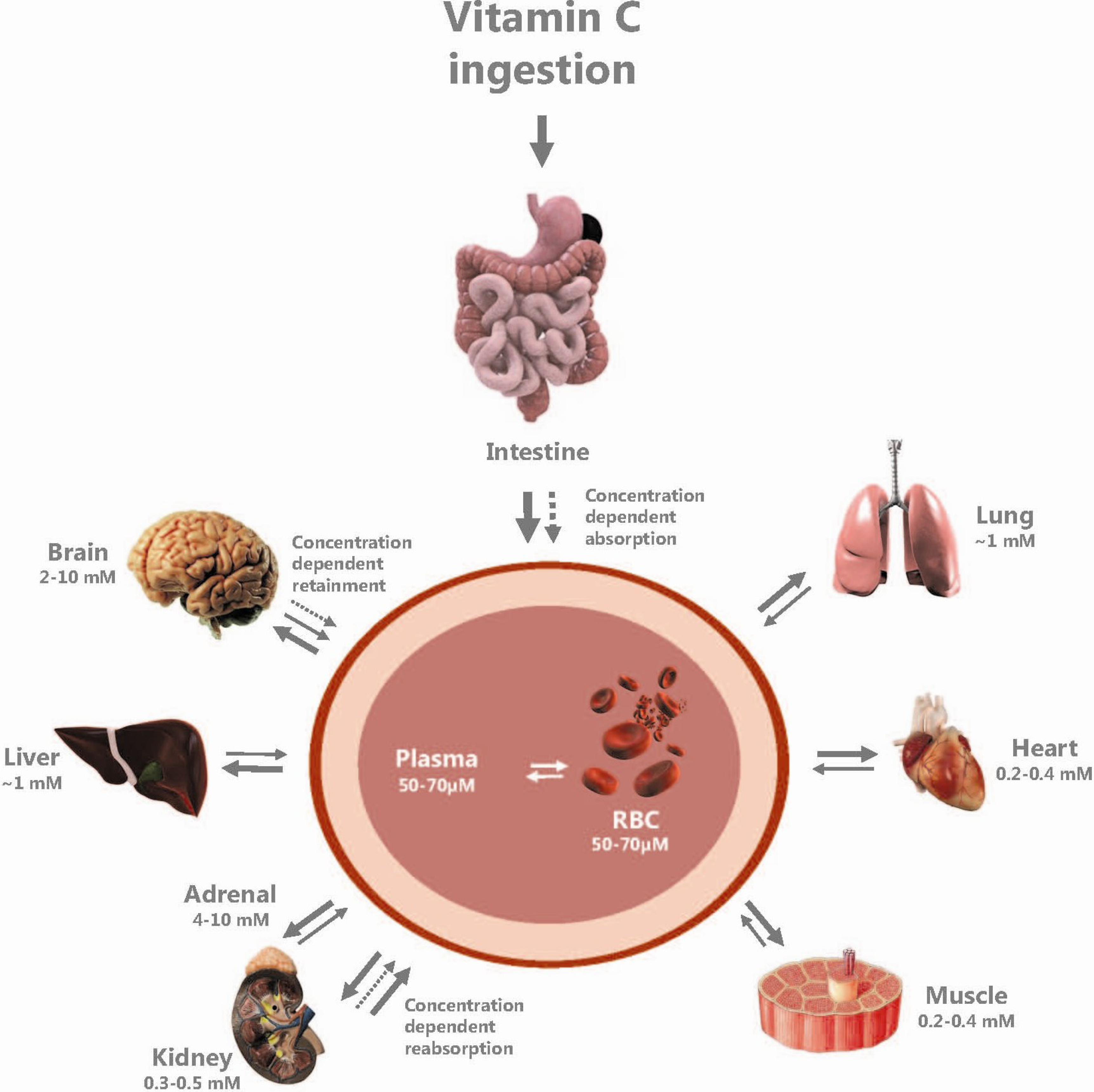
2.5. Vitamin C Distribution
3. Regulation of VitC Transport by Substrate Concentrations

{kind=link}
{kind=link}
| Cell line | Deficiency regimen | Principal findings |
|---|---|---|
| Human intestinal cell line (Caco-2 TC7) [125] | Culture medium was supplemented with ASC at concentrations of 45 μg/mL, 450 μg/mL or 4.5 mg/mL. | Exposure to 4.5 mg/mL ASC significantly reduced the ASC uptake by 50% and expression of SVCT1 mRNA by 77% compared to control conditions. |
| Primary human platelets [124] | Culture medium’s ASC concentration was reduced to 30% of standard levels. | Vmax increased by 240% in response to the reduction of ASC concentration. A subsequent increase in SVCT2 protein level was reported. An ASC supplement of 500 μM only slightly decreased SVCT2 levels. |
| Human hepatic cell line (HepG2) [127] | Cells were incubated with 10% fetal bovine serum containing 10 mM ASC (supplemented), 0.7 μM ASC (control), 0 μM ASC (depleted). | ASC-supplemented cells responded with a reduced transport of ASC and a coherent reduced SVCT1 expression (mRNA and protein). Depleted cells displayed increased ASC transport and increased SVCT1 expression. No changes were found for SVCT2. |
| Primary rat astrocytes [131] | Astrocytes were incubated with ASC (from 0 to 300 μM) in culture medium prior to measurements of uptake rates. | ASC depletion of culture medium increased the Vmax by 15% after one hour and 20% after 6 h. ASC repletion resulted in a 20% decrease after one hour and 30% after 18 h. |
| Rat osteosarcoma cell line (ROS 17/2.8) [132] | Cells were incubated with ASC (from 0 to 300 μM) in culture medium prior to measurements of uptake rates. | ASC depletion of culture medium increased the Vmax by 41% after six hours. ASC repletion resulted in a 40% decrease after six hours. |
| Porcine proximal tubule cell line (LLC-PK1) [128] | Cells were incubated with increasing concentrations of ASC in culture medium (10, 25, 50 and 100 μM). | Increasing concentrations in ASC reduced apical SVCT1 expression and induced translocation of SVCT1 to the cytoplasm before the signal was diminished. |
| Animals species | Vitamin C regimen | Principal findings |
|---|---|---|
| Guinea pig [129] | ASC content in diet was increased by five- and 25-times compared to standard diets. | A reduction in ASC influx across the ileum by 32%–52% in animals fed high ASC diet compared to controls (standard). |
| Guinea pig [130] | Animals received either high (5000 mg/kg diet), low (0 mg/kg diet) or control (maintenance) (200 mg/kg diet) levels of vitC. | A high vitC level (hypervitaminosis) reduced the ASC rate of uptake across the intestinal brush border by 25%–50% compared to controls. Hypovitaminotic animals were not found to be different from controls. |
| Guinea pig [31] | Young and old animals, long-term on either control (325 mg vitC/kg) or deficient (100 mg vitC/kg) diets. | No effect of dietary vitC regimen on the expression of SVCT1 or SVCT2 mRNA in liver or brain. |
| Knockout mice (smp30/gnl−/−) [30] | Effects of vitC depletion vs. control (1.5 g vitC/L water) in wild-type (WT) and knockout (KO) mice. | In KO, mice vitC depletion increased SVCT1 and SVCT2 mRNA expression in the liver (by 21 and 55%, respectively) and increased SVCT1 by 55% in the small intestine compared to control counterparts. No changes were found in the kidney or cerebellum. In WT-mice, depletion increased SVCT2 expression in the small intestine by 43%. |
| Knockout mice (gulo−/−) [29] | Gulo−/− mice exposed to different ASC levels (drinking water): high 3.33 g/L; standard 0.33 g/L; low 0.033 g/L and depletion 0 g/L. WT mice were included as controls. | Depletion resulted in an increased mRNA expression of SVCT2 in the liver compared to WT controls. A trend towards increased protein levels of SVCT2 in liver and cerebellum was reported, although it did not reach a statistical level of significance. |
4. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Frei, B.; Birlouez-Aragon, I.; Lykkesfeldt, J. Authors’ perspective: What is the optimum intake of vitamin C in humans? Crit. Rev. Food Sci. Nutr. 2012, 52, 815–829. [Google Scholar] [CrossRef]
- Agarwal, M.; Mehta, P.K.; Dwyer, J.H.; Dwyer, K.M.; Shircore, A.M.; Nordstrom, C.K.; Sun, P.; Paul-Labrador, M.; Yang, Y.; Merz, C.N.B. Differing relations to early atherosclerosis between vitamin C from supplements vs. FOOD in the Los Angeles atherosclerosis study: A prospective cohort study. Open Cardiovasc. Med. J. 2012, 6, 113–121. [Google Scholar]
- Langlois, M.; Duprez, D.; Delanghe, J.; de Buyzere, M.; Clement, D.L. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation 2001, 103, 1863–1868. [Google Scholar] [CrossRef]
- Myint, P.K.; Luben, R.N.; Welch, A.A.; Bingham, S.A.; Wareham, N.J.; Khaw, K.-T. Plasma vitamin C concentrations predict risk of incident stroke over 10 y in 20,649 participants of the European Prospective Investigation into Cancer–Norfolk prospective population study. Am. J. Clin. Nutr. 2008, 87, 64–69. [Google Scholar]
- Simon, J.A.; Hudes, E.S.; Browner, W.S. Serum ascorbic acid and cardiovascular disease prevalence in US adults. Epidemiology 1998, 9, 316–321. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency increase lifestyle-associated vascular disease progression? Evidence based on experimental and clinical studies. Antioxid. Redox Signal. 2013, in press. [Google Scholar]
- Tveden-Nyborg, P.; Johansen, L.K.; Raida, Z.; Villumsen, C.K.; Larsen, J.O.; Lykkesfeldt, J. Vitamin C deficiency in early postnatal life impairs spatial memory and reduces the number of hippocampal neurons in guinea pigs. Am. J. Clin. Nutr. 2009, 90, 540–546. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal vitamin C deficiency during pregnancy persistently impairs hippocampal neurogenesis in offspring of guinea pigs. PLoS One 2012, 7, e48488. [Google Scholar] [CrossRef] [Green Version]
- Harrison, F.E.; Dawes, S.M.; Meredith, M.E.; Babaev, V.R.; Li, L.; May, J.M. Low vitamin C and increased oxidative stress and cell death in mice that lack the sodium-dependent vitamin C transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef]
- Smith, J.L.; Hodges, R.E. Serum levels of vitamin C in relation to dietary and supplemental intake of vitamin C in smokers and nonsmokers. Ann. N. Y. Acad. Sci. 1987, 498, 144–152. [Google Scholar] [CrossRef]
- Touvier, M.; Lioret, S.; Vanrullen, I.; Bocle, J.-C.; Boutron-Ruault, M.-C.; Berta, J.-L.; Volatier, J.-L. Vitamin and mineral inadequacy in the French population: Estimation and application for the optimization of food fortification. Int. J. Vitam. Nutr. Res. 2006, 76, 343–351. [Google Scholar] [CrossRef]
- Schleicher, R.L.; Carroll, M.D.; Ford, E.S.; Lacher, D.A. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am. J. Clin. Nutr. 2009, 90, 1252–1263. [Google Scholar] [CrossRef]
- Simon, J.A.; Hudes, E.S. Serum ascorbic acid and cardiovascular disease prevalence in US adults: The Third National Health and Nutrition Examination Survey (NHANES III). Ann. Epidemiol. 1999, 9, 358–365. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Priemé, H.; Loft, S.; Poulsen, H.E. Effect of smoking cessation on plasma ascorbic acid concentration. BMJ 1996, 313, 91. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Smoking depletes vitamin C: Should smokers be recommended to take supplements? Cigar. Smoke Oxid. Stress 2006, 237–260. [Google Scholar] [CrossRef]
- Wei, W.; Kim, Y.; Boudreau, N. Association of smoking with serum and dietary levels of antioxidants in adults: NHANES III, 1988–1994. Am. J. Public Health 2001, 91, 258–264. [Google Scholar] [CrossRef]
- Frikke-Schmidt, H.; Tveden-Nyborg, P.; Lykkesfeldt, J. Vitamin C in Human Nutrition. In Vitamins in the Prevention of Human Diseases; Herrmann, W., Obeid, R., Eds.; de Gruyter, Walter: Berlin, Germany, 2011. [Google Scholar]
- Lykkesfeldt, J.; Loft, S.; Nielsen, J.B.; Poulsen, H.E. Ascorbic acid and dehydroascorbic acid as biomarkers of oxidative stress caused by smoking. Am. J. Clin. Nutr. 1997, 65, 959–963. [Google Scholar]
