Potential Health-modulating Effects of Isoflavones and Metabolites via Activation of PPAR and AhR

Abstract
:1. Introduction
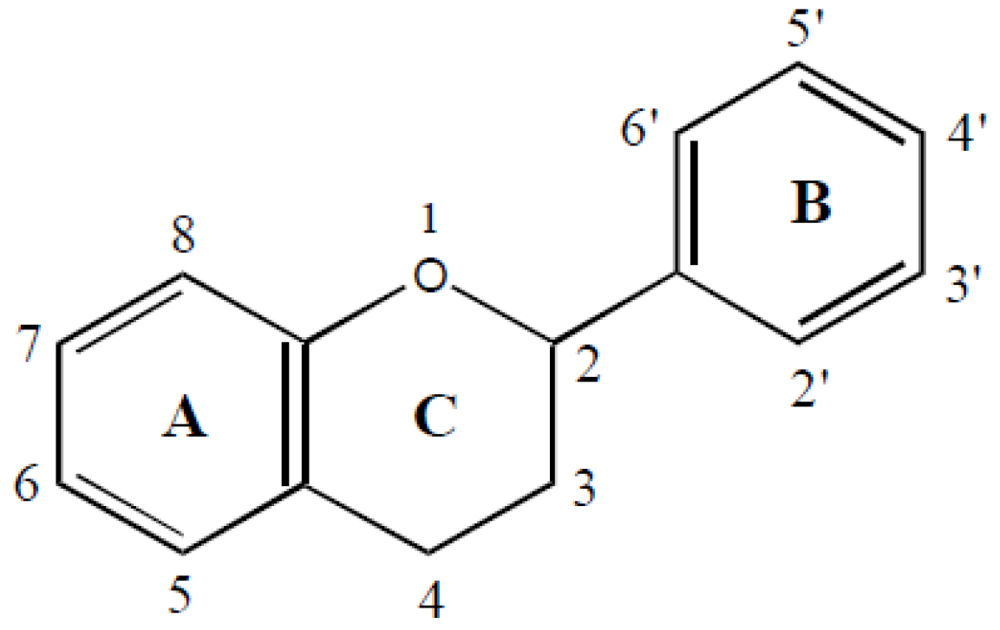
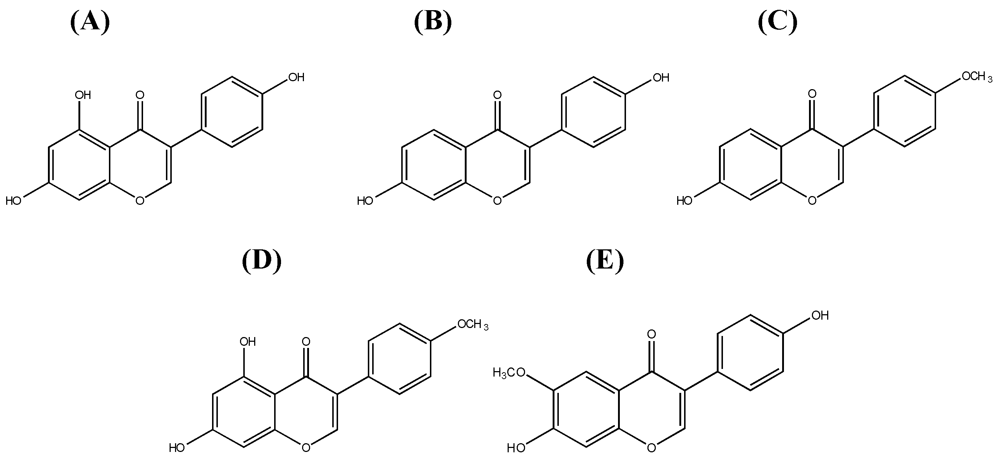
1.1. Systematics of Isoflavones

1.2. Dietary Sources and Intake of Isoflavones
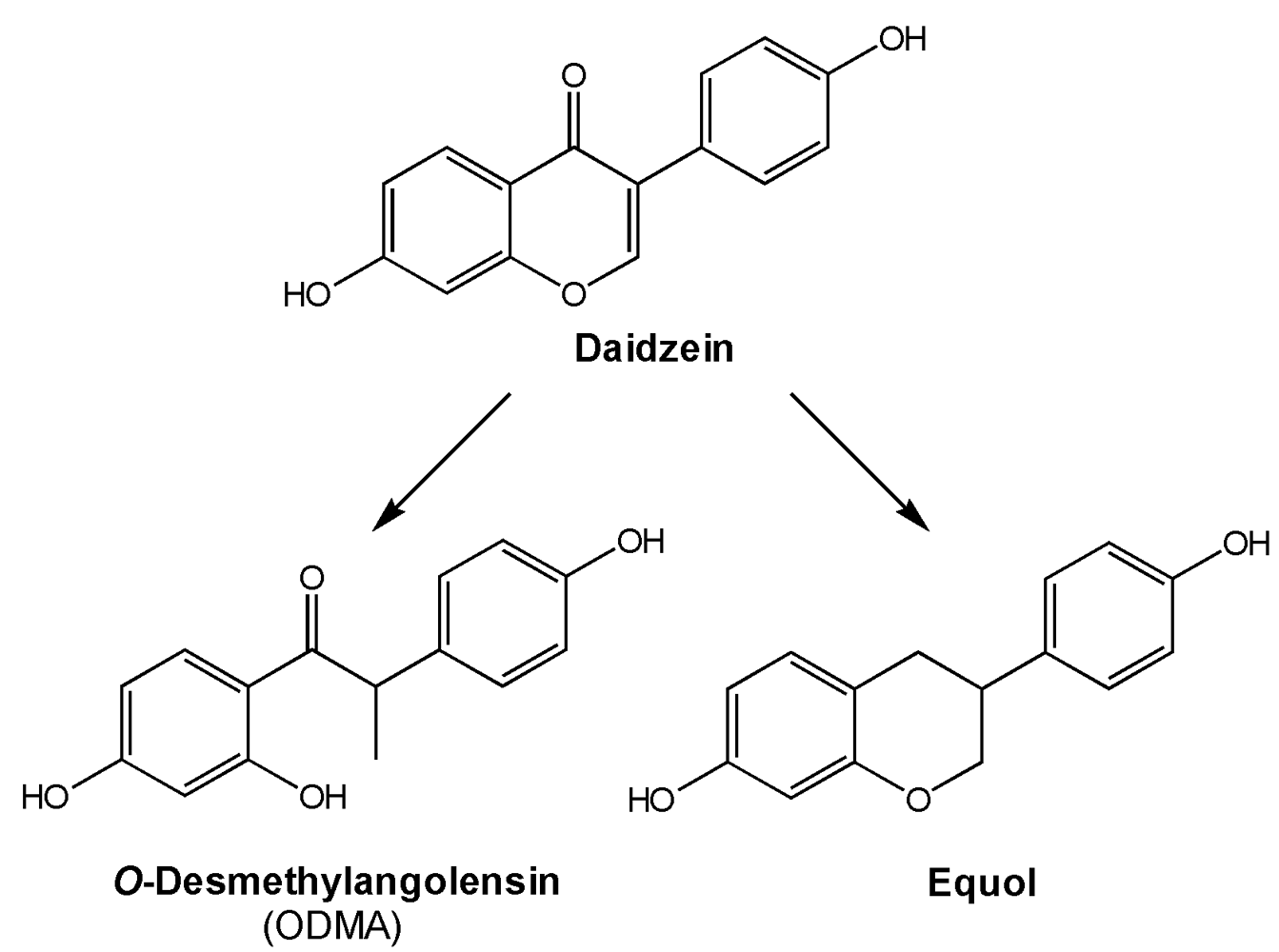
1.3. Metabolism and Bioavailability of Isoflavones

1.4. Metabolic Diseases
1.4.1. Peroxisome proliferator-activated receptors α and γ
1.4.2. Inflammation and Atherosclerosis
1.4.3. PPAR Activation in in vitro Assays

{kind=link}
{kind=link}
{kind=link}
| PPARα Transactivation | PPARγ Ligands | PPARγ Transactivation | Ref |
|---|---|---|---|
| biochanin A, genistein, daidzein, equol | [116] | ||
| genistein | genistein | [117] | |
| daidzein | daidzein | [118] | |
| genistein | [119] | ||
| daidzein | [120] | ||
| genistein, daidzein | genistein, daidzein | [121] | |
| biochanin A, genistein, daidzein, equol, ODMA, 6-hydroxydaidzein, 3´-hydroxygenistein, 6´-hydroxy-ODMA, angolensin, dihydrogenistein, dihydrobiochaninA, dihydroformononetin, dihydrodaidzein, p-ethylphenol | biochanin A, genistein, daidzein, equol, ODMA, 6-hydroxydaidzein, 3´-hydroxygenistein, 6´-hydroxy-ODMA, dihydrogenistein, dihydrodaidzein | [115] | |
| biochanin A, genistein, daidzein, ODMA, 6-hydroxydaidzein, 3´-hydroxygenistein | [114] | ||
| genistein, daidzein | genistein, daidzein, glycitein | [122] | |
| daidzein, equol | [123] | ||
| biochanin A, formononetin, genistein | biochanin A, genistein, daidzein | biochanin A, formononetin, genistein | [124] |
| Compounds | Cell line | Downregulated pro-inflammatory mediators | Upregulated anti-inflammatory mediators | Ref. |
|---|---|---|---|---|
| genistein, equol | RAW 264.7 | NO, PGE2 | [136] | |
| genistein, | RAW 264.7 | TNFα, IL-6, iNOS, NFκB | IL-10 | [114] |
| daidzein, | TNFα, IL-6, iNOS, NFκB | |||
| formononetin | iNOS | |||
| biochanin A | TNFα, IL-6, iNOS, NFκB, Cox-2 | IL-10 | ||
| equol | TNFα, IL-6, COX-2 | |||
| ODMA | TNFα, IL-6 | |||
| genistein | HBMEC | TNFα, IL-1β, monocyte chemoattractant protein-1, IL-8, intercellular adhesion molecule-1 | [137] | |
| genistein, daidzein | murine J774 macrophages | iNOS, NO | [138] | |
| genistein | Human chondrocytes | COX-2, NO | [139] | |
| biochanin A | MC3T3-E1 cells | TNFα, IL-6, NO | [140] | |
| genistein | PBLs | TNFα, IL-8 | [141] | |
| genistein | mesencephalic neuron-glia cultures | TNFα, NO, superoxide | [142] | |
| daidzein, formononetin | mesencephalic neuron-glia cultures | TNFα, NO, superoxide | [143] | |
| biochanin A | mesencephalic neuron-glia cultures | TNFα, NO, superoxide | [144] | |
| genistein | alveolar macrophages | TNFα | [145] | |
| daidzein | PBMC | higher concentrations reduced IL-10 and IFN-γ levels | low concentration increased IL-2, IL-4,and IFN-γ | [146] |
| genistein | IL-2, IL-4, IL-10, IFN-γ mRNA and protein | |||
| genistein | RAW 264.7 | NO, PGE2 | [147] | |
| genistein | RAW 264.7 | PGE2, iNOS, COX-2 | [148] | |
| genistein, daidzein, glycetein | RAW 264.7 | NO, iNOS | [149] | |
| genistein, daidzein, equol | MCF-7 cells | COX-2 | [150] |
1.4.4. PPAR activation by isoflavones and its health effects
1.5. Xenobiotic Metabolism and Cell Cycle Control
1.5.1. The aryl hydrocarbon receptor
1.5.2. AhR in vitro assays
| Agonistic effects | Antagonistic effects | Assay | Ref. |
|---|---|---|---|
| Dai(+)* | Dai(-), Gen(-) | Gel mobility shift assay (agonistic effects) | [220] |
| LBA (rat hepatic cytosol) (antagonistic effects) | |||
| Dai(-), Gen(+), Gly(-), Equ(+) | LBA (mammalian liver cell cytosol) | [218] | |
| Dai(+), Gen(+), Gly(+), Equ(-) | CALUX (mouse hepatoma cells) | [217] | |
| Gen(-) | LBA (rat hepatic cytosol) | [224] | |
| Dai(+)*,Gen (-) Dai(-),Gen (-) | SW-ELISA (Hepa-1c1c7) | [225] | |
| Dai(+)*,Gen (-) Dai(-),Gen (-) | CALUX (HepG2 cells) | ||
| Dai(+), Gen(+) | Transactivation assay (Hepa-1 cells) | [190] | |
| Dai(-), Gen(-) | Transactivation assay (HepG2 cells) | ||
| Dai(-), Gen(-) | Transactivation assay (MCF-7 cells) | ||
| Dai(-), Gen(-) | LBA (rat hepatic cytosol) | [191] | |
| Dai(+)*, Gen(+)* | Dai(+), Gen(+) | CYP1A1 expression in HepG2 cells | [226] |
| Bio(+) | Bio(+) | CYP1A1 expression in MCF-7 cells | [227] |
| LBA (rat hepatic cytosol) | |||
| Bio(+)* | Bio(+) | CALUX (MCF-7 cells) | [228] |
| CYP1A1 and CYP1B1 expression in MCF-7 cells | |||
| Bio(+)#, Dai(-), Equ(+)*, For(+)#, Gen(-) | Transactivation assay (yeast) | [189] |
1.5.3. Cytochrome P450 enzyme CYP1A1
1.5.4. Cell cycle control
| Effect on cell cycle(cell type) | Further effects | Tested isoflavone (concentration) | Ref. |
|---|---|---|---|
| G2/M arrest(colon cancer)a | Genistein (111 µM) | [258] | |
| G2/M arrest(prostate cancer)b | Concomitant decrease of cyclin B | Isoflavones from soybean cake; genistein most efficient (30–50 µM) | [259] |
| G2/M arrest(bladder cancer)c | Inhibition of cdc2 kinase activity | Genistein (37 or 185 µM) | [260] |
| Direct induction of apoptosis without alteration of cell cycle distribution | Daidzein (39.3 or 196.7 µM) and biochanin A (35.2 or 175.9 µM) | ||
| Suppression of tumor growth in vivo (xenograft model; mice) | Genistein and combined isoflavones | ||
| G2/M arrest(prostate cancer)d | Genistein (18.5–74 µM) | [261] | |
| G2/M arrest(breast cancer cells overexpressing Bcl-2)e1 | Genistein (50 µM) | [262] | |
| G0/G1 arrest(control breast cancer cells)e2 | Genistein (50 µM) | ||
