Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases


Abstract
:Abbreviations
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis, also known as Lou Gehrig’s disease |
| ARE | Antioxidant Response Element |
| EAAC1 | Excitatory amino acid transporter C1 |
| EAAT | Excitatory amino acid transporter |
| ERE | Electrophile Response Element |
| FA | Friedreich’s axtaia |
| GCLC (heavy subunit of GCS), GCS | γ-glutamylcysteine synthetase |
| Grx | glutaredoxin |
| GSSG | glutathione disulfide |
| GPx | glutathione peroxidase |
| GST | glutathione S-transferase |
| GS | glutathione synthetase |
| GR | glutathione reductase |
| HD | Huntington’s disease |
| MS | multiple sclerosis |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| PD | Parkinson’s disease |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| GSH | reduced glutathione |
| Xc− | cystine/glutamate transport system |
1. Introduction
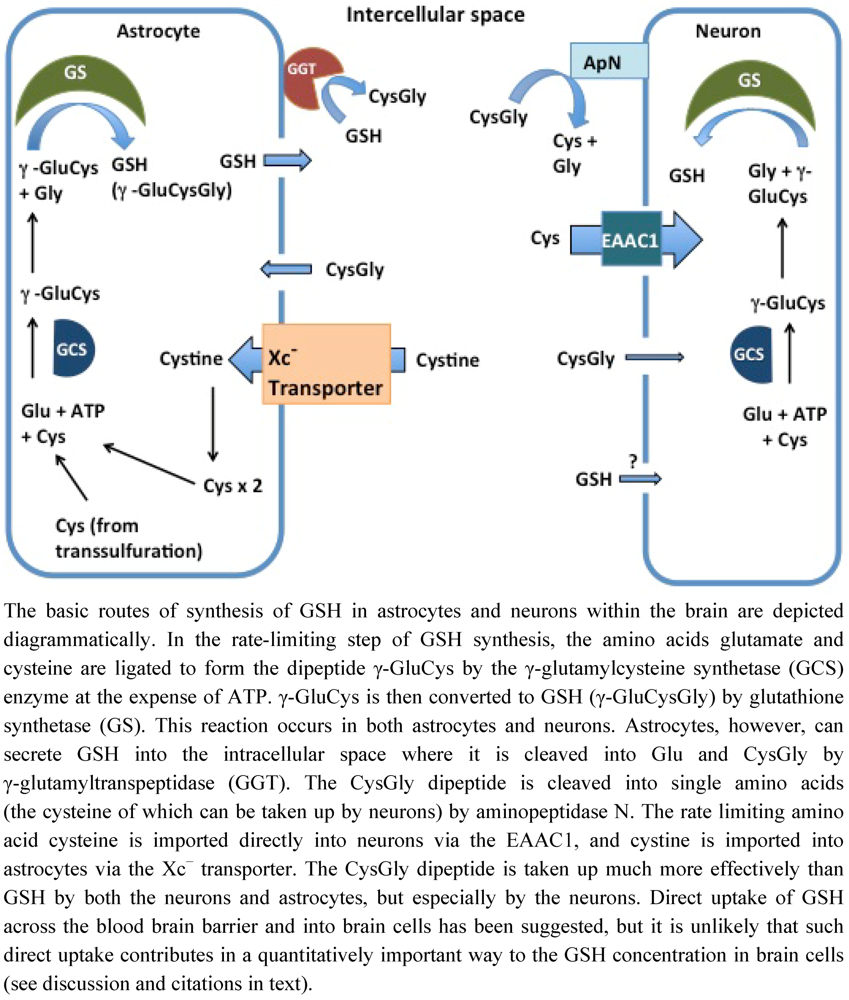
2. Intracellular Synthesis and Transport of GSH in Brain

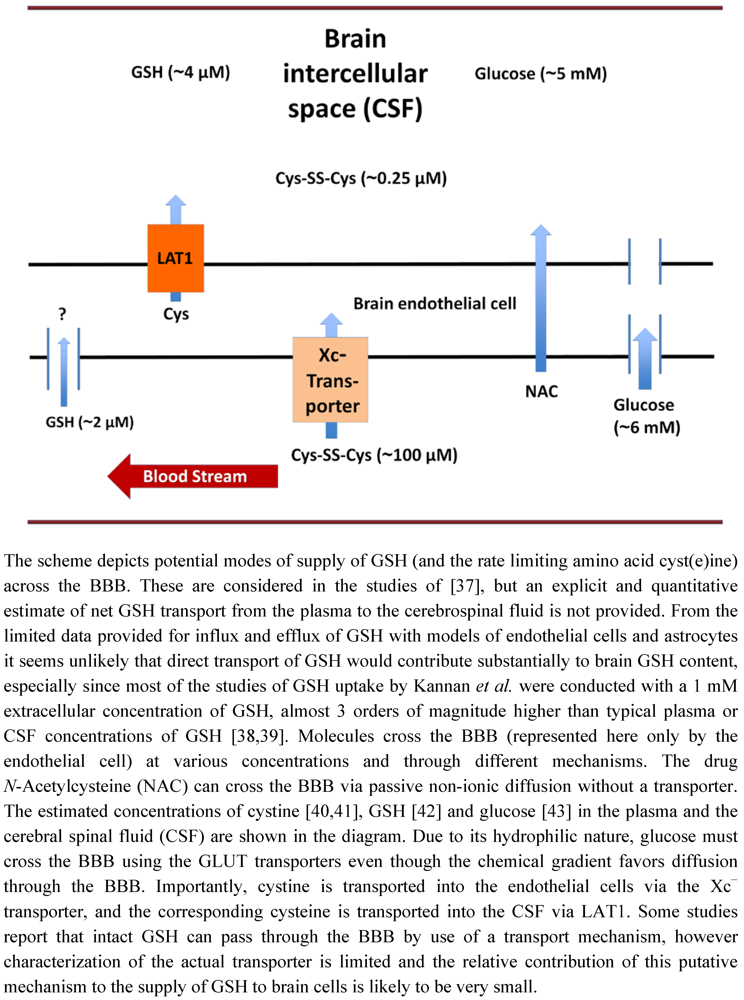
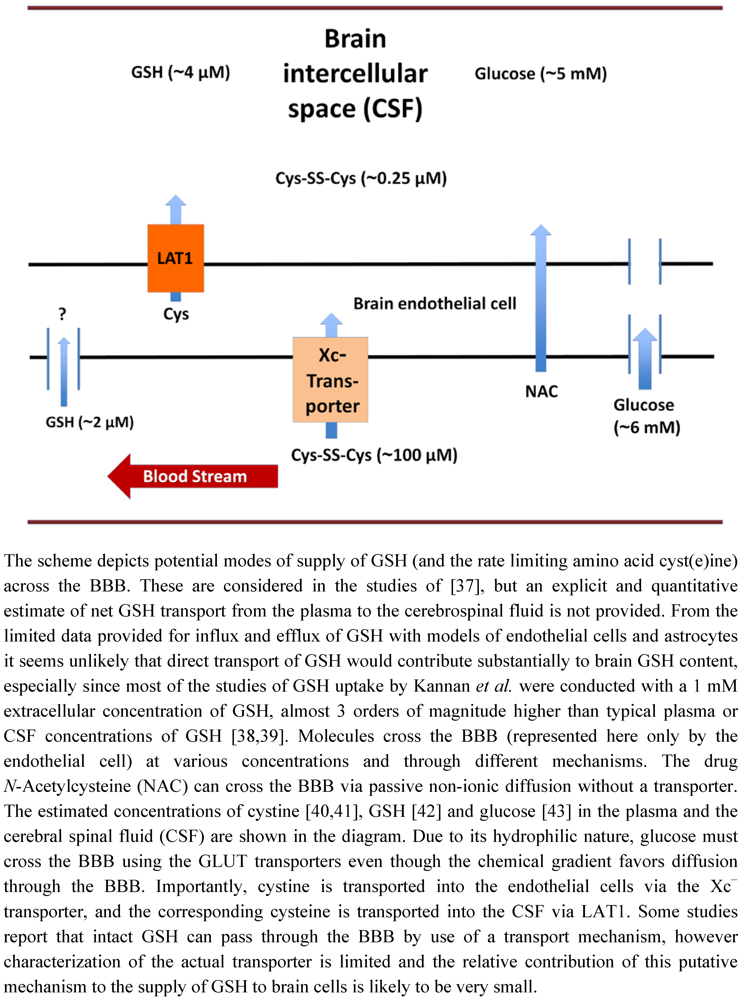