- Lykkesfeldt, J.; Poulsen, H.E. Is vitamin C supplementation beneficial? Lessons learned from randomised controlled trials. Br. J. Nutr. 2010, 103, 1251–1259. [Google Scholar] [CrossRef]
- Wilson, J.X. Vitamin C Transport in Animals and Plants. In Vitamin C—Functions and Biochemistry in Animals and Plants; Asard, H., May, J.M., Smirnoff, N., Eds.; BIOS Scientific Publishers: London, UK, 2004. [Google Scholar]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1997, 82, 1213–1223. [Google Scholar] [CrossRef]
- Frikke-Schmidt, H.; Tveden-Nyborg, P.; Birck, M.M.; Lykkesfeldt, J. High dietary fat and cholesterol exacerbates chronic vitamin C deficiency in guinea pigs. Br. J. Nutr. 2011, 105, 54–61. [Google Scholar] [CrossRef]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef]
- Levine, M.; Padayatty, S.J.; Espey, M.G. Vitamin C: A concentration-function approach yields pharmacology and therapeutic discoveries. Adv. Nutr. 2011, 2, 78–88. [Google Scholar] [CrossRef]
- Levine, M.; Rumsey, S.C.; Daruwala, R.; Park, J.B.; Wang, Y. Criteria and recommendations for vitamin C intake. JAMA 1999, 281, 1415–1423. [Google Scholar] [CrossRef]
- Harris, L.J.; Ray, S.N.; Ward, A. The excretion of vitamin C in human urine and its dependence on the dietary intake. Biochem. J. 1933, 27, 2011–2015. [Google Scholar]
- Meredith, M.E.; Harrison, F.E.; May, J.M. Differential regulation of the ascorbic acid transporter SVCT2 during development and in response to ascorbic acid depletion. Biochem. Biophys. Res. Commun. 2011, 414, 737–742. [Google Scholar] [CrossRef]
- Amano, A.; Aigaki, T.; Maruyama, N.; Ishigami, A. Ascorbic acid depletion enhances expression of the sodium-dependent vitamin C transporters, SVCT1 and SVCT2, and uptake of ascorbic acid in livers of SMP30/GNL knockout mice. Arch. Biochem. Biophys. 2010, 496, 38–44. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Hasselholt, S.; Miyashita, N.; Moos, T.; Poulsen, H.E.; Lykkesfeldt, J. Chronic vitamin C deficiency does not accelerate oxidative stress in ageing brains of guinea pigs. Basic Clin. Pharmacol. Toxicol. 2012, 110, 524–529. [Google Scholar] [CrossRef]
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for l-gulono-gamma-lactone oxidase, the enzyme for l-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688. [Google Scholar]
- Chatterjee, I. Evolution and the biosynthesis of ascorbic acid. Science 1973, 182, 1271–1272. [Google Scholar]
- Nandi, A.; Mukhopadhyay, C.K.; Ghosh, M.K.; Chattopadhyay, D.J.; Chatterjee, I.B. Evolutionary significance of vitamin C biosynthesis in terrestrial vertebrates. Free Radic. Biol. Med. 1997, 22, 1047–1054. [Google Scholar] [CrossRef]
- Malo, C.; Wilson, J.X. Glucose modulates vitamin C transport in adult human small intestinal brush border membrane vesicles. J. Nutr. 2000, 130, 63–69. [Google Scholar]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Trueba, G.P.; Poulsen, H.E.; Christen, S. Vitamin C deficiency in weanling guinea pigs: Differential expression of oxidative stress and DNA repair in liver and brain. Br. J. Nutr. 2007, 98, 1116–1119. [Google Scholar]
- Hughes, R.E.; Hurley, R.J.; Jones, P.R. The retention of ascorbic acid by guinea-pig tissues. Br. J. Nutr. 1971, 26, 433–438. [Google Scholar] [CrossRef]
- Kuo, C.-H.; Yonehara, N.; Yoshida, H. Subcellular ascorbic acid in scorbutic guinea pig brain. J. Nutr. Sci. Vitaminol. 1979, 25, 9–13. [Google Scholar] [CrossRef]
- Wilson, J.X. Regulation of vitamin C transport. Annu. Rev. Nutr. 2005, 25, 105–125. [Google Scholar] [CrossRef]
- Rose, R.C. Solubility properties of reduced and oxidized ascorbate as determinants of membrane permeation. Biochim. Biophys. Acta 1987, 924, 254–256. [Google Scholar] [CrossRef]
- Wilson, J.X.; Dixon, S.J. High-affinity sodium-dependent uptake of ascorbic acid by rat osteoblasts. J. Membr. Biol. 1989, 111, 83–91. [Google Scholar] [CrossRef]