| G2/M arrest(bladder cancer)f | Reduction of tumor volume in vivo (xenograft model; mice) | Genistein (50 µM) | [263] |
| G2/M arrest(androgen-insensitive prostate cancer)g1 | Induction of tumor suppressor gene expression (p21, p16) | Genistein (10 or 25 µM) | [264] |
| G0/G1 arrest(androgen-sensitive prostate cancer)g2 | Induction of apoptosis(only in androgen-insensitive cells) | Genistein (10 or 25 µM) | |
| G2/M arrest(liver cancer)h | Induction of tumor suppressor genes expression (p21),Accumulation of p53 protein | Genistein (37–111 µM) | [265] |
| G2/M arrest(leukemia cells)i | Stimulates Raf-1 activation, Decreases Akt activation, Induction of p21 and cyclin B expression, Induction of apoptosis | Genistein (10 or 25 µM) | [266] |
| G2/M arrest(prostate cancer)j | Increased p21 expression, Decreased cyclin B expression, Decreased NFκB activity | Genistein (15 or 30 µM) | [267] |
| G1 cell arrest(androgen-sensitive prostate cancer)k | Increased p27 and p21 expression | Genistein (≤20 µM) | [268] |
| Induction of apoptosis | Genistein (40–80 µM) | ||
| G2/M arrest(non-tumorigenic breast cells)l | Enhanced expression of p21 and p53, but not p27 | Genistein (30 µM) | [269] |
| G2/M arrest(prostate cancer)m | Genistein (20–100 µM) | [270] | |
| G2/M arrest(B cell leukemia)n | Decreased IL-10 secretion, Upregulation of IFNγ | Genistein (7.5–60 µM) | [271] |
| G2/M arrest(breast cancer)o | Increased cyclin B | Genistein (15 or 30 µM) | [272] |
| G2/M arrest(eye cancer; choroidal melanoma)p | Induction of p21, but not required for cell cycle arrest | Genistein (30 or 60 µM) | [273] |
| G2/M arrest(eye cancer; choroidal melanoma)q | Upregulation of CDK1 and p21, but no effect of CDK2 and p27 | Genistein (30 µM) | [274] |
| G1 cell arrest(eye cancer; choroidal melanoma)q | Upregulation of CDK2 and weakly p21 and p27 | Daidzein (150 µM) | |
| G2/M arrest(eye cancer; choroidalmelanoma)r | Impairment of CDK1 dephosphorylation, Weak accumulation of p53 protein | Genistein (60 µM) | [275] |
| G2/M arrest(metastatic melanoma)s | Genistein (60 µM) | [276] | |
| G2/M arrest(gastric cancer)t | Genistein (25 or 60 µM) | [277] | |
| G1 cell arrest(gastric cancer)t | Daidzein (25 or 60 µM) | ||
| G2/M arrest(metastatic melanoma)u | Genistein (60 µM) | [278] | |
| S phase arrest(metastatic melanoma)u | Daidzein (60 µM) | ||
| G0/G1 arrest(colon cancer)v | Biphasic effect on cell growth | Daidzein (5–100 µM) | [279] |
1.5.5. AhR activation by isoflavones and health effects
2. General Conclusion
References
- Haslam, E. Practical Polyphenolics: from Structure to Molecular Recognition and Physiological Action; Cambridge Univ. Press: Cambridge, UK, 1998; Volume XV, p. 422. [Google Scholar]
- Di Carlo, G.; Mascolo, N.; Izzo, A.A.; Capasso, F. Flavonoids: old and new aspects of a class of natural therapeutic drugs. Life Sci. 1999, 65, 337–353. [Google Scholar]
- Peterson, J.; Dwyer, J. Flavonoids: Dietary occurrence and biochemical activity. Nutrition Res. 1998, 18, 1995–2018. [Google Scholar]
- Mazur, W.; Adlercreutz, H. Overview of naturally occurring endocrine-active substances in the human diet in relation to human health. Nutrition 2000, 16, 654–658. [Google Scholar]
- Liggins, J.; Bluck, L.J.; Runswick, S.; Atkinson, C.; Coward, W.A.; Bingham, S.A. Daidzein and genistein content of fruits and nuts. J. Nutr. Biochem. 2000, 11, 326–331. [Google Scholar]
- Dwyer, J.T.; Goldin, B.R.; Saul, N.; Gualtieri, L.; Barakat, S.; Adlercreutz, H. Tofu and soy drinks contain phytoestrogens. J. Am. Diet. Assoc. 1994, 94, 739–743. [Google Scholar]
- Zhang, Y.C.; Albrecht, D.; Bomser, J.; Schwartz, S.J.; Vodovotz, Y. Isoflavone profile and biological activity of soy bread. J. Agric. Food Chem. 2003, 51, 7611–7616. [Google Scholar]
- Clarke, D.B.; Bailey, V.; Lloyd, A.S. Determination of phytoestrogens in dietary supplements by LC-MS/MS. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2008, 25, 534–547. [Google Scholar]
- Reiter, E.; Beck, V.; Medjakovic, S.; Mueller, M.; Jungbauer, A. Comparison of hormonal activity of isoflavone-containing supplements used to treat menopausal complaints. Menopause 2009, 16, 1049–1060. [Google Scholar]
- Arai, Y.; Watanabe, S.; Kimira, M.; Shimoi, K.; Mochizuki, R.; Kinae, N. Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL cholesterol concentration. J. Nutr. 2000, 130, 2243–2250. [Google Scholar]
- Kim, J.S.; Kwon, C.S. Estimated dietary isoflavone intake of Korean population based on national nutrition survey. Nutr. Res. 2001, 21, 947–953. [Google Scholar]
- Lee, S.A.; Wen, W.; Xiang, Y.B.; Barnes, S.; Liu, D.; Cai, Q.; Zheng, W.; Xiao, O.S. Assessment of dietary isoflavone intake among middle-aged Chinese men. J. Nutr. 2007, 137, 1011–1016. [Google Scholar]
- Liu, Z.; Li, W.; Sun, J.; Liu, C.; Zeng, Q.; Huang, J.; Yu, B.; Huo, J. Intake of soy foods and soy isoflavones by rural adult women in China. Asia Pac. J. Clin. Nutr. 2004, 13, 204–209. [Google Scholar]
- Surh, J.; Kim, M.J.; Koh, E.; Kim, Y.K.L.; Kwon, H. Estimated intakes of isoflavones and coumestrol in Korean population. Int. J. Food Sci. Nutr. 2006, 57, 325–344. [Google Scholar]
- Takata, Y.; Maskarinec, G.; Franke, A.; Nagata, C.; Shimizu, H. A comparison of dietary habits among women in Japan and Hawaii. Public Health Nutr. 2004, 7, 319–326. [Google Scholar]