3. Glutathione Transport across the Blood Brain Barrier
4. Experimental Manipulation of Cellular GSH Content
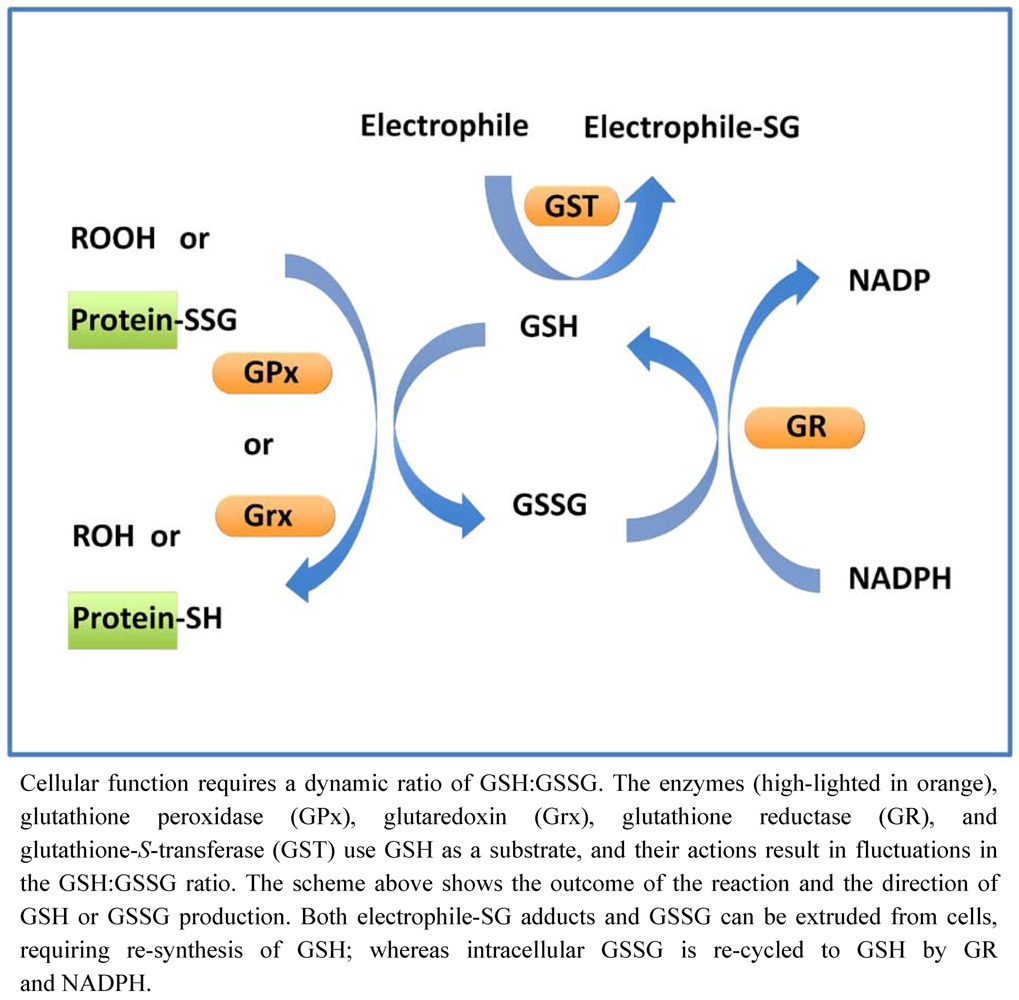
5. Glutathione Cycle
6. Glutathione-Dependent Enzymes
6.1. Glutathione Peroxidases
6.2. Glutathione S-Transferases
6.3. Glutathione Reductases
6.4. Glutaredoxins
7. Oxidative Stress and Dysregulation of Thiol Homeostasis

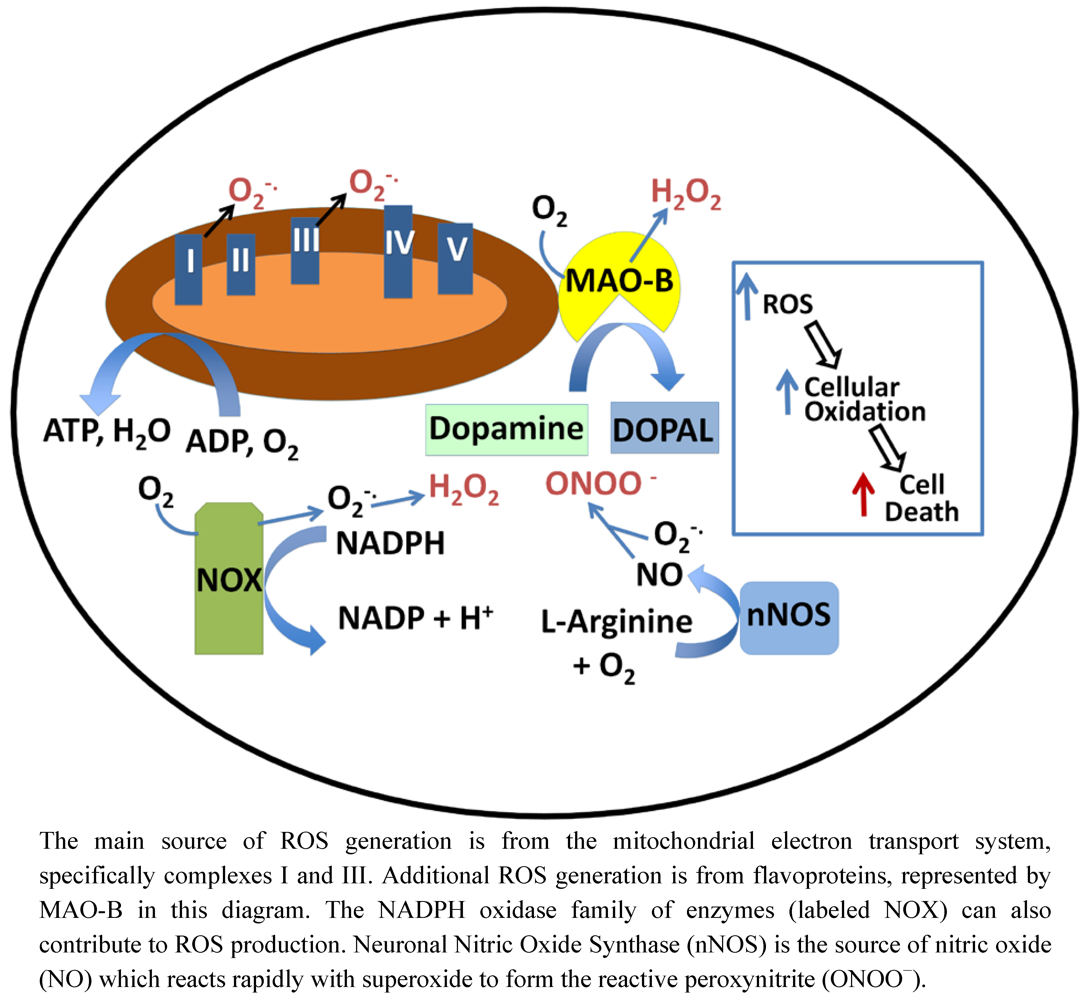
8. Mitochondrial Glutathione: Transport and Regulation of Apoptosis
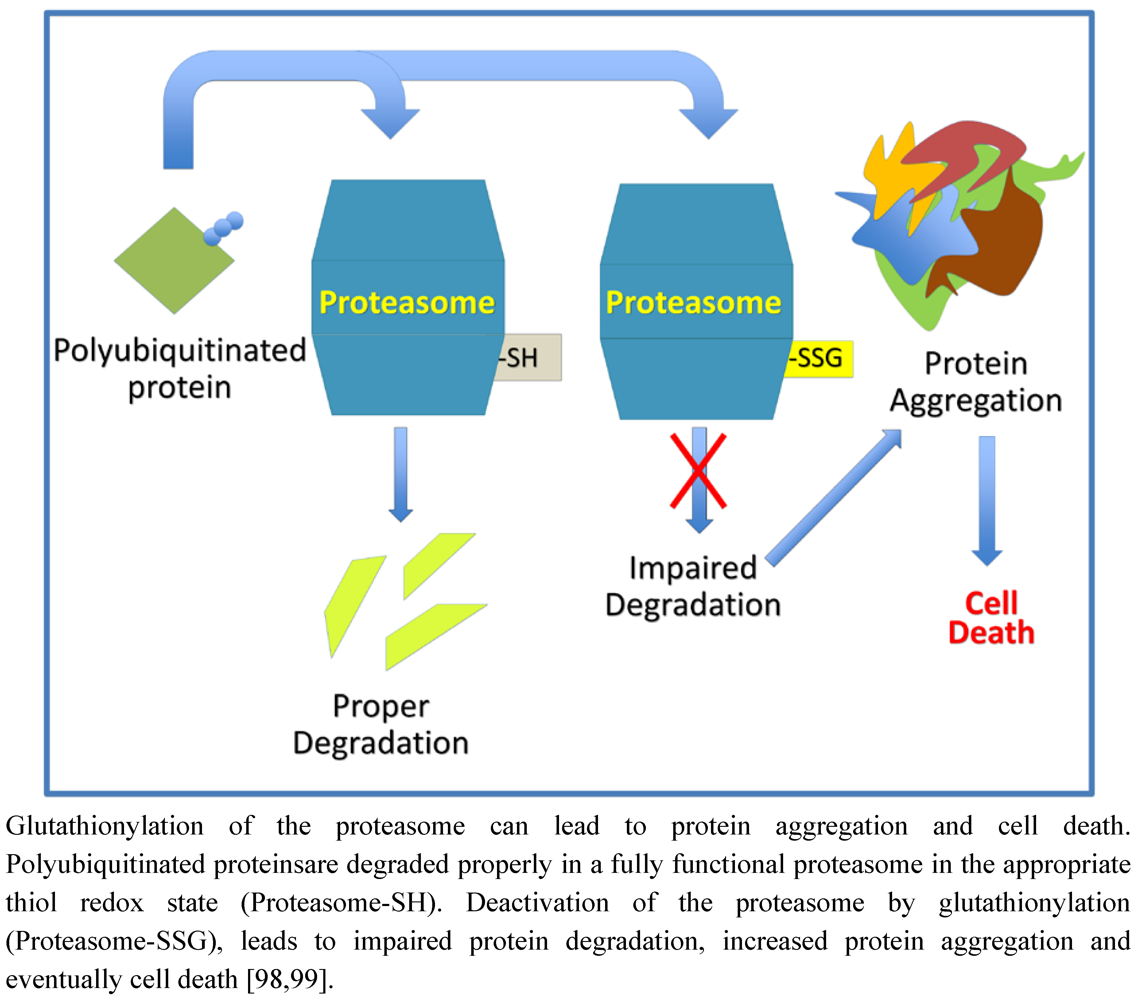
9. Protein Degradation and Aggregation
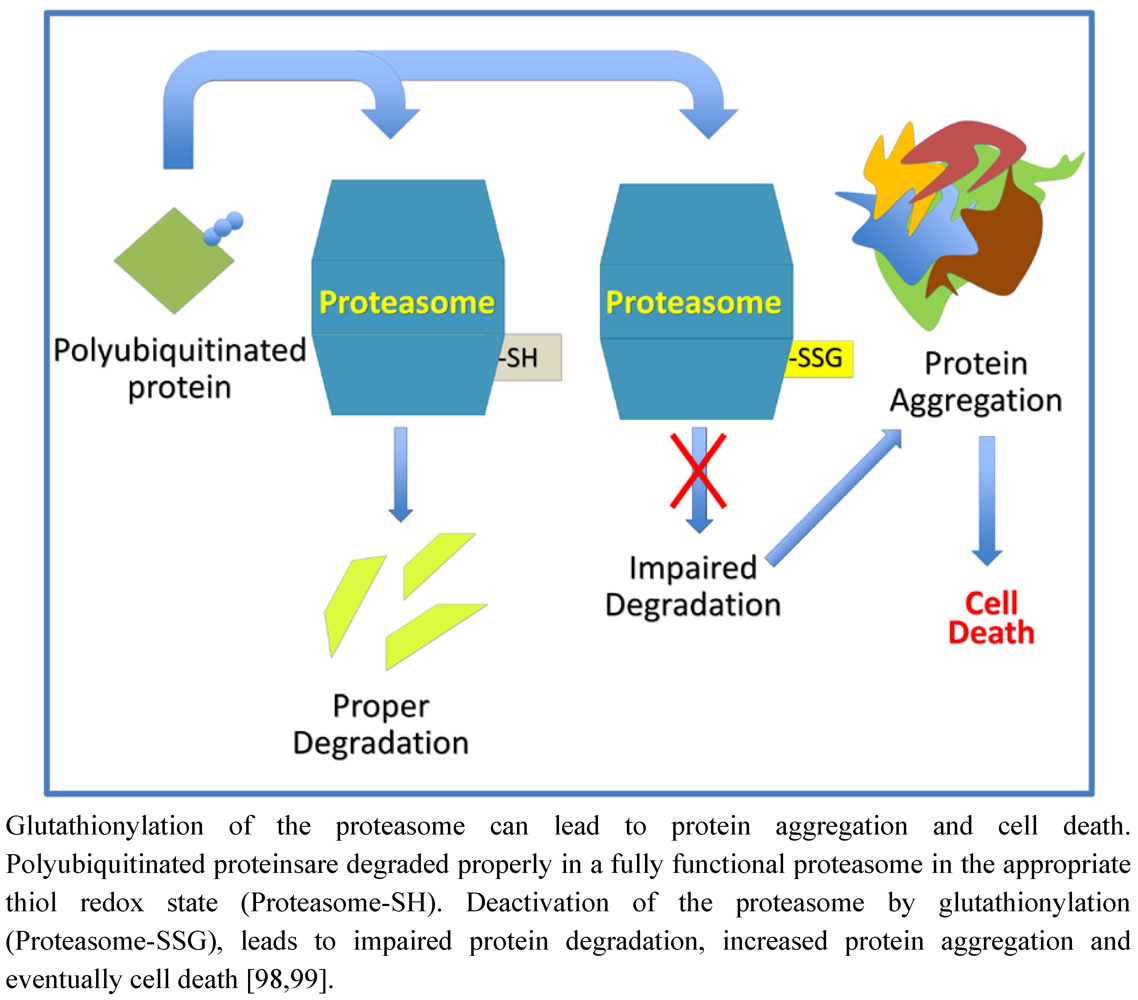
10. Protein S-Glutathionylation in Cellular Homeostasis and Regulation
11. Impairment of Glutathione Homeostasis in Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Reported Changes in Patient Glutathione Homeostasis | Reported Alterations in Glutathione Related Enzymes | ROS Implicated in Disease | Reported Protein Aggregation Associated with Disease | Reported Protein Glutathionylation Associated with Disease |
|---|---|---|---|---|---|
| Parkinson’s Disease | Decrease in substantia nigra GSH [106] | GST-P1 (pi) mutation is associated with increased PD susceptibility [111] | Environmental factors that cause PD lead to oxidative damage [64] | α-Synuclein aggregates [92] | NADP-dependent isocitrate dehydrogenase [115] |
| Alzheimer’s Disease | Decrease in erythrocyte glutathione [107] | GPx1 mutation may be a risk factor for AD [112] | Beta amyloid may lead to mitochondrial instability, leading to increased ROS production [63] | Beta-amyloid, Tau aggregates [93] | Tau [116] |
| Huntington’s Disease | Decrease in plasma GSH [108] | Decreased GPx activity in erythrocytes [113] | Increased ROS production in mutant huntingtin-containing cells treated with thapsigargin [61] | Huntingtin aggregates [94] | NR |
| ALS | Decrease in erythrocyte GSH [109] | Decreased GST pi (P1) expression in motor brain cortex [114] | Mutated SOD1 increases ROS levels [62] | SOD1 aggregates [95] | SOD1 [117] |
| Friedreich’s Ataxia | Decrease in free glutathione in erythrocytes [110] | NR | FA cells show an increased sensitivity to oxidative damage [60] | NR | Actin [118] |