- Wagner, E.S.; White, W.; Jennings, M.; Bennett, K. The entrapment of [14C] ascorbic acid in human erythrocytes. Biochim. Biophys. Acta 1987, 902, 133–136. [Google Scholar] [CrossRef]
- Goldenberg, H.; Schweinzer, E. Transport of vitamin C in animal and human cells. J. Bioenerg. Biomembr. 1994, 26, 359–367. [Google Scholar] [CrossRef]
- Washko, P.W.; Wang, Y.; Levine, M. Ascorbic acid recycling in human neutrophils. J. Biol. Chem. 1993, 268, 15531–15535. [Google Scholar]
- Hughes, R.; Maton, S. The passage of vitamin C across the erythrocyte membrane. Br. J. Haematol. 1968, 14, 247–253. [Google Scholar] [CrossRef]
- Rumsey, S.C.; Daruwala, R.; Al-Hasani, H.; Zarnowski, M.J.; Simpson, I.A.; Levine, M. Dehydroascorbic acid transport by GLUT4 in Xenopus Oocytes and isolated rat adipocytes. J. Biol. Chem. 2000, 275, 28246–28253. [Google Scholar]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar]
- Vera, J.C.; Rivas, C.I.; Fischbarg, J.; Golde, D.W. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 1993, 364, 79–82. [Google Scholar] [CrossRef]
- Mardones, L.; Ormazabal, V.; Romo, X.; Jaña, C.; Binder, P.; Peña, E.; Vergara, M.; Zúñiga, F.A. The glucose transporter-2 (GLUT2) is a low affinity dehydroascorbic acid transporter. Biochem. Biophys. Res. Commun. 2011, 410, 7–12. [Google Scholar] [CrossRef]
- Zhao, F.-Q.; Keating, A.F. Functional properties and genomics of glucose transporters. Curr. Genomics 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Liang, W.-J.; Johnson, D.; Jarvis, S.M. Vitamin C transport systems of mammalian cells. Mol. Membr. Biol. 2001, 18, 87–95. [Google Scholar] [CrossRef]
- Vera, J.C.; Rivas, C.I.; Velásquez, F.V.; Zhang, R.H.; Concha, I.I.; Golde, D.W. Resolution of the facilitated transport of dehydroascorbic acid from its intracellular accumulation as ascorbic acid. J. Biol. Chem. 1995, 270, 23706–23712. [Google Scholar]
- Dhariwal, K.R.; Hartzell, W.O.; Levine, M. Ascorbic acid and dehydroascorbic acid measurements in human plasma and serum. Am. J. Clin. Nutr. 1991, 54, 712–716. [Google Scholar]
- Rumsey, S.C.; Levine, M. Absorption, transport, and disposition of ascorbic acid in humans. J. Nutr. Biochem. 1998, 9, 116–130. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Increased oxidative damage in vitamin C deficiency is accompanied by induction of ascorbic acid recycling capacity in young but not mature guinea pigs. Free Radic. Res. 2002, 36, 567–574. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Viscovich, M.; Poulsen, H.E. Ascorbic acid recycling in human erythrocytes is induced by smoking in vivo. Free Radic. Biol. Med. 2003, 35, 1439–1447. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.-C.; Whitesell, R.R.; Cobb, C.E. Ascorbate recycling in human erythrocytes: Role of GSH in reducing dehydroascorbate. Free Radic. Biol. Med. 1996, 20, 543–551. [Google Scholar] [CrossRef]
- May, J.M.; Mendiratta, S.; Hill, K.E.; Burk, R.F. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J. Biol. Chem. 1997, 272, 22607–22610. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.-C.; Mendiratta, S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch. Biochem. Biophys. 1998, 349, 281–289. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.-C.; Morrow, J.D.; Cobb, C.E. Ascorbate-dependent protection of human erythrocytes against oxidant stress generated by extracellular diazobenzene sulfonate. Biochem. Pharmacol. 2000, 60, 47–53. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.-C.; Cobb, C.E. Extracellular reduction of the ascorbate free radical by human erythrocytes. Biochem. Biophys. Res. Commun. 2000, 267, 118–123. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.-C.; Morrow, J.D. Mechanisms of ascorbic acid recycling in human erythrocytes. Biochim. Biophys. Acta 2001, 1528, 159–166. [Google Scholar]
- May, J.M.; Qu, Z.-C.; Cobb, C.E. Recycling of the ascorbate free radical by human erythrocyte membranes. Free Radic. Biol. Med. 2001, 31, 117–124. [Google Scholar] [CrossRef]