- Wakai, K.; Egami, I.; Kato, K.; Kawamura, T.; Tamakoshi, A.; Lin, Y.; Nakayama, T.; Wada, M.; Ohno, Y. Dietary intake and sources of isoflavones among Japanese. Nutr. Cancer 1999, 33, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Boker, L.K.; Van der Schouw, Y.T.; De Kleijn, M.J.J.; Jacques, P.F.; Grobbee, D.E.; Peeters, P.H.M. Intake of dietary phytoestrogens by Dutch women. J. Nutr. 2002, 132, 1319. [Google Scholar]
- Chun, O.K.; Chung, S.J.; Song, W.O. Estimated dietary flavonoid intake and major food sources of U.S. adults. J. Nutr. 2007, 137, 1244–1252. [Google Scholar] [PubMed]
- De Kleijn, M.J.J.; Van der Schouw, Y.T.; Wilson, P.W.F.; Adlercreutz, H.; Mazur, W.; Grobbee, D.E.; Jacques, P.F. Intake of dietary phytoestrogens is low in postmenopausal women in the United States: The framingham study. J. Nutr. 2001, 131, 1826. [Google Scholar]
- Horn-Ross, P.L.; John, E.M.; Canchola, A.J.; Stewart, S.L.; Lee, M.M. Phytoestrogen intake and endometrial cancer risk. J. Natl. Cancer Inst. 2003, 95, 1158–1164. [Google Scholar]
- Mulligan, A.A.; Welch, A.A.; McTaggart, A.A.; Bhaniani, A.; Bingham, S.A. Intakes and sources of soya foods and isoflavones in a UK population cohort study (EPIC-Norfolk). Eur. J. Clin. Nutr. 2007, 61, 248–254. [Google Scholar]
- Setchell, K.D.R.; Brown, N.M.; Zimmer-Nechemias, L.; Brashear, W.T.; Wolfe, B.E.; Kirschner, A.S.; Heubi, J.E. Evidence for lack of absorption of soy isoflavone glycosides in humans, supporting the crucial role of intestinal metabolism for bioavailability. Am. J. Clin. Nutr. 2002, 76, 447–453. [Google Scholar]
- Richelle, M.; Pridmore-Merten, S.; Bodenstab, S.; Enslen, M.; Offord, E.A. Hydrolysis of isoflavone glycosides to aglycones by beta-glycosidase does not alter plasma and urine isoflavone pharmacokinetics in postmenopausal women. J. Nutr. 2002, 132, 2587–2592. [Google Scholar]
- Zheng, V.; Lee, S.O.; Murphy, P.A.; Hendrich, S.; Verbruggen, M.A. The apparent absorptions of isoflavone glucosides and aglucons are similar in women and are increased by rapid gut transit time and low fecal isoflavone degradation. J. Nutr. 2004, 134, 2534. [Google Scholar]
- Zubik, L.; Meydani, M. Bioavailability of soybean isoflavones from aglycone and glucoside forms in American women. Am. J. Clin. Nutr. 2003, 77, 1459–1465. [Google Scholar]
- Izumi, T.; Piskula, M.K.; Osawa, S.; Obata, A.; Tobe, K.; Saito, M.; Kataoka, S.; Kubota, Y.; Kikuchi, M. Soy isoflavone aglycones are absorbed faster and in higher amounts than their glucosides in humans. J. Nutr. 2000, 130, 1695–1699. [Google Scholar]
- Kano, M.; Takayanagi, T.; Harada, K.; Sawada, S.; Ishikawa, F. Bioavailability of isoflavones after ingestion of soy beverages in healthy adults. J. Nutr. 2006, 136, 2291–2296. [Google Scholar]
- de Pascual-Teresa, S.; Hallund, J.; Talbot, D.; Schroot, J.; Williams, C.M.; Bugel, S.; Cassidy, A. Absorption of isoflavones in humans: effects of food matrix and processing. J. Nutr. Biochem. 2006, 17, 257–264. [Google Scholar] [Green Version]
- Tsunoda, N.; Pomeroy, S.; Nestel, P. Absorption in humans of isoflavones from soy and red clover is similar. J. Nutr. 2002, 132, 2199. [Google Scholar]
- Day, A.J.; Dupont, M.S.; Rhodes, M.J.C.; Morgan, M.R.A.; Williamson, G.; Ridley, S.; Rhodes, M. Deglycosylation of flavonoid and isoflavonoid glycosides by human small intestine and liver β-glucosidase activity. FEBS Lett. 1998, 436, 71. [Google Scholar]
- Akaza, H.; Miyanaga, N.; Takashima, N.; Naito, S.; Hirao, Y.; Tsukamoto, T.; Fujioka, T.; Mori, M.; Kim, W.J.; Song, J.M.; Pantuck, A.J. Comparisons of percent equol producers between prostate vancer patients and controls: Case-controlled studies of isoflavones in Japanese, Korean and American residents. Jpn. J. Clin. Oncol. 2004, 34, 86–89. [Google Scholar]
- Akaza, H.; Miyanaga, N.; Takashima, N.; Naito, S.; Hirao, Y.; Tsukamoto, T.; Mori, M. Is daidzein non-metabolizer a high risk for prostate cancer? A case-controlled study of serum soybean isoflavone concentration. Jpn. J. Clin. Oncol. 2002, 32, 296–300. [Google Scholar]
- Ingram, D.; Sanders, K.; Kolybaba, M.; Lopez, D. Case-control study of phyto-oestrogens and breast cancer. Lancet 1997, 350, 990–994. [Google Scholar]
- Duncan, A.M.; Merz-Demlow, B.E.; Xu, X.; Phipps, W.R.; Kurzer, M.S. Premenopausal equol excretors show plasma hormone profiles associated with lowered risk of breast cancer. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 581–586. [Google Scholar]
- Marugame, T.; Katanoda, K. International comparisons of cumulative risk of breast and prostate cancer, from Cancer Incidence in Five Continents Vol. VIII. Jpn. J. Clin. Oncol. 2006, 36, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Althuis, M.D.; Dozier, J.M.; Anderson, W.F.; Devesa, S.S.; Brinton, L.A. Global trends in breast cancer incidence and mortality 1973-1997. Intern. J. Epidem. 2005, 34, 405–412. [Google Scholar]
- Nagata, C.; Kawakami, N.; Shimizu, H. Trends in the incidence rate and risk factors for breast cancer in Japan. Breast Cancer Res. Treat. 1997, 44, 75–82. [Google Scholar]
- Shen, Y.C.; Chang, C.J.; Hsu, C.; Cheng, C.C.; Chiu, C.F.; Cheng, A.L. Significant difference in the trends of female breast cancer incidence between Taiwanese and Caucasian Americans: implications from age-period-cohort analysis. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 1986–1990. [Google Scholar]