11.1. Parkinson’s Disease
11.2. Alzheimer’s Disease
11.3. Huntington’s Disease
11.4. Amyotrophic Lateral Sclerosis
11.5. Friedreich’s Ataxia
12. Glutathione in Food/Supplements
13. Glutathione as a Therapeutic Agent
14. Glutathione as a Biomarker?
15. Conclusions
Acknowledgments
Author Disclosure Statement
References
- Anderson, M.E. Glutathione: An overview of biosynthesis and modulation. Chem. Biol. Interact. 1998, 111-112, 1–14. [Google Scholar] [CrossRef]
- Morales, A.; Miranda, M.; Sanchez-Reyes, A.; Colell, A.; Biete, A.; Fernandez-Checa, J.C. Transcriptional regulation of the heavy subunit chain of gamma-glutamylcysteine synthetase by ionizing radiation. FEBS Lett. 1998, 427, 15–20. [Google Scholar] [CrossRef]
- Rahman, I.; Smith, C.A.; Lawson, M.F.; Harrison, D.J.; MacNee, W. Induction of gamma-glutamylcysteine synthetase by cigarette smoke is associated with AP-1 in human alveolar epithelial cells. FEBS Lett. 1996, 396, 21–25. [Google Scholar] [CrossRef]
- Wild, A.C.; Moinova, H.R.; Mulcahy, R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 1999, 274, 33627–33636. [Google Scholar]
- Liu, R.M.; Gao, L.; Choi, J.; Forman, H.J. Gamma-glutamylcysteine synthetase: mRNA stabilization and independent subunit transcription by 4-hydroxy-2-nonenal. Am. J. Physiol. 1998, 275, L861–L869. [Google Scholar]
- Sun, W.M.; Huang, Z.Z.; Lu, S.C. Regulation of gamma-glutamylcysteine synthetase by protein phosphorylation. Biochem. J. 1996, 320, 321–328. [Google Scholar]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 2008, 108, 227–238. [Google Scholar] [CrossRef]
- Aoyama, K.; Watabe, M.; Nakaki, T. Modulation of neuronal glutathione synthesis by EAAC1 and its interacting protein GTRAP3-18. Amino Acids 2012, 42, 163–169. [Google Scholar] [CrossRef]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Gros, C.; Hirrlinger, J. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J. Neurosci. Res. 2001, 66, 1003–1008. [Google Scholar] [CrossRef]
- Thompson, G.A.; Meister, A. Utilization of l-cystine by the gamma-glutamyl transpeptidase-gamma-glutamyl cyclotransferase pathway. Proc. Natl. Acad. Sci. USA 1975, 72, 1985–1988. [Google Scholar] [CrossRef]
- Anderson, M.E.; Meister, A. Transport and direct utilization of gamma-glutamylcyst(e)ine for glutathione synthesis. Proc. Natl. Acad. Sci. USA 1983, 80, 707–711. [Google Scholar] [CrossRef]
- Kranich, O.; Dringen, R.; Sandberg, M.; Hamprecht, B. Utilization of cysteine and cysteine precursors for the synthesis of glutathione in astroglial cultures: Preference for cystine. Glia 1998, 22, 11–18. [Google Scholar] [CrossRef]
- Fournier, K.M.; Gonzalez, M.I.; Robinson, M.B. Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J. Biol. Chem. 2004, 279, 34505–34513. [Google Scholar]
- Watabe, M.; Aoyama, K.; Nakaki, T. Regulation of glutathione synthesis via interaction between glutamate transport-associated protein 3-18 (GTRAP3-18) and excitatory amino acid carrier-1 (EAAC1) at plasma membrane. Mol. Pharmacol. 2007, 72, 1103–1110. [Google Scholar] [CrossRef]
- Himi, T.; Ikeda, M.; Yasuhara, T.; Nishida, M.; Morita, I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J. Neural Transm. 2003, 110, 1337–1348. [Google Scholar] [CrossRef]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- La Bella, V.; Valentino, F.; Piccoli, T.; Piccoli, F. Expression and developmental regulation of the cystine/glutamate exchanger (Xc−) in the rat. Neurochem. Res. 2007, 32, 1081–1090. [Google Scholar] [CrossRef]
- Bannai, S.; Sato, H.; Ishii, T.; Sugita, Y. Induction of cystine transport activity in human fibroblasts by oxygen. J. Biol. Chem. 1989, 264, 18480–18484. [Google Scholar]
- Bannai, S.; Sato, H.; Ishii, T.; Taketani, S. Enhancement of glutathione levels in mouse peritoneal macrophages by sodium arsenite, cadmium chloride and glucose/glucose oxidase. Biochim. Biophys. Acta 1092, 175–179. [Google Scholar]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system Xc−: cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P.; Methner, A. Regulation of xCT expression and system Xc− function in neuronal cells. Amino Acids 2012, 42, 171–179. [Google Scholar] [CrossRef]
- McBean, G.J. The transsulfuration pathway: A source of cysteine for glutathione in astrocytes. Amino Acids 2012, 42, 199–205. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Thomas, M.; Ghorpade, A.; Gendelman, H.E.; Banerjee, R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J. Biol. Chem. 2006, 281, 35785–35793. [Google Scholar]
- Rosado, J.O.; Salvador, M.; Bonatto, D. Importance of the trans-sulfuration pathway in cancer prevention and promotion. Mol. Cell. Biochem. 2007, 301, 1–12. [Google Scholar] [CrossRef]
- Kandil, S.; Brennan, L.; McBean, G.J. Glutathione depletion causes a JNK and p38MAPK-mediated increase in expression of cystathionine-gamma-lyase and upregulation of the transsulfuration pathway in C6 glioma cells. Neurochem. Int. 2010, 56, 611–619. [Google Scholar] [CrossRef]
- Qin, S.; Colin, C.; Hinners, I.; Gervais, A.; Cheret, C.; Mallat, M. System Xc− and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J. Neurosci. 2006, 26, 3345–3356. [Google Scholar]
- Gras, G.; Samah, B.; Hubert, A.; Leone, C.; Porcheray, F.; Rimaniol, A.C. EAAT expression by macrophages and microglia: Still more questions than answers. Amino Acids 2012, 42, 221–229. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Gutterer, J.M.; Kussmaul, L.; Hamprecht, B.; Dringen, R. Microglial cells in culture express a prominent glutathione system for the defense against reactive oxygen species. Dev. Neurosci. 2000, 22, 384–392. [Google Scholar] [CrossRef]
- Persson, M.; Ronnback, L. Microglial self-defence mediated through GLT-1 and glutathione. Amino Acids 2012, 42, 207–219. [Google Scholar] [CrossRef]
- Flagg, E.W.; Coates, R.J.; Jones, D.P.; Eley, J.W.; Gunter, E.W.; Jackson, B.; Greenberg, R.S. Plasma total glutathione in humans and its association with demographic and health-related factors. Br. J. Nutr. 1993, 70, 797–808. [Google Scholar] [CrossRef]
- Jones, D.P.; Carlson, J.L.; Samiec, P.S.; Sternberg, P., Jr.; Mody, V.C., Jr.; Reed, R.L.; Brown, L.A. Glutathione measurement in human plasma. Evaluation of sample collection, storage and derivatization conditions for analysis of dansyl derivatives by HPLC. Clin. Chim. Acta 1998, 275, 175–184. [Google Scholar] [CrossRef]
- Dunn-Meynell, A.A.; Sanders, N.M.; Compton, D.; Becker, T.C.; Eiki, J.; Zhang, B.B.; Levin, B.E. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J. Neurosci. 2009, 29, 7015–7022. [Google Scholar]
- Duelli, R.; Maurer, M.H.; Staudt, R.; Sokoloff, L.; Kuschinsky, W. Correlation between local glucose transporter densities and local 3-O-methylglucose transport in rat brain. Neurosci. Lett. 2001, 310, 101–104. [Google Scholar] [CrossRef]
- Kannan, R.; Chakrabarti, R.; Tang, D.; Kim, K.J.; Kaplowitz, N. GSH transport in human cerebrovascular endothelial cells and human astrocytes: Evidence for luminal localization of Na+-dependent GSH transport in HCEC. Brain Res. 2000, 852, 374–382. [Google Scholar] [CrossRef]
- Kannan, R.; Kuhlenkamp, J.F.; Jeandidier, E.; Trinh, H.; Ookhtens, M.; Kaplowitz, N. Evidence for carrier-mediated transport of glutathione across the blood-brain barrier in the rat. J. Clin. Invest. 1990, 85, 2009–2013. [Google Scholar] [CrossRef]
- Kannan, R.; Kuhlenkamp, J.F.; Ookhtens, M.; Kaplowitz, N. Transport of glutathione at blood-brain barrier of the rat: Inhibition by glutathione analogs and age-dependence. J. Pharmacol. Exp. Ther. 1992, 263, 964–970. [Google Scholar]
- Valdovinos-Flores, C.; Gonsebatt, M.E. The role of amino acid transporters in GSH synthesis in the blood-brain barrier and central nervous system. Neurochem. Int. 2012, 61, 405–414. [Google Scholar] [CrossRef]