- Mendiratta, S.; Qu, Z.-C.; May, J.M. Erythrocyte ascorbate recycling: Antioxidant effects in blood. Free Radic. Biol. Med. 1998, 24, 789–797. [Google Scholar] [CrossRef]
- Mendiratta, S.; Qu, Z.-C.; May, J.M. Enzyme-dependent ascorbate recycling in human erythrocytes: Role of thioredoxin reductase. Free Radic. Biol. Med. 1998, 25, 221–228. [Google Scholar] [CrossRef]
- Winkler, B.S.; Orselli, S.M.; Rex, T.S. The redox couple between glutathione and ascorbic acid: A chemical and physiological perspective. Free Radic. Biol. Med. 1994, 17, 333–349. [Google Scholar] [CrossRef]
- Park, J.B.; Levine, M. Purification, cloning and expression of dehydroascorbic acid-reducing activity from human neutrophils: Identification as glutaredoxin. Biochem. J. 1996, 315, 931–938. [Google Scholar]
- Wells, W.W.; Xu, D.P. Dehydroascorbate reduction. J. Bioenerg. Biomembr. 1994, 26, 369–377. [Google Scholar] [CrossRef]
- Wells, W.W.; Xu, D.P.; Yang, Y.; Rocque, P.A. Mammalian thioltransferase (glutaredoxin) and protein disulfide isomerase have dehydroascorbate reductase activity. J. Biol. Chem. 1990, 265, 15361–15364. [Google Scholar]
- Del Bello, B.; Maellaro, E.; Sugherini, L.; Santucci, A.; Comporti, M.; Casini, A.F. Purification of NADPH-dependent dehydroascorbate reductase from rat liver and its identification with 3 alpha-hydroxysteroid dehydrogenase. Biochem. J. 1994, 304, 385–390. [Google Scholar]
- Buettner, G. The pecking order of free radicals and antioxidants: Lipid peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Linster, C.L.; van Schaftingen, E. Vitamin C. FEBS J. 2006, 274, 1–22. [Google Scholar] [CrossRef]
- Diliberto, E.J., Jr.; Heckman, G.D.; Daniels, A.J. Characterization of ascorbic acid transport by adrenomedullary chromaffin cells. Evidence for Na+-dependent co-transport. J. Biol. Chem. 1983, 258, 12886–12894. [Google Scholar]
- Welch, R.W.; Wang, Y.; Crossman, A., Jr.; Park, J.B.; Kirk, K.L.; Levine, M. Accumulation of vitamin C (ascorbate) and its oxidized metabolite dehydroascorbic acid occurs by separate mechanisms. J. Biol. Chem. 1995, 270, 12584–12592. [Google Scholar]
- Welch, R.W.; Bergsten, P.; Butler, J.D.; Levine, M. Ascorbic acid accumulation and transport in human fibroblasts. Biochem. J. 1993, 294, 505–510. [Google Scholar]
- Washko, P.; Rotrosen, D.; Levine, M. Ascorbic acid transport and accumulation in human neutrophils. J. Biol. Chem. 1989, 264, 18996–19002. [Google Scholar]
- Padh, H.; Aleo, J.J. Characterization of the ascorbic acid transport by 3T6 fibroblasts. Biochim. Biophys. Acta 1987, 901, 283–290. [Google Scholar] [CrossRef]
- Zhou, A.; Nielsen, J.H.; Farver, O.; Thorn, N.A. Transport of ascorbic acid and dehydroascorbic acid by pancreatic islet cells from neonatal rats. Biochem. J. 1991, 274, 739–744. [Google Scholar]
- Bergsten, P.; Yu, R.; Kehrl, J.; Levine, M. Ascorbic acid transport and distribution in human B lymphocytes. Arch. Biochem. Biophys. 1995, 317, 208–214. [Google Scholar] [CrossRef]
- Wright, J.; Castranova, V.; Colby, H.; Miles, P. Ascorbate uptake by isolated rat lung cells. J. Appl. Physiol. 1981, 51, 1477–1483. [Google Scholar]
- Stevenson, N.R. Active transport of l-ascorbic acid in the human ileum. Gastroenterology 1974, 67, 952–956. [Google Scholar]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.-Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent l-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Wilson, J.X.; Jaworski, E.M.; Dixon, S.J. Evidence for electrogenic sodium-dependent ascorbate transport in rat astroglia. Neurochem. Res. 1991, 16, 73–78. [Google Scholar] [CrossRef]
- Maffia, M.; Ahearn, G.; Vilella, S.; Zonno, V.; Storelli, C. Ascorbic acid transport by intestinal brush-border membrane vesicles of the teleost Anguilla anguilla. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1993, 264, R1248–R1253. [Google Scholar]
- Helbig, H.; Korbmacher, C.; Wohlfarth, J.; Berweck, S.; Kuhner, D.; Wiederholt, M. Electrogenic Na+-ascorbate cotransport in cultured bovine pigmented ciliary epithelial cells. Am. J. Physiol. Cell Physiol. 1989, 256, C44–C49. [Google Scholar]