- Chia, K.S.; Reilly, M.; Tan, C.S.; Lee, J.; Pawitan, Y.; Adami, H.O.; Hall, P.; Mow, B. Profound changes in breast cancer incidence may reflect changes into a Westernized lifestyle: a comparative population-based study in Singapore and Sweden. Int. J. Cancer 2005, 113, 302–306. [Google Scholar]
- Adlercreutz, H.; Honjo, H.; Higashi, A.; Fotsis, T.; Hamalainen, E.; Hasegawa, T.; Okada, H. Urinary excretion of lignans and isoflavonoid phytoestrogens in Japanese men and women consuming a traditional Japanese diet. Am. J. Clin. Nutr. 1991, 54, 1093–1100. [Google Scholar]
- Blakesmith, S.J.; Samman, S.; Lyons-Wall, P.M.; Joannou, G.E.; Petocz, P. Urinary isoflavonoid excretion is inversely associated with the ratio of protein to dietary fibre intake in young women. Eur. J. Clin. Nutr. 2005, 59, 284. [Google Scholar]
- Hall, M.C.; O'Brien, B.; McCormack, T. Equol producer status, salivary estradiol profile and urinary excretion of isoflavones in Irish Caucasian women, following ingestion of soymilk. Steroids 2007, 72, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, T.E.; Maroni, P.D.; Ferucci, P.G.; Dayton, R.; Barnes, S.; Jones, K.; Moore, R.; Ogden, L.G.; Wähälä, K.; Sackett, H.M.; Gray, K.J. Long-term dietary habits affect soy isoflavone metabolism and accumulation in prostatic fluid in Caucasian men. J. Nutr. 2005, 135, 1400–1406. [Google Scholar]
- Lu, L.J.W.; Anderson, K.E. Sex and long-term soy diets affect the metabolism and excretion of soy isoflavones in humans. Am. J. Clin. Nutr. 1998, 68, 1500S–1504S. [Google Scholar]
- Nagata, C.; Iwasa, S.; Shiraki, M.; Ueno, T.; Uchiyama, S.; Urata, K.; Sahashi, Y.; Shimizu, H. Associations among maternal soy intake, isoflavone levels in urine and blood samples, and maternal and umbilical hormone concentrations (Japan). CCC 2006, 17, 1107–1113. [Google Scholar] [PubMed]
- Rafii, F.; Davis, C.; Park, M.; Heinze, T.M.; Beger, R.D. Variations in metabolism of the soy isoflavonoid daidzein by human intestinal microfloras from different individuals. Arch. Microbiol. 2003, 180, 11–16. [Google Scholar]
- Setchell, K.D.R.; Maynard Brown, N.; Zimmer-Nechimias, L.; Wolfe, B.; Creutzinger, V.; Heubi, J.E.; Desai, P.B.; Jakate, A.S. Bioavailability, disposition, and dose-response effects of soy isoflavones when consumed by healthy women at physiologically typical dietary intakes. J. Nutr. 2003, 133, 1027. [Google Scholar] [PubMed]
- Song, K.B.; Atkinson, C.; Frankenfeld, C.L.; Jokela, T.; Wähälä, K.; Thomas, W.K.; Lampe, J.W. Prevalence of daidzein-metabolizing phenotypes differs between Caucasian and Korean American women and girls. J. Nutr. 2006, 136, 1347–1351. [Google Scholar]
- Todaka, E.; Sakurai, K.; Fukata, H.; Miyagawa, H.; Uzuki, M.; Omori, M.; Osada, H.; Ikezuki, Y.; Tsutsumi, O.; Iguchi, T.; Mori, C. Fetal exposure to phytoestrogens - The difference in phytoestrogen status between mother and fetus. Environ. Res. 2005, 99, 195–203. [Google Scholar]
- Hwang, C.S.; Kwak, H.S.; Lim, H.J.; Lee, S.H.; Kang, Y.S.; Choe, T.B.; Hur, H.G.; Han, K.O. Isoflavone metabolites and their in vitro dual functions: They can act as an estrogenic agonist or antagonist depending on the estrogen concentration. J. Steroid Biochem. Mol. Biol. 2006, 101, 246–253. [Google Scholar]
- Cassidy, A.; Hanley, B.; Lamuela-Raventos, R.M. Isoflavones, lignans and stilbenes - Origins, metabolism and potential importance to human health. J. Sci. Food Agric. 2000, 80, 1044. [Google Scholar] [CrossRef]
- Heinonen, S.M.; Hoikkala, A.; Wähälä, K.; Adlercreutz, H. Metabolism of the soy isoflavones daidzein, genistein and glycitein in human subjects. Identification of new metabolites having an intact isoflavonoid skeleton. J. Steroid Biochem. Mol. Biol. 2003, 87, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Joannou, G.E.; Kelly, G.E.; Reeder, A.Y.; Waring, M.; Nelson, C. A urinary profile study of dietary phytoestrogens. The identification and mode of metabolism of new isoflavonoids. J. Steroid Biochem. Mol. Biol. 1995, 54, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Larkin, T.; Price, W.E.; Astheimer, L. The key importance of soy isoflavone bioavailability to understanding health benefits. Crit. Rev. Food Sci. Nutr. 2008, 48, 538–552. [Google Scholar]
- Espin, J.C.; Garcia-Conesa, M.T.; Tomas-Barberan, F.A. Nutraceuticals: facts and fiction. Phytochemi. 2007, 68, 2986–3008. [Google Scholar]
- Nielsen, I.L.; Williamson, G. Review of the factors affecting bioavailability of soy isoflavones in humans. Nutr. Cancer 2007, 57, 1–10. [Google Scholar]
- Cassidy, A. Factors affecting the bioavailability of soy isoflavones in humans. J. AOAC Int. 2006, 89, 1182–1188. [Google Scholar]
- Hendrich, S. Bioavailability of isoflavones. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2002, 777, 203–210. [Google Scholar]
- World Health Organization. Cardiovascular diseases (CVDs). Fact sheet No. 317. Available online: http://www.who.int/mediacentre/factsheets/fs317/en/index.html (accessed: July 8th, 2009).
- Hajer, G.R.; Van Haeften, T.W.; Visseren, F.L.J. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Europ. Heart J. 2008, 29, 2959–2971. [Google Scholar] [CrossRef]
- Zalesin, K.C.; Franklin, B.A.; Miller, W.M.; Peterson, E.D.; McCullough, P.A. Impact of Obesity on Cardiovascular Disease. Endocrinol. Metab. Clin. North Am. 2008, 37, 663–684. [Google Scholar]
- Alberti, G.; Zimmet, P.; Shaw, J.; Grundy, S.M. The IDF Consensus worldwide definition of the metabolic syndrome. 2006. Available online: http://www.idf.org/home/index.cfm?node=1429 (accessed: July 8th,2009).