- Yanai, Y.; Shibasaki, T.; Kohno, N.; Mitsui, T.; Nakajima, H. Concentrations of sulfur-containing free amino acids in lumbar cerebrospinal fluid from patients with consciousness disturbances. Acta Neurol. Scand. 1983, 68, 386–393. [Google Scholar]
- Goudas, L.C.; Langlade, A.; Serrie, A.; Matson, W.; Milbury, P.; Thurel, C.; Sandouk, P.; Carr, D.B. Acute decreases in cerebrospinal fluid glutathione levels after intracerebroventricular morphine for cancer pain. Anesth. Analg. 1999, 89, 1209–1215. [Google Scholar] [CrossRef]
- Guerra-Romero, L.; Tauber, M.G.; Fournier, M.A.; Tureen, J.H. Lactate and glucose concentrations in brain interstitial fluid, cerebrospinal fluid, and serum during experimental pneumococcal meningitis. J. Infect. Dis. 1992, 166, 546–550. [Google Scholar] [CrossRef]
- Cornford, E.M.; Braun, L.D.; Crane, P.D.; Oldendorf, W.H. Blood-brain barrier restriction of peptides and the low uptake of enkephalins. Endocrinology 1978, 103, 1297–1303. [Google Scholar] [CrossRef]
- William, R.J.B.; Abbott, A.; Meister, A. Extracellular metabolism of glutathione accounts for its disapperance from the basolateral circulation of the kidney. J. Biol. Chem. 1984, 259, 15393–15400. [Google Scholar]
- Muruganandam, A.; Herx, L.M.; Monette, R.; Durkin, J.P.; Stanimirovic, D.B. Development of immortalized human cerebromicrovascular endothelial cell line as an in vitro model of the human blood-brain barrier. FASEB J. 1997, 11, 1187–1197. [Google Scholar]
- More, S.S.; Vince, R. Design, synthesis and biological evaluation of glutathione peptidomimetics as components of anti-Parkinson prodrugs. J. Med. Chem. 2008, 51, 4581–4588. [Google Scholar] [CrossRef]
- Hosoya, K.; Tomi, M.; Ohtsuki, S.; Takanaga, H.; Saeki, S.; Kanai, Y.; Endou, H.; Naito, M.; Tsuruo, M.; Terasaki, T. Enhancement of l-cystine transport activity and its relation to xCT gene induction at the blood-brain barrier by diethyl maleate treatment. J. Pharmacol. Exp. Ther. 2002, 302, 225–231. [Google Scholar] [CrossRef]
- Wade, L.A.; Brady, H.M. Cysteine and cystine transport at the blood-brain barrier. J. Neurochem. 1981, 37, 730–734. [Google Scholar]
- Killian, D.M.; Chikhale, P.J. Predominant functional activity of the large, neutral amino acid transporter (LAT1) isoform at the cerebrovasculature. Neurosci. Lett. 2001, 306, 1–4. [Google Scholar] [CrossRef]
- Anderson, M.E.; Powrie, F.; Puri, R.N.; Meister, A. Glutathione monoethyl ester: Preparation, uptake by tissues, and conversion to glutathione. Arch. Biochem. Biophys. 1985, 239, 538–548. [Google Scholar] [CrossRef]
- Kosower, N.S.; Kosower, E.M. Diamide: An oxidant probe for thiols. Methods Enzymol. 1995, 251, 123–133. [Google Scholar]
- Sabens, E.A.; Distler, A.M.; Mieyal, J.J. Levodopa deactivates enzymes that regulate thiol-disulfide homeostasis and promotes neuronal cell death: Implications for therapy of Parkinson’s disease. Biochemistry 2010, 49, 2715–2724. [Google Scholar]
- Guan, J.; Lo, M.; Dockery, P.; Mahon, S.; Karp, C.M.; Buckley, A.R.; Lam, S.; Gout, P.W.; Wang, Y.Z. The Xc− cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: Use of sulfasalazine. Cancer Chemother. Pharmacol. 2009, 64, 463–472. [Google Scholar] [CrossRef]
- Shukla, K.; Thomas, A.G.; Ferraris, D.V.; Hin, N.; Sattler, R.; Alt, J.; Rojas, C.; Slusher, B.S.; Tsukamoto, T. Inhibition of Xc− transporter-mediated cystine uptake by sulfasalazine analogs. Bioorg. Med. Chem. Lett. 2011, 21, 6184–6187. [Google Scholar]
- Mitozo, P.A.; de Souza, L.F.; Loch-Neckel, G.; Flesch, S.; Maris, A.F.; Figueiredo, C.P.; Dos Santos, A.R.; Farina, M.; Dafre, A.L. A study of the relative importance of the peroxiredoxin-, catalase-, and glutathione-dependent systems in neural peroxide metabolism. Free Radic. Biol. Med. 2011, 51, 69–77. [Google Scholar] [CrossRef]
- Awasthi, Y.C.; Chaudhary, P.; Vatsyayan, R.; Sharma, A.; Awasthi, S.; Sharma, R. Physiological and pharmacological significance of glutathione-conjugate transport. J. Toxicol. Environ. Health B Crit. Rev. 2009, 12, 540–551. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Starke, D.W.; Mieyal, J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox Signal. 2009, 11, 1059–1081. [Google Scholar] [CrossRef]
- Melo, A.; Monteiro, L.; Lima, R.M.; Oliveira, D.M.; Cerqueira, M.D.; El-Bacha, R.S. Oxidative stress in neurodegenerative diseases: Mechanisms and therapeutic perspectives. Oxid. Med. Cell. Longev. 2011, 467180. [Google Scholar]
- Armstrong, J.S.; Khdour, O.; Hecht, S.M. Does oxidative stress contribute to the pathology of Friedreich’s ataxia? A radical question. FASEB J. 2010, 24, 2152–2163. [Google Scholar] [CrossRef]
- Jin, Y.N.; Johnson, G.V. The interrelationship between mitochondrial dysfunction and transcriptional dysregulation in Huntington disease. J. Bioenerg. Biomembr. 2010, 42, 199–205. [Google Scholar] [CrossRef]
- Goldsteins, G.; Keksa-Goldsteine, V.; Ahtoniemi, T.; Jaronen, M.; Arens, E.; Akerman, K.; Chan, P.H.; Koistinaho, J. Deleterious role of superoxide dismutase in the mitochondrial intermembrane space. J. Biol. Chem. 2008, 283, 8446–8452. [Google Scholar]
- Eckert, A.; Keil, U.; Marques, C.A.; Bonert, A.; Frey, C.; Schussel, K.; Muller, W.E. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer’s disease. Biochem. Pharmacol. 2003, 66, 1627–1634. [Google Scholar] [CrossRef]
- Miller, R.L.; James-Kracke, M.; Sun, G.Y.; Sun, A.Y. Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem. Res. 2009, 34, 55–65. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef]
- Kumar, C.; Igbaria, A.; D’Autreaux, B.; Planson, A.G.; Junot, C.; Godat, E.; Bachhawat, A.K.; Delaunay-Moisan, A.; Toledano, M.B. Glutathione revisited: A vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011, 30, 2044–2056. [Google Scholar] [CrossRef]
- Kann, O.; Kovacs, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H(2)O(2) signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Adam-Vizi, V. Production of reactive oxygen species in brain mitochondria: Contribution by electron transport chain and non-electron transport chain sources. Antioxid. Redox Signal. 2005, 7, 1140–1149. [Google Scholar] [CrossRef]
- Nagatsu, T.; Sawada, M. Molecular mechanism of the relation of monoamine oxidase B and its inhibitors to Parkinson’s disease: Possible implications of glial cells. J. Neural Transm. Suppl. 2006, 71, 53–65. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Beckman, J.S. Peroxynitrite versus hydroxyl radical: The role of nitric oxide in superoxide-dependent cerebral injury. Ann. N. Y. Acad. Sci. 1994, 738, 69–75. [Google Scholar] [CrossRef]
- Jones, D.P.; Go, Y.S. Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 2010, 12, 116–125. [Google Scholar] [CrossRef]
- Wadey, A.L.; Muyderman, H.; Kwek, P.T.; Sims, N.R. Mitochondrial glutathione uptake: Characterization in isolated brain mitochondria and astrocytes in culture. J. Neurochem. 2009, 109, 101–108. [Google Scholar] [CrossRef]
- Garcia, J.; Han, D.; Sancheti, H.; Yap, L.P.; Kaplowitz, N.; Cadenas, E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 2010, 285, 39646–39654. [Google Scholar]
- Lash, L.H. Mitochondrial glutathione transport: Physiological, pathological and toxicological implications. Chem. Biol. Interact. 2006, 163, 54–67. [Google Scholar] [CrossRef]
- Kamga, C.K.; Zhang, S.X.; Wang, Y. Dicarboxylate carrier-mediated glutathione transport is essential for reactive oxygen species homeostasis and normal respiration in rat brain mitochondria. Am. J. Physiol. Cell Physiol. 2010, 299, C497–C505. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Marquardt, K.; Lash, L.H.; Linseman, D.A. Bcl-2 is a novel interacting partner for the 2-oxoglutarate carrier and a key regulator of mitochondrial glutathione. Free Radic. Biol. Med. 2012, 52, 410–419. [Google Scholar] [CrossRef]