- Daruwala, R.; Song, J.; Koh, W.S.; Rumsey, S.C.; Levine, M. Cloning and functional characterization of the human sodium-dependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999, 460, 480–484. [Google Scholar] [CrossRef]
- Eck, P.; Erichsen, H.C.; Taylor, J.G.; Yeager, M.; Hughes, A.L.; Levine, M.; Chanock, S.J. Comparison of the genomic structure and variation in the two human sodium-dependent vitamin C transporters, SLC23A1 and SLC23A2. Hum. Genet. 2004, 115, 285–294. [Google Scholar]
- May, J.M. The SLC23 family of ascorbate transporters: Ensuring that you get and keep your daily dose of vitamin C. Br. J. Pharmacol. 2011, 164, 1793–1801. [Google Scholar] [CrossRef]
- Wang, Y.; Mackenzie, B.; Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Human vitamin C (l-ascorbic acid) transporter SVCT1. Biochem. Biophys. Res. Commun. 2000, 267, 488–494. [Google Scholar] [CrossRef]
- Takanaga, H.; Mackenzie, B.; Hediger, M.A. Sodium-dependent ascorbic acid transporter family SLC23. Pflüg. Arch. Eur. J. Physiol. 2004, 447, 677–682. [Google Scholar]
- Wang, H.; Dutta, B.; Huang, W.; Devoe, L.D.; Leibach, F.H.; Ganapathy, V.; Prasad, P.D. Human Na+-dependent vitamin C transporter 1 (hSVCT1): Primary structure, functional characteristics and evidence for a non-functional splice variant. Biochim. Biophys. Acta 1999, 1461, 1–9. [Google Scholar] [CrossRef]
- Kuo, S.-M.; MacLean, M.E.; McCormick, K.; Wilson, J.X. Gender and sodium-ascorbate transporter isoforms determine ascorbate concentrations in mice. J. Nutr. 2004, 134, 2216–2221. [Google Scholar]
- Liang, W.-J.; Johnson, D.; Ma, L.-S.; Jarvis, S.M. Regulation of the human vitamin C transporters expressed in COS-1 cells by protein kinase C. Am. J. Physiol. Cell Physiol. 2002, 283, C1696–C1704. [Google Scholar] [CrossRef]
- Maulén, N.P.; Henrı́quez, E.A.; Kempe, S.; Cárcamo, J.G.; Schmid-Kotsas, A.; Bachem, M.; Grünert, A.; Bustamante, M.E.; Nualart, F.; Vera, J.C. Up-regulation and polarized expression of the sodium-ascorbic acid transporter SVCT1 in post-confluent differentiated CaCo-2 cells. J. Biol. Chem. 2003, 278, 9035–9041. [Google Scholar] [CrossRef]
- Godoy, A.; Ormazabal, V.; Moraga-Cid, G.; Zúñiga, F.A.; Sotomayor, P.; Barra, V.; Vasquez, O.; Montecinos, V.; Mardones, L.; Guzmán, C. Mechanistic insights and functional determinants of the transport cycle of the ascorbic acid transporter SVCT2—Activation by sodium and absolute dependence on bivalent cations. J. Biol. Chem. 2007, 282, 615–624. [Google Scholar]
- Savini, I.; Rossi, A.; Pierro, C.; Avigliano, L.; Catani, M.V. SVCT1 and SVCT2: Key proteins for vitamin C uptake. Amino Acids 2008, 34, 347–355. [Google Scholar] [CrossRef]
- Rivas, C.I.; Zuniga, F.A.; Salas-Burgos, A.; Mardones, L.; Ormazabal, V.; Vera, J.C. Vitamin C transporters. J. Physiol. Biochem. 2008, 64, 357–375. [Google Scholar] [CrossRef]
- Boyer, J.C.; Campbell, C.E.; Sigurdson, W.J.; Kuo, S.M. Polarized localization of vitamin C transporters, SVCT1 and SVCT2, in epithelial cells. Biochem. Biophys. Res. Commun. 2005, 334, 150–156. [Google Scholar] [CrossRef]
- Varma, S.; Sobey, K.; Campbell, C.E.; Kuo, S.-M. Hierarchal contribution of N-and C-terminal sequences to the differential localization of homologous sodium-dependent vitamin C transporters, SVCT1 and SVCT2, in Epithelial Cells. Biochemistry 2009, 48, 2969–2980. [Google Scholar] [CrossRef]
- Subramanian, V.S.; Subramanya, S.B.; Ghosal, A.; Marchant, J.S.; Harada, A.; Said, H.M. Modulation of function of sodium-dependent vitamin C transporter 1 (SVCT1) by Rab8a in intestinal epithelial cells: Studies utilizing Caco-2 cells and Rab8a knockout mice. Dig. Dis. Sci. 2012, 58, 641–649. [Google Scholar]
- Michels, A.J.; Hagen, T.M.; Frei, B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Ann. Rev. Nutr. 2013, 33, 45–70. [Google Scholar] [CrossRef]