- Gurnell, M.; Chatterjee, V.K.K.; O'Rahilly, S.; Savage, D.B. The metabolic syndrome: Peroxisome proliferator-activated receptor gamma and its therapeutic modulation. J. Clin. Endocrinol. Metab. 2003, 88, 2412–2421. [Google Scholar]
- Howard, B.V.; Criqui, M.H.; Curb, J.D.; Rodabough, R.; Safford, M.M.; Santoro, N.; Wilson, A.C.; Wylie-Rosett, J. Risk factor clustering in the insulin resistance syndrome and its relationship to cardiovascular disease in postmenopausal White, Black, Hispanic, and Asian/Pacific Islander women. Metab. Clin. Exp. 2003, 52, 362–371. [Google Scholar] [PubMed]
- Lakka, H.-M.; Laaksonen, D.E.; Lakka, T.A.; Niskanen, L.K.; Kumpusalo, E.; Tuomilehto, J.; Salonen, J.T. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. J. Am. Med. Assoc. 2002, 288, 2709–2716. [Google Scholar]
- McNeill, A.M.; Rosamond, W.D.; Girman, C.J.; Golden, S.H.; Schmidt, M.I.; East, H.E.; Ballantyne, C.M.; Heiss, G. The metabolic syndrome and 11-year risk of incident cardiovascular disease in the atherosclerosis risk in communities study. Diabetes Care 2005, 28, 385–390. [Google Scholar]
- Ridker, P.M.; Buring, J.E.; Cook, N.R.; Rifai, N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003, 107, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar]
- Rahmouni, K.; Mark, A.L.; Haynes, W.G.; Sigmund, C.D. Adipose depot-specific modulation of angiotensinogen gene expression in diet-induced obesity. Am. J. Physiol. Endocrinol. Metabol. 2004, 286, E891–E895. [Google Scholar]
- Shimomura, I.; Funahashi, T.; Takahashi, M.; Maeda, K.; Kotani, K.; Nakamura, T.; Yamashita, S.; Miura, M.; Fukuda, Y.; Takemura, K.; Tokunaga, K.; Matsuzawa, Y. Enhanced expression of PAI-1 in visceral fat: Possible contributor to vascular disease in obesity. Nat. Med. 1996, 2, 800–803. [Google Scholar]
- Combs, T.P.; Wagner, J.A.; Berger, J.; Doebber, T.; Wang, W.J.; Zhang, B.B.; Tanen, M.; Berg, A.H.; O'Rahilly, S.; Savage, D.B.; Chatterjee, K.; Weiss, S.; Larson, P.J.; Gottesdiener, K.M.; Gertz, B.J.; Charron, M.J.; Scherer, P.E.; Moller, D.E. Induction of adipocyte complement-related protein of 30 kilodaltons by PPARγ agonists: A potential mechanism of insulin sensitization. Endocrinology 2002, 143, 998–1007. [Google Scholar]
- You, T.; Nicklas, B.J.; Ding, J.; Penninx, B.W.J.H.; Goodpaster, B.H.; Bauer, D.C.; Tylavsky, F.A.; Harris, T.B.; Kritchevsky, S.B. The metabolic syndrome is associated with circulating adipokines in older adults across a wide range of adiposity. J. Gerontol.- Series A Biol. Sci. Med. Sci. 2008, 63, 414–419. [Google Scholar]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; Evans, R.M. The nuclear receptor super-family: The second decade. Cell 1995, 83, 835–839. [Google Scholar]
- Fajas, L.; Auboeuf, D.; Raspe, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; Vidal-Puig, A.; Flier, J.; Briggs, M.R.; Staels, B.; Vidal, H.; Auwerx, J. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000, 49, 497–505. [Google Scholar]
- Chinetti, G.; Griglio, S.; Antonucci, M.; Torra, I.P.; Delerive, P.; Majd, Z.; Fruchart, J.C.; Chapman, J.; Najib, J.; Staels, B. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 1998, 273, 25573–25580. [Google Scholar]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; Lehmann, J.M. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 4318–4323. [Google Scholar]
- Lehmann, J.M.; Lenhard, J.M.; Oliver, B.B.; Ringold, G.M.; Kliewer, S.A. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1997, 272, 3406–3410. [Google Scholar]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 4312–4317. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-deoxy-delta12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARgamma. Cell 1995, 83, 803–812. [Google Scholar]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPARgamma). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding adipocyte differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPARgamma2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; Ezaki, O.; Akanuma, Y.; Gavrilova, O.; Vinson, C.; Reitman, M.L.; Kagechika, H.; Shudo, K.; Yoda, M.; Nakano, Y.; Tobe, K.; Nagai, R.; Kimura, S.; Tomita, M.; Froguel, P.; Kadowaki, T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar]
- Kallen, C.B.; Lazar, M.A. Antidiabetic thiazolidinediones inhibit leptin (ob) gene expression in 3T3-L1 adipocytes. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 5793–5796. [Google Scholar]
- Takata, Y.; Kitami, Y.; Yang, Z.H.; Nakamura, M.; Okura, T.; Hiwada, K. Vascular inflammation is negatively autoregulated by interaction between CCAAT/enhancer-binding protein-δ and peroxisome proliferator-activated receptor-γ. Circ. Res. 2002, 91, 427–433. [Google Scholar]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar]
- Laplante, M.; Festuccia, W.T.; Soucy, G.; Gelinas, Y.; Lalonde, J.; Berger, J.P.; Deshaies, Y. Mechanisms of the depot specificity of peroxisome proliferator-activated receptor gamma action on adipose tissue metabolism. Diabetes 2006, 55, 2771–2778. [Google Scholar]
- Guan, H.P.; Yong, L.; Jensen, M.V.; Newgard, C.B.; Steppan, C.M.; Lazar, M.A. A futile metabolic cycle activated in adipocytes by antidiabetic agents. Nat. Med. 2002, 8, 1122–1128. [Google Scholar]
- Knight, B.L.; Hebbachi, A.; Hauton, D.; Brown, A.M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421. [Google Scholar]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Yoshikawa, T.; Hasty, A.H.; Tamura, Y.; Osuga, J.; Okazaki, H.; Iizuka, Y.; Takahashi, A.; Sone, H.; Gotoda, T.; Ishibashi, S.; Yamada, N. Dual regulation of mouse Delta(5)- and Delta(6)-desaturase gene expression by SREBP-1 and PPARalpha. J. Lipid Res. 2002, 43, 107–114. [Google Scholar]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar]
- Schoonjans, K.; Peinado-Onsurbe, J.; Lefebvre, A.M.; Heyman, R.A.; Briggs, M.; Deeb, S.; Staels, B.; Auwerx, J. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996, 15, 5336–5348. [Google Scholar]
- Staels, B.; Vu-Dac, N.; Kosykh, V.A.; Saladin, R.; Fruchart, J.C.; Dallongeville, J.; Auwerx, J. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J. Clin. Invest. 1995, 95, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Vu-Dac, N.; Schoonjans, K.; Kosykh, V.; Dallongeville, J.; Fruchart, J.C.; Staels, B.; Auwerx, J. Fibrates increase human apolipoprotein A-II expression through activation of the peroxisome proliferator-activated receptor. J. Clin. Invest. 1995, 96, 741–750. [Google Scholar]
- Vu-Dac, N.; Schoonjans, K.; Laine, B.; Fruchart, J.C.; Auwerx, J.; Staels, B. Negative regulation of the human apolipoprotein A-I promoter by fibrates can be attenuated by the interaction of the peroxisome proliferator-activated receptor with its response element. J. Biol. Chem. 1994, 269, 31012–31018. [Google Scholar]
- Ross, R. Atherosclerosis - An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–32. [Google Scholar]
- Stoll, G.; Bendszus, M. Inflammation and atherosclerosis: Novel insights into plaque formation and destabilization. Stroke 2006, 37, 1923–1932. [Google Scholar]
- Laine, P.; Kaartinen, M.; Penttila, A.; Panula, P.; Paavonen, T.; Kovanen, P.T. Association between myocardial infarction and the mast cells in the adventitia of the infarct-related coronary artery. Circulation 1999, 99, 361–369. [Google Scholar]
- Shah, P.K.; Falk, E.; Badimon, J.J.; Fernandez-Ortiz, A.; Mailhac, A.; Villareal-Levy, G.; Fallon, J.T.; Regnstrom, J.; Fuster, V. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation 1995, 92, 1565–1569. [Google Scholar] [PubMed]