- Colell, A.; Garcia-Ruiz, C.; Miranda, M.; Ardite, E.; Mari, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernandez-Checa, J.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998, 115, 1541–1551. [Google Scholar] [CrossRef]
- Muyderman, H.; Nilsson, M.; Sims, N.R. Highly selective and prolonged depletion of mitochondrial glutathione in astrocytes markedly increases sensitivity to peroxynitrite. J. Neurosci. 2004, 24, 8019–8028. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Cell death: Protein misfolding and neurodegenerative diseases. Apoptosis 2009, 14, 455–468. [Google Scholar] [CrossRef]
- Kumar, P.; Pradhan, K.; Karunya, R.; Ambasta, R.K.; Querfurth, H.W. Cross-functional E3 ligases Parkin and C-terminus Hsp70-interacting protein in neurodegenerative disorders. J. Neurochem. 2012, 120, 350–370. [Google Scholar] [CrossRef]
- Vali, S.; Chinta, S.J.; Peng, J.; Sultana, Z.; Singh, N.; Sharma, P.; Sharada, S.; Andersen, J.K.; Bharath, M.M. Insights into the effects of alpha-synuclein expression and proteasome inhibition on glutathione metabolism through a dynamic in silico model of Parkinson’s disease: Validation by cell culture data. Free Radic. Biol. Med. 2008, 45, 1290–1301. [Google Scholar] [CrossRef]
- Allen, E.M.; Mieyal, J.J. Protein-thiol oxidation and cell death: Regulatory role of glutaredoxins. Antioxid. Redox Signal. 2012. [Google Scholar] [CrossRef]
- Dennissen, F.J.; Kholod, N.; van Leeuwen, F.W. The ubiquitin proteasome system in neurodegenerative diseases: Culprit, accomplice or victim? Prog. Neurobiol. 2012, 96, 190–207. [Google Scholar] [CrossRef]
- Garcia-Arencibia, M.; Hochfeld, W.E.; Toh, P.P.; Rubinsztein, D.C. Autophagy, a guardian against neurodegeneration. Semin. Cell Dev. Biol. 2010, 21, 691–698. [Google Scholar]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar]
- Dixon, B.M.; Heath, S.H.; Kim, R.; Suh, J.H.; Hagen, T.M. Assessment of endoplasmic reticulum glutathione redox status is confounded by extensive ex vivo oxidation. Antioxid. Redox Signal. 2008, 10, 963–972. [Google Scholar]
- Stefani, I.C.; Wright, D.; Polizzi, K.M.; Kontoravdi, C. The role of ER stress-induced apoptosis in neurodegeneration. Curr. Alzheimer Res. 2012, 9, 373–387. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Gotz, J.; Eckert, A.; Matamales, M.; Ittner, L.M.; Liu, X. Modes of Abeta toxicity in Alzheimer’s disease. Cell. Mol. Life Sci. 2011, 68, 3359–3375. [Google Scholar] [CrossRef]
- Hatters, D.M. Protein misfolding inside cells: The case of huntingtin and Huntington’s disease. IUBMB Life 2008, 60, 724–728. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar]
- Gonzalez-Dosal, R.; Horan, K.A.; Rahbek, S.H.; Ichijo, H.; Chen, Z.J.; Mieyal, J.J.; Hartmann, R.; Paludan, S.R. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: Role for S-glutathionylation of TRAF3 and 6. PLoS Pathog. 2011, 7, e1002250. [Google Scholar]
- Zucchelli, S.; Codrich, M.; Marcuzzi, F.; Pinto, M.; Vilotti, S.; Biagioli, M.; Ferrer, I.; Gustincich, S. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 2010, 19, 3759–3770. [Google Scholar] [CrossRef]
- Silva, G.M.; Netto, L.E.; Discola, K.F.; Piassa-Filho, G.M.; Pimenta, D.C.; Barcena, J.A.; Demasi, M. Role of glutaredoxin 2 and cytosolic thioredoxins in cysteinyl-based redox modification of the 20S proteasome. FEBS J. 2008, 275, 2942–2955. [Google Scholar] [CrossRef]
- Silva, G.M.; Netto, L.E.; Simoes, V.; Santos, L.F.; Gozzo, F.C.; Demasi, M.A.; Oliveira, C.L.; Bicev, R.N.; Klitzke, C.F.; Sogayar, M.C.; Demasi, M. Redox control of 20S proteasome gating. Antioxid. Redox Signal. 2012, 16, 1183–1194. [Google Scholar] [CrossRef]
- Mieyal, J.J.; Chock, P.B. Posttranslational modification of cysteine in redox signaling and oxidative stress: Focus on S-glutathionylation. Antioxid. Redox Signal. 2012, 16, 471–475. [Google Scholar] [CrossRef]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef]
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases-focus on S-glutathionylation. Antioxid. Redox Signal. 2012, 16, 543–566. [Google Scholar] [CrossRef]
- Shelton, M.D.; Chock, P.B.; Mieyal, J.J. Glutaredoxin: Role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 2005, 7, 348–366. [Google Scholar] [CrossRef]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef]
- Cristalli, D.O.; Arnal, N.; Marra, F.A.; de Alaniz, M.J.; Marra, C.A. Peripheral markers in neurodegenerative patients and their first-degree relatives. J. Neurol. Sci. 2012, 314, 48–56. [Google Scholar] [CrossRef]
- Klepac, N.; Relja, M.; Klepac, R.; Hecimovic, S.; Babic, T.; Trkulja, V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J. Neurol. 2007, 254, 1676–1683. [Google Scholar] [CrossRef] [Green Version]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Singh, R.L.; Kalita, J.; Misra, U.K. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Invest. 2001, 31, 1007–1011. [Google Scholar] [CrossRef]
- Vilar, R.; Coelho, H.; Rodrigues, E.; Gama, M.J.; Rivera, I.; Taioli, E.; Lechner, E.C. Association of A313 G polymorphism (GSTP1*B) in the glutathione-S-transferase P1 gene with sporadic Parkinson’s disease. Eur. J. Neurol. 2007, 14, 156–161. [Google Scholar] [CrossRef]
- Paz-y-Mino, C.; Carrera, C.; Lopez-Cortes, A.; Munoz, M.J.; Cumbal, N.; Castro, B.; Cabrera, A.; Sanchez, M.E. Genetic polymorphisms in apolipoprotein E and glutathione peroxidase 1 genes in the Ecuadorian population affected with Alzheimer’s disease. Am. J. Med. Sci. 2010, 340, 373–377. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, Y.R.; Cheng, M.L.; Liu, J.L.; Lee, Y.M.; Lee, P.W.; Soong, B.W.; Chiu, D.T. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef]
- Usarek, E.; Gajewska, B.; Kazmierczak, B.; Kuzma, M.; Dziewulska, D.; Baranczyk-Kuzma, A. A study of glutathione S-transferase pi expression in central nervous system of subjects with amyotrophic lateral sclerosis using RNA extraction from formalin-fixed, paraffin-embedded material. Neurochem. Res. 2005, 30, 1003–1007. [Google Scholar] [CrossRef]
- Kil, I.S.; Park, J.W. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 2005, 280, 10846–10854. [Google Scholar] [CrossRef]
- Dinoto, L.; Deture, M.A.; Purich, D.L. Structural insights into Alzheimer filament assembly pathways based on site-directed mutagenesis and S-glutathionylation of three-repeat neuronal Tau protein. Microsc. Res. Tech. 2005, 67, 156–163. [Google Scholar] [CrossRef]
- Wilcox, K.C.; Zhou, L.; Jordon, J.K.; Huang, Y.; Yu, Y.; Redler, R.L.; Chen, X.; Caplow, M.; Dokholyan, N.V. Modifications of superoxide dismutase (SOD1) in human erythrocytes: A possible role in amyotrophic lateral sclerosis. J. Biol. Chem. 2009, 284, 13940–13947. [Google Scholar]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; Di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; Piemonte, F. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar]
- Martin, H.L.; Teismann, P. Glutathione-A review on its role and significance in Parkinson’s disease. FASEB J. 2009, 23, 3263–3272. [Google Scholar] [CrossRef]
- Ridet, J.L.; Bensadoun, J.C.; Deglon, N.; Aebischer, P.; Zurn, A.D. Lentivirus-mediated expression of glutathione peroxidase: Neuroprotection in murine models of Parkinson’s disease. Neurobiol. Dis. 2006, 21, 29–34. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Paik, S.R.; Lee, D.; Cho, H.J.; Lee, E.N.; Chang, C.S. Oxidized glutathione stimulated the amyloid formation of α-synuclein. FEBS Lett. 2003, 537, 63–67. [Google Scholar] [CrossRef]
- Trinh, K.; Moore, K.; Wes, P.D.; Muchowski, P.J.; Dey, J.; Andrews, L.; Pallanck, L.J. Induction of the phase II detoxification pathway suppresses neuron loss in Drosophila models of Parkinson’s disease. J. Neurosci. 2008, 28, 465–472. [Google Scholar]