- Corpe, C.P.; Tu, H.; Eck, P.; Wang, J.; Faulhaber-Walter, R.; Schnermann, J.; Margolis, S.; Padayatty, S.; Sun, H.; Wang, Y.; et al. Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation, and perinatal survival in mice. J. Clin. Investig. 2010, 120, 1069–1083. [Google Scholar] [CrossRef]
- Mellors, A.J.; Nahrwold, D.L.; Rose, R.C. Ascorbic acid flux across mucosal border of guinea pig and human ileum. Am. J. Physiol. 1977, 233, E374–E379. [Google Scholar]
- Harris, D.S.; Slot, J.W.; Geuze, H.J.; James, D.E. Polarized distribution of glucose transporter isoforms in Caco-2 cells. Proc. Natl. Acad. Sci. USA 1992, 89, 7556–7560. [Google Scholar] [CrossRef]
- Helliwell, P.A.; Richardson, M.; Affleck, J.; Kellett, G.L. Stimulation of fructose transport across the intestinal brush-border membrane by PMA is mediated by GLUT2 and dynamically regulated by protein kinase C. Biochem. J. 2000, 350, 149–154. [Google Scholar] [CrossRef]
- Mesonero, J.; Mahraoui, L.; Matosin, M.; Rodolosse, A.; Rousset, M.; Brot-Laroche, E. Expression of the hexose transporters GLUTI-GLUT5 and SGLTI in clones of Caco-2 cells. Biochem. Soc. Trans. 1994, 22, 681–684. [Google Scholar]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J. Clin. Investig. 1997, 100, 2842–2848. [Google Scholar] [CrossRef]
- Qiao, H.; May, J.M. Development of ascorbate transporters in brain cortical capillary endothelial cells in culture. Brain Res. 2008, 1208, 79–86. [Google Scholar]
- García, M.D.L.A.; Salazar, K.; Millán, C.; Rodríguez, F.; Montecinos, H.; Caprile, T.; Silva, C.; Cortes, C.; Reinicke, K.; Vera, J.C. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia 2005, 50, 32–47. [Google Scholar] [CrossRef]
- Mun, G.H.; Kim, M.J.; Lee, J.H.; Kim, H.J.; Chung, Y.H.; Chung, Y.B.; Kang, J.S.; Hwang, Y.I.; Oh, S.H.; Kim, J.-G.; et al. Immunohistochemical study of the distribution of sodium‐dependent vitamin C transporters in adult rat brain. J. Neurosci. Res. 2006, 83, 919–928. [Google Scholar] [CrossRef]
- Farrell, C.L.; Yang, J.; Pardridge, W.M. GLUT-1 glucose transporter is present within apical and basolateral membranes of brain epithelial interfaces and in microvascular endothelia with and without tight junctions. J. Histochem. Cytochem. 1992, 40, 193–199. [Google Scholar] [CrossRef]
- Hediger, M.A. New view at C. Nat. Med. 2002, 8, 445–446. [Google Scholar] [CrossRef]
- Berger, U.V.; Hediger, M.A. The vitamin C transporter SVCT2 is expressed by astrocytes in culture but not in situ. Neuroreport 2000, 11, 1395–1399. [Google Scholar] [CrossRef]
- Ralli, E.P.; Friedman, G.J.; Rubin, S.H. The mechanism of the excretion of vitamin C by the human kidney. J. Clin. Investig. 1938, 17, 765–770. [Google Scholar] [CrossRef]
- Rose, R.C. Ascorbic acid transport in mammalian kidney. Am. J. Physiol. 1986, 250, F627–F632. [Google Scholar]
- Bowers-Komro, D.M.; McCormick, D.B. Characterization of ascorbic acid uptake by isolated rat kidney cells. J. Nutr. 1991, 121, 57–64. [Google Scholar]
- Subramanian, V.S.; Marchant, J.S.; Boulware, M.J.; Said, H.M. A C-terminal region dictates the apical plasma membrane targeting of the human sodium-dependent vitamin C transporter-1 in polarized epithelia. J. Biol. Chem. 2004, 279, 27719–27728. [Google Scholar]
- Lee, J.H.; Oh, C.S.; Mun, G.H.; Kim, J.H.; Chung, Y.H.; Hwang, Y.I.; Shin, D.H.; Lee, W.J. Immunohistochemical localization of sodium-dependent l-ascorbic acid transporter 1 protein in rat kidney. Histochem. Cell Biol. 2006, 126, 491–494. [Google Scholar] [CrossRef]
- Martin, M.; Ferrier, B.; Roch-Ramel, F. Renal excretion of ascorbic acid in the rat: A micropuncture study. Am. J. Physiol. 1983, 244, F335–F341. [Google Scholar]
- Bianchi, J.; Rose, R.C. Transport of l-ascorbic acid and dehydro-l-ascorbic acid across renal cortical basolateral membrane vesicles. Biochim. Biophys.Acta 1985, 820, 265–273. [Google Scholar] [CrossRef]
- Bianchi, J.; Rose, R.C. Na+-independent dehydro-l-ascorbic acid uptake in renal brush-border membrane vesicles. Biochim. Biophys. Acta 1985, 819, 75–82. [Google Scholar] [CrossRef]