- Fuster, V.; Moreno, P.R.; Fayad, Z.A.; Corti, R.; Badimon, J.J. Atherothrombosis and high-risk plaque: Part I: Evolving concepts. J. Am. Coll. Cardiol. 2005, 46, 937–954. [Google Scholar]
- Marx, N.; Duez, H.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors and atherogenesis: Regulators of gene expression in vascular cells. Circ. Res. 2004, 94, 1168–1178. [Google Scholar]
- Zhou, M.; Xu, H.; Pan, L.; Wen, J.; Liao, W.; Chen, K. Rosiglitazone promotes atherosclerotic plaque stability in fat-fed ApoE-knockout mice. Eur. J. Pharmacol. 2008, 590, 297–302. [Google Scholar]
- Marx, N.; Sukhova, G.; Murphy, C.; Libby, P.; Plutzky, J. Macrophages in human atheroma contain PPARgamma: Differentiation-dependent peroxisomal proliferator-activated receptor gamma (PPARgamma) expression and reduction of MMP-9 activity through PPARgamma activation in mononuclear phagocytes in vitro. Am. J. Pathol. 1998, 153, 17–23. [Google Scholar]
- Li, A.C.; Binder, C.J.; Gutierrez, A.; Brown, K.K.; Plotkin, C.R.; Pattison, J.W.; Valledor, A.F.; Davis, R.A.; Willson, T.M.; Witztum, J.L.; Palinski, W.; Glass, C.K. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J. Clin. Invest. 2004, 114, 1564–1576. [Google Scholar] [PubMed]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9774–9779. [Google Scholar]
- Chinetti, G.; Lestavel, S.; Bocher, V.; Remaley, A.T.; Neve, B.; Torra, I.P.; Teissier, E.; Minnich, A.; Jaye, M.; Duverger, N.; Brewer, H.B.; Fruchart, J.C.; Clavey, V.; Staels, B. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001, 7, 53–58. [Google Scholar]
- Souissi, I.J.; Billiet, L.; Cuaz-Perolin, C.; Slimane, M.N.; Rouis, M. Matrix metalloproteinase-12 gene regulation by a PPAR alpha agonist in human monocyte-derived macrophages. Exp. Cell Res. 2008, 314, 3405–3414. [Google Scholar]
- Jeanpierre, E.; Le Tourneau, T.; Zawadzki, C.; Van Belle, E.; Mouquet, F.; Susen, S.; Ezekowitz, M.D.; Staels, B.; Jude, B.; Corseaux, D. Beneficial effects of fenofibrate on plaque thrombogenicity and plaque stability in atherosclerotic rabbits. Cardiovasc. Pathol. 2009, 18, 140–147. [Google Scholar]
- Mueller, M.; Jungbauer, A. Red clover extract – a source for substances that activate PPARalpha and ameliorate cytokine secretion profile of LPS-stimulated macrophages. Menopause 2010. In press.. [Google Scholar]
- Mueller, M.; Jungbauer, A. Red clover extract: A putative source for simultaneous treatment of menopausal disorders and the metabolic syndrome. Menopause 2008, 15, 1120–1131. [Google Scholar]
- Chacko, B.K.; Chandler, R.T.; D'Alessandro, T.L.; Mundhekar, A.; Khoo, N.K.H.; Botting, N.; Barnes, S.; Patel, R.P. Anti-inflammatory effects of isoflavones are dependent on flow and human endothelial cell PPARgamma. J. Nutr. 2007, 137, 351–356. [Google Scholar]
- Dang, Z.C.; Audinot, V.; Papapoulos, S.E.; Boutin, J.A.; Lowik, C.W. Peroxisome proliferator-activated receptor gamma (PPARgamma ) as a molecular target for the soy phytoestrogen genistein. J. Biol. Chem. 2003, 278, 962–967. [Google Scholar]
- Dang, Z.; Löwik, C.W.G.M. The balance between concurrent activation of ERs and PPARs determines daidzein-induced osteogenesis and adipogenesis. J. Bone Miner. Res. 2004, 19, 853–861. [Google Scholar]
- Kim, S.; Shin, H.J.; Kim, S.Y.; Kim, J.H.; Lee, Y.S.; Kim, D.H.; Lee, M.O. Genistein enhances expression of genes involved in fatty acid catabolism through activation of PPARalpha. Mol. Cell. Endocrinol. 2004, 220, 51–58. [Google Scholar]
- Kwon, Y.I.; Vattem, D.A.; Shetty, K. Evaluation of clonal herbs of Lamiaceae species for management of diabetes and hypertension. Asia Pac. J. Clin. Nutr. 2006, 15, 107–118. [Google Scholar]
- Mezei, O.; Banz, W.J.; Steger, R.W.; Peluso, M.R.; Winters, T.A.; Shay, N. Soy isoflavones exert antidiabetic and hypolipidemic effects through the PPAR pathways in obese Zucker rats and murine RAW 264.7 cells. J. Nutr. 2003, 133, 1238–1243. [Google Scholar] [PubMed]
- Ricketts, M.L.; Moore, D.D.; Banz, W.J.; Mezei, O.; Shay, N.F. Molecular mechanisms of action of the soy isoflavones includes activation of promiscuous nuclear receptors. A review. J. Nutr. Biochem. 2005, 16, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.W.; Lee, O.H.; Banz, W.J.; Moustaid-Moussa, N.; Shay, N.F.; Kim, Y.C. Daidzein and the daidzein metabolite, equol, enhance adipocyte differentiation and PPARgamma transcriptional activity. J. Nutr. Biochem. 2009.
- Shen, P.; Liu, M.H.; Ng, T.Y.; Chan, Y.H.; Yong, E.L. Differential effects of isoflavones, from Astragalus Membranaceus and Pueraria Thomsonii, on the activation of PPARalpha, PPARgamma, and adipocyte differentiation in vitro. J. Nutr. 2006, 136, 899–905. [Google Scholar] [PubMed]
- Yeh, W.C.; Cao, Z.; Classon, M.; McKnight, S.L. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995, 9, 168–181. [Google Scholar]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar]
- Lin, F.T.; Lane, M.D. CCAAT/enhancer binding protein alpha is sufficient to initiate the 3T3-L1 adipocyte differentiation program. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 8757–8761. [Google Scholar]
- Park, H.J.; Della-Fera, M.A.; Hausman, D.B.; Rayalam, S.; Ambati, S.; Baile, C.A. Genistein inhibits differentiation of primary human adipocytes. J. Nutr. Biochem. 2009, 20, 140–148. [Google Scholar]
- Phrakonkham, P.; Viengchareun, S.; Belloir, C.; Lombès, M.; Artur, Y.; Canivenc-Lavier, M.C. Dietary xenoestrogens differentially impair 3T3-L1 preadipocyte differentiation and persistently affect leptin synthesis. J. Steroid Biochem. Mol. Biol. 2008, 110, 95–103. [Google Scholar]
- Zhang, M.; Ikeda, K.; Xu, J.W.; Yamori, Y.; Gao, X.M.; Zhang, B.L. Genistein suppresses adipogenesis of 3T3-L1 cells via multiple signal pathways. Phytotherapy Res. 2009, 23, 713–718. [Google Scholar]
- Liao, Q.C.; Li, Y.L.; Qin, Y.F.; Quarles, L.D.; Xu, K.K.; Li, R.; Zhou, H.H.; Xiao, Z.S. Inhibition of adipocyte differentiation by phytoestrogen genistein through a potential downregulation of extracellular signal-regulated kinases 1/2 activity. J. Cell. Biochem. 2008, 104, 1853–1864. [Google Scholar]
- Hwang, J.T.; Park, I.J.; Shin, J.I.; Lee, Y.K.; Lee, S.K.; Baik, H.W.; Ha, J.; Park, O.J. Genistein, EGCG, and capsaicin inhibit adipocyte differentiation process via activating AMP-activated protein kinases. Biochem. Biophys. Res. Commun. 2005, 338, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Nelson-Dooley, C.; Della-Fera, M.A.; Yang, J.Y.; Zhang, W.; Duan, J.; Hartzell, D.L.; Hamrick, M.W.; Baile, C.A. Genistein decreases food intake, body weight, and fat pad weight and causes adipose tissue apoptosis in ovariectomized female mice. J. Nutr. 2006, 136, 409–414. [Google Scholar] [PubMed]
- Hanada, T.; Yoshimura, A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002, 13, 413–421. [Google Scholar]
- Makarov, S.S. NF-kappaB as a therapeutic target in chronic inflammation: recent advances. Mol. Med. Today 2000, 6, 441–448. [Google Scholar]
- Blay, M.; Espinel, A.E.; Delgado, M.A.; Baiges, I.; Bladé, C.; Arola, L.; Salvadó, J. Isoflavone effect on gene expression profile and biomarkers of inflammation. J. Pharm. Biomed. Anal. 2010, 51, 382–390. [Google Scholar]
- Lee, Y.W.; Lee, W.H. Protective effects of genistein on proinflammatory pathways in human brain microvascular endothelial cells. J. Nutr. Biochem. 2008, 19, 819–825. [Google Scholar]