- Gorner, K.; Holtorf, E.; Odoy, S.; Nuscher, B.; Yamamoto, A.; Regula, J.T.; Beyer, K.; Haass, C.; Kahle, P.J. Differential effects of Parkinson’s disease-associated mutations on stability and folding of DJ-1. J. Biol. Chem. 2004, 279, 6943–6951. [Google Scholar]
- Saeed, U.; Ray, A.; Valli, R.K.; Kumar, A.M.; Ravindranath, V. DJ-1 loss by glutaredoxin but not glutathione depletion triggers Daxx translocation and cell death. Antioxid. Redox Signal. 2010, 13, 127–144. [Google Scholar] [CrossRef]
- Chung, K.K.; Dawson, V.L.; Dawson, T.M. S-nitrosylation in Parkinson’s disease and related neurodegenerative disorders. Methods Enzymol. 2005, 396, 139–150. [Google Scholar]
- Wu, Y.; Fan, Y.; Xue, B.; Luo, L.; Shen, J.; Zhang, S.; Jiang, Y.; Yin, Z. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene 2006, 25, 5787–5800. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Keating, A.F. Protective role for ovarian glutathione S-transferase isoform pi during 7,12-dimethylbenz[a]anthracene-induced ovotoxicity. Toxicol. Appl. Pharmacol. 2012, 260, 201–208. [Google Scholar] [CrossRef]
- Dusinska, M.; Staruchova, M.; Horska, A.; Smolkova, B.; Collins, A.; Bonassi, S.; Volkovova, K. Are glutathione S transferases involved in DNA damage signalling? Interactions with DNA damage and repair revealed from molecular epidemiology studies. Mutat. Res. 2012, 736, 130–137. [Google Scholar] [CrossRef]
- Reddy, P.; Naidoo, R.N.; Robins, T.G.; Mentz, G.; Li, H.; London, S.J.; Batterman, S. GSTM1 and GSTP1 gene variants and the effect of air pollutants on lung function measures in South African children. Am. J. Ind. Med. 2012. [Google Scholar] [CrossRef]
- Garrido, M.; Tereshchenko, Y.; Zhevtsova, Z.; Taschenberger, G.; Bahr, M.; Kugler, S. Glutathione depletion and overproduction both initiate degeneration of nigral dopaminergic neurons. Acta Neuropathol. 2011, 121, 475–485. [Google Scholar] [CrossRef]
- Liedhegner, E.A.; Steller, K.M.; Mieyal, J.J. Levodopa activates apoptosis signaling kinase 1 (ASK1) and promotes apoptosis in a neuronal model: Implications for the treatment of Parkinson’s disease. Chem. Res. Toxicol. 2011, 24, 1644–1652. [Google Scholar]
- Jung, K.H.; Park, J.W. Suppression of mitochondrial NADP(+)-dependent isocitrate dehydrogenase activity enhances curcumin-induced apoptosis in HCT116 cells. Free Radic. Res. 2011, 45, 431–438. [Google Scholar] [CrossRef]
- Kil, I.S.; Jung, K.H.; Nam, W.S.; Park, J.W. Attenuated mitochondrial NADP+-dependent isocitrate dehydrogenase activity enhances EGCG-induced apoptosis. Biochimie 2011, 93, 1808–1815. [Google Scholar] [CrossRef]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Kontush, A. Amyloid-beta: An antioxidant that becomes a pro-oxidant and critically contributes to Alzheimer’s disease. Free Radic. Biol. Med. 2001, 31, 1120–1131. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Hamanishi, T.; Furuta, H.; Kato, H.; Doi, A.; Tamai, M.; Shimomura, H.; Sakagashira, S.; Nishi, M.; Sasaki, H.; Sanke, T.; Nanjo, K. Functional variants in the glutathione peroxidase-1 (GPx-1) gene are associated with increased intima-media thickness of carotid arteries and risk of macrovascular diseases in japanese type 2 diabetic patients. Diabetes 2004, 53, 2455–2460. [Google Scholar] [CrossRef]
- Padurariu, M.; Ciobica, A.; Hritcu, L.; Stoica, B.; Bild, W.; Stefanescu, C. Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2010, 469, 6–10. [Google Scholar] [CrossRef]
- Spalletta, G.; Bernardini, S.; Bellincampi, L.; Federici, G.; Trequattrini, A.; Ciappi, F.; Bria, P.; Caltagirone, C.; Bossu, P. Glutathione S-transferase P1 and T1 gene polymorphisms predict longitudinal course and age at onset of Alzheimer disease. Am. J. Geriatr. Psychiatry 2007, 15, 879–887. [Google Scholar] [CrossRef]
- Savonenko, A.; Xu, G.M.; Melnikova, T.; Morton, J.L.; Gonzales, V.; Wong, M.P.; Price, D.L.; Tang, F.; Markowska, A.L.; Borchelt, D.R. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: Relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 2005, 18, 602–617. [Google Scholar] [CrossRef]
- Zhang, C.; Rodriguez, C.; Spaulding, J.; Aw, T.Y.; Feng, J. Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 28, 655–666. [Google Scholar]
- Fu, A.L.; Dong, Z.H.; Sun, M.J. Protective effect of N-acetyl-l-cysteine on amyloid beta-peptide-induced learning and memory deficits in mice. Brain Res. 1109, 201–206. [Google Scholar]
- Huang, Q.; Aluise, C.D.; Joshi, G.; Sultana, R.; St Clair, D.K.; Markesbery, W.R.; Butterfield, D.A. Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: Toward therapeutic modulation of mild cognitive impairment. J. Neurosci. Res. 2010, 88, 2618–2629. [Google Scholar]
- Xu, Y.; Hou, X.Y.; Liu, Y.; Zong, Y.Y. Different protection of K252a and N-acetyl-L-cysteine against amyloid-beta peptide-induced cortical neuron apoptosis involving inhibition of MLK3-MKK7-JNK3 signal cascades. J. Neurosci. Res. 2009, 87, 918–927. [Google Scholar] [CrossRef]
- Studer, R.; Baysang, G.; Brack, C. N-Acetyl-l-Cystein downregulates beta-amyloid precursor protein gene transcription in human neuroblastoma cells. Biogerontology 2001, 2, 55–60. [Google Scholar] [CrossRef]
- Adair, J.C.; Knoefel, J.E.; Morgan, N. Controlled trial of N-acetylcysteine for patients with probable Alzheimer’s disease. Neurology 2001, 57, 1515–1517. [Google Scholar] [CrossRef]
- McCaddon, A.; Davies, G. Co-administration of N-acetylcysteine, vitamin B12 and folate in cognitively impaired hyperhomocysteinaemic patients. Int. J. Geriatr. Psychiatry 2005, 20, 998–1000. [Google Scholar]
- McCaddon, A.; Hudson, P.R. l-methylfolate, methylcobalamin, and N-acetylcysteine in the treatment of Alzheimer’s disease-related cognitive decline. CNS Spectr. 2010, 15, 2–5. [Google Scholar]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative stress in Huntington’s disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar]
- Del Hoyo, P.; Garcia-Redondo, A.; de Bustos, F.; Molina, J.A.; Sayed, Y.; Alonso-Navarro, H.; Caballero, L.; Arenas, J.; Jimenez-Jimenez, F.J. Oxidative stress in skin fibroblasts cultures of patients with Huntington’s disease. Neurochem. Res. 2006, 31, 1103–1109. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; Bates, G.P. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Choo, Y.S.; Mao, Z.; Johnson, G.V.; Lesort, M. Increased glutathione levels in cortical and striatal mitochondria of the R6/2 Huntington’s disease mouse model. Neurosci. Lett. 2005, 386, 63–68. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Protective effect of sesamol against 3-nitropropionic acid-induced cognitive dysfunction and altered glutathione redox balance in rats. Basic Clin. Pharmacol. Toxicol. 2010, 107, 577–582. [Google Scholar] [CrossRef]
- Mao, Z.; Choo, Y.S.; Lesort, M. Cystamine and cysteamine prevent 3-NP-induced mitochondrial depolarization of Huntington’s disease knock-in striatal cells. Eur. J. Neurosci. 2006, 23, 1701–1710. [Google Scholar] [CrossRef]
- Carri, M.T.; Cozzolino, M. SOD1 and mitochondria in ALS: A dangerous liaison. J. Bioenerg. Biomembr. 2011, 43, 593–599. [Google Scholar] [CrossRef]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; Cleveland, D.W. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar]
- Bruijn, L.I.; Beal, M.F.; Becher, M.W.; Schulz, J.B.; Wong, P.C.; Price, D.L.; Cleveland, D.W. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc. Natl. Acad. Sci. USA 1997, 94, 7606–7611. [Google Scholar]
- Andrus, P.K.; Fleck, T.J.; Gurney, M.E.; Hall, E.D. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 2041–2048. [Google Scholar]
- Rothstein, J.D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 2009, 65, S3–S9. [Google Scholar] [CrossRef]