- Nualart, F.; Castro, T.; Low, M.; Henriquez, J.P.; Oyarce, K.; Cisternas, P.; Garcia, A.; Yanez, A.J.; Bertinat, R.; Montecinos, V.P.; et al. Dynamic expression of the sodium-vitamin C co-transporters, SVCT1 and SVCT2, during perinatal kidney development. Histochem. Cell Biol. 2013, 139, 233–247. [Google Scholar] [CrossRef]
- Savini, I.; Catani, M.V.; Arnone, R.; Rossi, A.; Frega, G.; del Principe, D.; Avigliano, L. Translational control of the ascorbic acid transporter SVCT2 in human platelets. Free Radic. Biol. Med. 2007, 42, 608–616. [Google Scholar] [CrossRef]
- MacDonald, L.; Thumser, A.E.; Sharp, P. Decreased expression of the vitamin C transporter SVCT1 by ascorbic acid in a human intestinal epithelial cell line. Br. J. Nutr. 2002, 87, 97–100. [Google Scholar] [CrossRef]
- Karaczyn, A.; Ivanov, S.; Reynolds, M.; Zhitkovich, A.; Kasprzak, K.S.; Salnikow, K. Ascorbate depletion mediates up‐regulation of hypoxia‐associated proteins by cell density and nickel. J. Cell. Biochem. 2006, 97, 1025–1035. [Google Scholar] [CrossRef]
- Reidling, J.C.; Rubin, S.A. Promoter analysis of the human ascorbic acid transporters SVCT1 and 2: Mechanisms of adaptive regulation in liver epithelial cells. J. Nutr. Biochem. 2011, 22, 344–350. [Google Scholar] [CrossRef]
- Castro, T.; Low, M.; Salazar, K.; Montecinos, H.; Cifuentes, M.; Yáñez, A.J.; Slebe, J.C.; Figueroa, C.D.; Reinicke, K.; de los Angeles García, M. Differential distribution of the Sodium-vitamin C cotransporter-1 along the proximal tubule of the mouse and human kidney. Kidney Int. 2008, 74, 1278–1286. [Google Scholar] [CrossRef]
- Rose, R.C.; Nahrwold, D.L. Intestinal ascorbic acid transport following diets of high or low ascorbic acid content. Int. J. Vitam. Nutr. Res. 1978, 48, 382–386. [Google Scholar]
- Karasov, W.H.; Darken, B.W.; Bottum, M.C. Dietary regulation of intestinal ascorbate uptake in guinea pigs. Am. J. Physiol. 1991, 260, G108–G118. [Google Scholar]
- Schjoldager, J.G.; Tveden-Nyborg, P.; Lykkesfeldt, J. Prolonged maternal vitamin C deficiency overrides preferential fetal ascorbate transport but does not influence perinatal survival in guinea pigs. Br. J. Nutr. 2013, in press. [Google Scholar]
- Wilson, J.X.; Jaworski, E.M.; Kulaga, A.; Dixon, S.J. Substrate regulation of ascorbate transport activity in astrocytes. Neurochem. Res. 1990, 15, 1037–1043. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef]
- Vissers, M.C.M.; Bozonet, S.M.; Pearson, J.F.; Braithwaite, L.J. Dietary ascorbate intake affects steady state tissue concentrations in vitamin C-deficient mice: Tissue deficiency after suboptimal intake and superior bioavailability from a food source (kiwifruit). Am. J. Clin. Nutr. 2011, 93, 292–301. [Google Scholar] [CrossRef]
- Carr, A.C.; Bozonet, S.M.; Pullar, J.M.; Simcock, J.W.; Vissers, M.C. Human skeletal muscle ascorbate is highly responsive to changes in vitamin C intake and plasma concentrations. Am. J. Clin. Nutr. 2013, 97, 800–807. [Google Scholar] [CrossRef]
- Jiménez-Fernández, E.; Ponce, M.; Zuasti, E.; Fernández-Díaz, C.; Manchado, M.; Infante, C. Molecular characterization and transcriptional regulation of the sodium-dependent vitamin C transporter genes (slc23a1 and slc23a2) in a teleost fish, the Senegalese sole (Solea senegalensis). Comp. Biochem. Physiol. B 2012, 161, 208–218. [Google Scholar]
- Dixon, S.J.; Wilson, J.X. Adaptive regulation of ascorbate transport in osteoblastic cells. J. Bone Miner. Res. 1992, 7, 675–681. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lindblad, M.; Tveden-Nyborg, P.; Lykkesfeldt, J. Regulation of Vitamin C Homeostasis during Deficiency. Nutrients 2013, 5, 2860-2879. https://doi.org/10.3390/nu5082860
Lindblad M, Tveden-Nyborg P, Lykkesfeldt J. Regulation of Vitamin C Homeostasis during Deficiency. Nutrients. 2013; 5(8):2860-2879. https://doi.org/10.3390/nu5082860
Chicago/Turabian StyleLindblad, Maiken, Pernille Tveden-Nyborg, and Jens Lykkesfeldt. 2013. "Regulation of Vitamin C Homeostasis during Deficiency" Nutrients 5, no. 8: 2860-2879. https://doi.org/10.3390/nu5082860