- Hämäläinen, M.; Nieminen, R.; Vuorela, P.; Heinonen, M.; Moilanen, E. Anti-inflammatory effects of flavonoids: Genistein, kaempferol, quercetin, and daidzein inhibit STAT-1 and NF-kB activations, whereas flavone, isorhamnetin, naringenin, and pelargonidin inhibit only NF-?B activation along with their inhibitory effect on iNOS expression and NO production in activated macrophages. Med. Inflamm. 2007, 2007, 1–10. [Google Scholar]
- Hooshmand, S.; Soung, D.Y.; Lucas, E.A.; Madihally, S.V.; Levenson, C.W.; Arjmandi, B.H. Genistein reduces the production of proinflammatory molecules in human chondrocytes. J. Nutr. Biochem. 2007, 18, 609–614. [Google Scholar]
- Lee, K.H.; Choi, E.M. Biochanin A stimulates osteoblastic differentiation and inhibits hydrogen peroxide-induced production of inflammatory mediators in MC3T3-E1 cells. Biol. Pharm. Bull. 2005, 28, 1948–1953. [Google Scholar]
- Richard, N.; Porath, D.; Radspieler, A.; Schwager, J. Effects of resveratrol, piceatannol, triacetoxystilbene, and genistein on the inflammatory response of human peripheral blood leukocytes. Mol. Nutr. Food Res. 2005, 49, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, S.; Ma, G.; Ye, M.; Lu, G. Genistein protects dopaminergic neurons by inhibiting microglial activation. NeuroReport 2005, 16, 267–270. [Google Scholar]
- Chen, H.Q.; Wang, X.J.; Jin, Z.Y.; Xu, X.M.; Zhao, J.W.; Xie, Z.J. Protective effect of isoflavones from Trifolium pratense on dopaminergic neurons. Neurosci. Res. 2008, 62, 123–130. [Google Scholar]
- Chen, H.Q.; Jin, Z.Y.; Li, G.H. Biochanin A protects dopaminergic neurons against lipopolysaccharide-induced damage through inhibition of microglia activation and proinflammatory factors generation. Neurosci. Letters 2007, 417, 112–117. [Google Scholar]
- Morris, P.E.; Olmstead, L.E.; Howard-Carroll, A.E.; Dickens, G.R.; Goltz, M.L.; Courtney-Shapiro, C.; Fanti, P. In vitro and in vivo effects of genistein on murine alveolar macrophage TNFα production. Inflammation 1999, 23, 231–239. [Google Scholar]
- Chan, Y.C.; Wu, C.C.; Chan, K.C.; Lin, Y.G.; Liao, J.W.; Wang, M.F.; Chang, Y.H.; Jeng, K.C. Nanonized black soybean enhances immune response in senescence-accelerated mice. Intern. J. Nanomed. 2009, 4, 27–35. [Google Scholar]
- Dia, V.P.; Berhow, M.A.; De Mejia, E.G. Bowman-birk inhibitor and genistein among soy compounds that synergistically inhibit nitric oxide and prostaglandin E2 pathways in lipopolysaccharide-induced macrophages. J. Agric. Food Chem. 2008, 56, 11707–11717. [Google Scholar]
- Liang, Y.C.; Huang, Y.T.; Tsai, S.H.; Lin-Shiau, S.Y.; Chen, C.F.; Lin, J.K. Suppression of inducible cyclooxygenase and inducible nitric oxide synthase by apigenin and related flavonoids in mouse macrophages. Carcinogenesis 1999, 20, 1945–1952. [Google Scholar]
- Sheu, F.; Lai, H.H.; Yen, G.C. Suppression effect of soy isoflavones on nitric oxide production in RAW 264.7 Macrophages. J. Agric. Food Chem. 2001, 49, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.Y.; Leung, L.K. Soya isoflavones suppress phorbol 12-myristate 13-acetate-induced COX-2 expression in MCF-7 cells. Br. J. Nutr. 2006, 96, 169–176. [Google Scholar]
- Bitto, A.; Altavilla, D.; Bonaiuto, A.; Polito, F.; Minutoli, L.; Di Stefano, V.; Giuliani, D.; Guarini, S.; Arcoraci, V.; Squadrito, F. Effects of aglycone genistein in a rat experimental model of postmenopausal metabolic syndrome. J. Endocrinol. 2009, 200, 367–376. [Google Scholar]
- Lee, Y.M.; Choi, J.S.; Kim, M.H.; Jung, M.H.; Lee, Y.S.; Song, J. Effects of dietary genistein on hepatic lipid metabolism and mitochondrial function in mice fed high-fat diets. Nutrition 2006, 22, 956–964. [Google Scholar]
- Ae Park, S.; Choi, M.S.; Cho, S.Y.; Seo, J.S.; Jung, U.J.; Kim, M.J.; Sung, M.K.; Park, Y.B.; Lee, M.K. Genistein and daidzein modulate hepatic glucose and lipid regulating enzyme activities in C57BL/KsJ-db/db mice. Life Sci. 2006, 79, 1207–1213. [Google Scholar]
- Jayagopal, V.; Albertazzi, P.; Kilpatrick, E.S.; Howarth, E.M.; Jennings, P.E.; Hepburn, D.A.; Atkin, S.L. Beneficial effects of soy phytoestrogen intake in postmenopausal women with type 2 diabetes. Diabetes Care 2002, 25, 1709–1714. [Google Scholar]
- Kojima, T.; Uesugi, T.; Toda, T.; Miura, Y.; Yagasaki, K. Hypolipidemic action of the soybean isoflavones genistein and genistin in glomerulonephritic rats. Lipids 2002, 37, 261–265. [Google Scholar]
- Baluchnejadmojarad, T.; Roghani, M. Chronic administration of genistein improves aortic reactivity of streptozotocin-diabetic rats: Mode of action. Vascular Pharm. 2008, 49, 1–5. [Google Scholar]
- Lee, C.S.; Kwon, S.J.; Na, S.Y.; Lim, S.P.; Lee, J.H. Genistein supplementation inhibits atherosclerosis with stabilization of the lesions in hypercholesterolemic rabbits. J. Korean Med. Sci. 2004, 19, 656–661. [Google Scholar]
- Asgary, S.; Moshtaghian, J.; Naderi, G.; Fatahi, Z.; Hosseini, M.; Dashti, G.; Adibi, S. Effects of dietary red clover on blood factors and cardiovascular fatty streak formation in hypercholesterolemic rabbits. Phytother. Res. 2007, 21, 768–770. [Google Scholar]
- Yamakoshi, J.; Piskula, M.K.; Izumi, T.; Tobe, K.; Saito, M.; Kataoka, S.; Obata, A.; Kikuchi, M. Isoflavone aglycone-rich extract without soy protein attenuates atherosclerosis development in cholesterol-fed rabbits. J. Nutr. 2000, 130, 1887–1893. [Google Scholar]
- Howes, J.B.; Tran, D.; Brillante, D.; Howes, L.G. Effects of dietary supplementation with isoflavones from red clover on ambulatory blood pressure and endothelial function in postmenopausal type 2 diabetes. Diabetes Obes. Metab. 2003, 5, 325–332. [Google Scholar]
- Howes, J.B.; Sullivan, D.; Lai, N.; Nestel, P.; Pomeroy, S.; West, L.; Eden, J.A.; Howes, L.G. The effects of dietary supplementation with isoflavones from red clover on the lipoprotein profiles of post menopausal women with mild to moderate hypercholesterolaemia. Atherosclerosis 2000, 152, 143–147. [Google Scholar]
- Lissin, L.W.; Oka, R.; Lakshmi, S.; Cooke, J.P. Isoflavones improve vascular reactivity in post-menopausal women with hypercholesterolemia. Vasc. Med. 2004, 9, 26–30. [Google Scholar]
- Taku, K.; Umegaki, K.; Sato, Y.; Taki, Y.; Endoh, K.; Watanabe, S. Soy isoflavones lower serum total and LDL cholesterol in humans: a meta-analysis of 11 randomized controlled trials. Am. J. Clin. Nutr. 2007, 85, 1148–1156. [Google Scholar]
- Davis, J.; Higginbotham, A.; O'Connor, T.; Moustaid-Moussa, N.; Tebbe, A.; Kim, Y.C.; Cho, K.W.; Shay, N.; Adler, S.; Peterson, R.; Banz, W. Soy protein and isoflavones influence adiposity and development of metabolic syndrome in the obese male ZDF rat. Ann. Nutr. Metab. 2007, 51, 42–52. [Google Scholar]
- Hidalgo, L.A.; Chedraui, P.A.; Morocho, N.; Ross, S.; San Miguel, G. The effect of red clover isoflavones on menopausal symptoms, lipids and vaginal cytology in menopausal women: A randomized, double-blind, placebo-controlled study. Gynecol. Endocrinol. 2005, 21, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sohn, I.; Lee, Y.S.; Lee, Y.S. Hepatic gene expression profiles are altered by genistein supplementation in mice with diet-induced obesity. J. Nutr. 2005, 135, 33–41. [Google Scholar]
- Kao, T.H.; Wu, W.M.; Hung, C.F.; Wu, W.B.; Chen, B.H. Anti-inflammatory effects of isoflavone powder produced from soybean cake. J. Agric. Food Chem. 2007, 55, 11068–11079. [Google Scholar]
- Fanti, P.; Asmis, R.; Stephenson, T.J.; Sawaya, B.P.; Franke, A.A. Positive effect of dietary soy in ESRD patients with systemic inflammation--correlation between blood levels of the soy isoflavones and the acute-phase reactants. Nephrol. Dial. Transplant. 2006, 21, 2239–2246. [Google Scholar]