- Panov, A.; Kubalik, N.; Zinchenko, N.; Hemendinger, R.; Dikalov, S.; Bonkovsky, H.L. Respiration and ROS production in brain and spinal cord mitochondria of transgenic rats with mutant G93a Cu/Zn-superoxide dismutase gene. Neurobiol. Dis. 2011, 44, 53–62. [Google Scholar] [CrossRef]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef]
- Cova, E.; Bongioanni, P.; Cereda, C.; Metelli, M.R.; Salvaneschi, L.; Bernuzzi, S.; Guareschi, S.; Rossi, B.; Ceroni, M. Time course of oxidant markers and antioxidant defenses in subgroups of amyotrophic lateral sclerosis patients. Neurochem. Int. 2010, 56, 687–693. [Google Scholar] [CrossRef]
- Bonnefont-Rousselot, D.; Lacomblez, L.; Jaudon, M.; Lepage, S.; Salachas, F.; Bensimon, G.; Bizard, C.; Doppler, V.; Delattre, J.; Meininger, V. Blood oxidative stress in amyotrophic lateral sclerosis. J. Neurol. Sci. 2000, 178, 57–62. [Google Scholar]
- Morahan, J.M.; Yu, B.; Trent, R.J.; Pamphlett, R. Genetic susceptibility to environmental toxicants in ALS. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144, 885–890. [Google Scholar]
- Tartari, S.; D’Alessandro, G.; Babetto, E.; Rizzardini, M.; Conforti, L.; Cantoni, L. Adaptation to G93Asuperoxide dismutase 1 in a motor neuron cell line model of amyotrophic lateral sclerosis: The role of glutathione. FEBS J. 2009, 276, 2861–2874. [Google Scholar] [CrossRef]
- Muyderman, H.; Hutson, P.G.; Matusica, D.; Rogers, M.L.; Rush, R.A. The human G93A-superoxide dismutase-1 mutation, mitochondrial glutathione and apoptotic cell death. Neurochem. Res. 2009, 34, 1847–1856. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1G93A mice model. Neurobiol. Dis. 2011, 43, 543–551. [Google Scholar] [CrossRef]
- Redler, R.L.; Wilcox, K.C.; Proctor, E.A.; Fee, L.; Caplow, M.; Dokholyan, N.V. Glutathionylation at Cys-111 induces dissociation of wild type and FALS mutant SOD1 dimers. Biochemistry 2011, 50, 7057–7066. [Google Scholar]
- Marmolino, D. Friedreich’s ataxia: Past, present and future. Brain Res. Rev. 2011, 67, 311–330. [Google Scholar] [CrossRef]
- Sparaco, M.; Gaeta, L.M.; Santorelli, F.M.; Passarelli, C.; Tozzi, G.; Bertini, E.; Simonati, A.; Scaravilli, F.; Taroni, F.; Duyckaerts, C.; et al. Friedreich’s ataxia: Oxidative stress and cytoskeletal abnormalities. J. Neurol. Sci. 2009, 287, 111–118. [Google Scholar] [CrossRef]
- Auchere, F.; Santos, R.; Planamente, S.; Lesuisse, E.; Camadro, J.M. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich’s ataxia. Hum. Mol. Genet. 2008, 17, 2790–2802. [Google Scholar] [CrossRef]
- Shoichet, S.A.; Baumer, A.T.; Stamenkovic, D.; Sauer, H.; Pfeiffer, A.F.; Kahn, C.R.; Muller-Wieland, D.; Richter, C.; Ristow, M. Frataxin promotes antioxidant defense in a thiol-dependent manner resulting in diminished malignant transformation in vitro. Hum. Mol. Genet. 2002, 11, 815–821. [Google Scholar] [CrossRef]
- Wang, J.; Boja, E.S.; Tan, W.; Tekle, E.; Fales, H.M.; English, S.; Mieyal, J.J.; Chock, P.B. Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 2001, 276, 47763–47766. [Google Scholar]
- Lee, S.B.; Bagley, J.A.; Lee, H.Y.; Jan, L.Y.; Jan, Y.N. Pathogenic polyglutamine proteins cause dendrite defects associated with specific actin cytoskeletal alterations in Drosophila. Proc. Natl. Acad. Sci. USA 2011, 108, 16795–16800. [Google Scholar]
- Jones, D.P.; Coates, R.J.; Flagg, E.W.; Eley, J.W.; Block, G.; Greenberg, R.S.; Gunter, E.W.; Jackson, B. Glutathione in foods listed in the National Cancer Institute’s Health Habits and History Food Frequency Questionnaire. Nutr. Cancer 1992, 17, 57–75. [Google Scholar] [CrossRef]
- Witschi, A.; Reddy, S.; Stofer, B.; Lauterburg, B.H. The systemic availability of oral glutathione. Eur. J. Clin. Pharmacol. 1992, 43, 667–669. [Google Scholar] [CrossRef]
- Valenzuela, A.; Aspillaga, M.; Vial, S.; Guerra, R. Selectivity of silymarin on the increase of the glutathione content in different tissues of the rat. Planta Med. 1989, 55, 420–422. [Google Scholar] [CrossRef]
- Reeta, K.H.; Mehla, J.; Gupta, Y.K. Curcumin is protective against phenytoin-induced cognitive impairment and oxidative stress in rats. Brain Res. 1301, 52–60. [Google Scholar]
- Jia, Z.; Hallur, S.; Zhu, H.; Li, Y.; Misra, H.P. Potent upregulation of glutathione and NAD(P)H:quinone oxidoreductase 1 by alpha-lipoic acid in human neuroblastoma SH-SY5Y cells: Protection against neurotoxicant-elicited cytotoxicity. Neurochem. Res. 2008, 33, 790–800. [Google Scholar] [CrossRef]
- Dickinson, D.A.; Iles, K.E.; Zhang, H.; Blank, V.; Forman, H.J. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. FASEB J. 2003, 17, 473–475. [Google Scholar]
- Singhal, N.K.; Srivastava, G.; Patel, D.K.; Jain, S.K.; Singh, M.P. Melatonin or silymarin reduces maneb- and paraquat-induced Parkinson’s disease phenotype in the mouse. J. Pineal Res. 2011, 50, 97–109. [Google Scholar]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar]
- Andreassen, O.A.; Dedeoglu, A.; Friedlich, A.; Ferrante, K.L.; Hughes, D.; Szabo, C.; Beal, M.F. Effects of an inhibitor of poly(ADP-ribose) polymerase, desmethylselegiline, trientine, and lipoic acid in transgenic ALS mice. Exp. Neurol. 2001, 168, 419–424. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Ferrante, R.J.; Dedeoglu, A.; Beal, M.F. Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. Neuroreport 2001, 12, 3371–3373. [Google Scholar] [CrossRef]
- Aebi, S.; Assereto, R.; Lauterburg, B.H. High-dose intravenous glutathione in man. Pharmacokinetics and effects on cyst(e)ine in plasma and urine. Eur. J. Clin. Invest. 1991, 21, 103–110. [Google Scholar] [CrossRef]
- Sechi, G.; Deledda, M.G.; Bua, G.; Satta, W.M.; Deiana, G.A.; Pes, G.M.; Rosati, G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1996, 20, 1159–1170. [Google Scholar] [CrossRef]
- Hauser, R.A.; Lyons, K.E.; McClain, T.; Carter, S.; Perlmutter, D. Randomized, double-blind, pilot evaluation of intravenous glutathione in Parkinson’s disease. Mov. Disord. 2009, 24, 979–983. [Google Scholar] [CrossRef]
- Okun, M.S.; Lan, A.; Jankovic, J. Based on the available randomized trial patients should say no to glutathione for Parkinson’s disease. Mov. Disord. 2010, 25, 961–962. [Google Scholar] [CrossRef]
- Naito, Y.; Matsuo, K.; Kokubo, Y.; Narita, Y.; Tomimoto, H. Higher-dose glutathione therapy for Parkinson’s disease in Japan: Is it really safe? Mov. Disord. 2010, 25, 962–963. [Google Scholar]
- Sechi, G.P. Reduced glutathione and Parkinson’s disease. Mov. Disord. 2010, 25, 2690–2691. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef]
- Clark, J.; Clore, E.L.; Zheng, K.; Adame, A.; Masliah, E.; Simon, D.K. Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in alpha-synuclein overexpressing mice. PLoS One 2010, 5, e12333. [Google Scholar]
- Andreassen, O.A.; Dedeoglu, A.; Klivenyi, P.; Beal, M.F.; Bush, A.I. N-acetyl-l-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. Neuroreport 2000, 11, 2491–2493. [Google Scholar] [CrossRef]
- Sandhir, R.; Sood, A.; Mehrotra, A.; Kamboj, S.S. N-Acetylcysteine reverses mitochondrial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener. Dis. 2012, 9, 145–157. [Google Scholar] [CrossRef]
- Louwerse, E.S.; Weverling, G.J.; Bossuyt, P.M.; Meyje, F.E.; de Jong, J.M. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch. Neurol. 1995, 52, 559–564. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399-1440. https://doi.org/10.3390/nu4101399
Johnson WM, Wilson-Delfosse AL, Mieyal JJ. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients. 2012; 4(10):1399-1440. https://doi.org/10.3390/nu4101399
Chicago/Turabian StyleJohnson, William M., Amy L. Wilson-Delfosse, and John. J. Mieyal. 2012. "Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases" Nutrients 4, no. 10: 1399-1440. https://doi.org/10.3390/nu4101399