- Seibel, J.; Molzberger, A.F.; Hertrampf, T.; Laudenbach-Leschowski, U.; Diel, P. Oral treatment with genistein reduces the expression of molecular and biochemical markers of inflammation in a rat model of chronic TNBS-induced colitis. Eur. J. Nutr. 2009, 48, 213–220. [Google Scholar]
- Wang, J.; Zhang, Q.; Jin, S.; He, D.; Zhao, S.; Liu, S. Genistein modulate immune responses in collagen-induced rheumatoid arthritis model. Maturitas 2008, 59, 405–412. [Google Scholar]
- Verdrengh, M.; Jonsson, I.M.; Holmdahl, R.; Tarkowski, A. Genistein as an anti-inflammatory agent. Inflamm.n Res. 2003, 52, 341–346. [Google Scholar]
- Kalhan, R.; Smith, L.J.; Nlend, M.C.; Nair, A.; Hixon, J.L.; Sporn, P.H.S. A mechanism of benefit of soy genistein in asthma: Inhibition of eosinophil p38-dependent leukotriene synthesis. Clin. Exp. Allergy 2008, 38, 103–112. [Google Scholar]
- Bloedon, L.T.; Jeffcoat, A.R.; Lopaczynski, W.; Schell, M.J.; Black, T.M.; Dix, K.J.; Thomas, B.F.; Albright, C.; Busby, M.G.; Crowell, J.A.; Zeisel, S.H. Safety and pharmacokinetics of purified soy isoflavones: single-dose administration to postmenopausal women. Am. J. Clin. Nutr. 2002, 76, 1126–1137. [Google Scholar]
- Adlercreutz, H.; Markkanen, H.; Watanabe, S. Plasma concentrations of phyto-oestrogens in Japanese men. Lancet 1993, 342, 1209–1210. [Google Scholar]
- Shimba, S.; Wada, T.; Tezuka, M. Arylhydrocarbon receptor (AhR) is involved in negative regulation of adipose differentiation in 3T3-L1 cells: AhR inhibits adipose differentiation independently of dioxin. J. Cell Sci. 2001, 114, 2809–2817. [Google Scholar]
- Morrow, D.; Qin, C.; Smith, R., 3rd; Safe, S. Aryl hydrocarbon receptor-mediated inhibition of LNCaP prostate cancer cell growth and hormone-induced transactivation. J. Steroid Biochem. Mol. Biol. 2004, 88, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Wormke, M. Inhibitory aryl hydrocarbon receptor-estrogen receptor α cross-talk and mechanisms of action. Chem. Res. Toxicol. 2003, 16, 807–816. [Google Scholar]
- Hahn, M.E.; Karchner, S.I.; Shapiro, M.A.; Perera, S.A. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 13743. [Google Scholar]
- Miller Iii, C.A. Expression of the human aryl hydrocarbon receptor complex in yeast. Activation of transcription by indole compounds. J. Biol. Chem. 1997, 272, 32824. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.D.; Winters, G.M.; Rogers, W.J.; Ziccardi, M.H.; Li, V.; Keser, B.; Denison, M.S. Activation of the Ah receptor signaling pathway by prostaglandins. J. Biochem. Mol. Toxicol. 2001, 15, 187–196. [Google Scholar]
- Schaldach, C.M.; Riby, J.; Bjeldanes, L.F. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry 1999, 38, 7594–7600. [Google Scholar]
- Phelan, D.; Winter, G.M.; Rogers, W.J.; Lam, J.C.; Denison, M.S. Activation of the Ah receptor signal transduction pathway by bilirubin and biliverdin. Arch. Biochem. Biophys. 1998, 357, 155. [Google Scholar]
- Sugihara, K.; Kitamura, S.; Yamada, T.; Okayama, T.; Ohta, S.; Yamashita, K.; Yasuda, M.; Fujii-Kuriyama, Y.; Saeki, K.; Matsui, S.; Matsuda, T. Aryl hydrocarbon receptor-mediated induction of microsomal drug-metabolizing enzyme activity by indirubin and indigo. Biochem. Biophys. Res. Commun. 2004, 318, 571–578. [Google Scholar]
- Adachi, J.; Mori, Y.; Matsui, S.; Takigami, H.; Fujino, J.; Kitagawa, H.; Matsuda, T.; Miller Iii, C.A.; Kato, T.; Saeki, K. Indirubin and Indigo are Potent Aryl Hydrocarbon Receptor Ligands Present in Human Urine. J. Biol. Chem. 2001, 276, 31475. [Google Scholar]
- Oesch-Bartlomowicz, B.; Huelster, A.; Wiss, O.; Antoniou-Lipfert, P.; Dietrich, C.; Arand, M.; Weiss, C.; Bockamp, E.; Oesch, F. Aryl hydrocarbon receptor activation by cAMP vs. dioxin: Divergent signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 9218. [Google Scholar] [PubMed]
- Jinno, A.; Maruyama, Y.; Ishizuka, M.; Kazusaka, A.; Nakamura, A.; Fujita, S. Induction of cytochrome P450-1A by the equine estrogen equilenin, a new endogenous aryl hydrocarbon receptor ligand. J. Steroid Biochem. Mol. Biol. 2006, 98, 48–55. [Google Scholar]
- Wei, Y.D.; Bergander, L.; Rannug, U.; Rannug, A. Regulation of CYP1A1 transcription via the metabolism of the tryptophan-derived 6-formylindolo[3,2-b]carbazole. Arch. Biochem. Biophys. 2000, 383, 99–107. [Google Scholar]
- Pohjanvirta, R.; Korkalainen, M.; McGuire, J.; Simanainen, U.; Juvonen, R.; Tuomisto, J.T.; Unkila, M.; Viluksela, M.; Bergman, J.; Poellinger, L.; Tuomisto, J. Comparison of acute toxicities of indolo[3,2-b]carbazole (ICZ) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in TCDD-sensitive rats. Food. Chem. Toxicol. 2002, 40, 1023. [Google Scholar]
- Medjakovic, S.; Jungbauer, A. Red clover isoflavones biochanin A and formononetin are potent ligands of the human aryl hydrocarbon receptor. J. Steroid Biochem. Mol. Biol. 2008, 108, 171–177. [Google Scholar]
- Zhang, S.; Qin, C.; Safe, S.H. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: Effects of structure and cell context. Environ. Health Perspect. 2003, 111, 1877. [Google Scholar]
- Ashida, H. Suppressive effects of flavonoids on dioxin toxicity. BioFactors 2000, 12, 201. [Google Scholar]
- Miller Iii, C.A. A human aryl hydrocarbon receptor signaling pathway constructed in yeast displays additive responses to ligand mixtures. Toxicol. Appl. Pharmacol. 1999, 160, 297. [Google Scholar]
- Chen, G.; Bunce, N.J. Polybrominated diphenyl ethers as Ah receptor agonists and antagonists. Toxicol. Sci. 2003, 76, 310–320. [Google Scholar]
- Chen, G.; Bunce, N.J. Interaction between halogenated aromatic compounds in the Ah receptor signal transduction pathway. Environ. Toxicol. 2004, 19, 480–489. [Google Scholar]
- Poland, A.; Glover, E.; Kende, A.S. Stereospecific, high affinity binding of 2,3,7,8 tetrachlorodibenzo p dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar] [PubMed]
- Mukai, M.; Tischkau, S.A. Effects of tryptophan photoproducts in the circadian timing system: searching for a physiological role for aryl hydrocarbon receptor. Toxicol. Sci. 2007, 95, 172–181. [Google Scholar]
- Heath-Pagliuso, S.; Rogers, W.J.; Tullis, K.; Seidel, S.D.; Denison, M.S.; Cenijn, P.H.; Brouwer, A. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry 1998, 37, 11508. [Google Scholar]
- McMillan, B.J.; Bradfield, C.A. The aryl hydrocarbon receptor is activated by modified low-density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1412–1417. [Google Scholar]
- Guengerich, F.P.; Martin, M.V.; McCormick, W.A.; Nguyen, L.P.; Glover, E.; Bradfield, C.A. Aryl hydrocarbon receptor response to indigoids in vitro and in vivo. Arch. Biochem. Biophys. 2004, 423, 309–316. [Google Scholar]
- Henry, E.C.; Bemis, J.C.; Henry, O.; Kende, A.S.; Gasiewicz, T.A. A potential endogenous ligand for the aryl hydrocarbon receptor has potent agonist activity in vitro and in vivo. Arch. Biochem. Biophys. 2006, 450, 67–77. [Google Scholar]
- Song, J.; Clagett-Dame, M.; Peterson, R.E.; Hahn, M.E.; Westler, W.M.; Sicinski, R.R.; DeLuca, H.F. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 14694. [Google Scholar]
- Savouret, J.F.; Antenos, M.; Quesne, M.; Xu, J.; Milgrom, E.; Casper, R.F. 7-ketocholesterol is an endogenous modulator for the arylhydrocarbon receptor. J. Biol. Chem. 2001, 276, 3054–3059. [Google Scholar]
- Yang, Y.M.; Huang, D.Y.; Liu, G.F.; Zhong, J.C.; Du, K.; Li, Y.F.; Song, X.H. Inhibitory effects of vitamin A on TCDD-induced cytochrome P-450 1A1 enzyme activity and expression. Toxicol. Sci. 2005, 85, 727–734. [Google Scholar]
- Nebert, D.W.; Karp, C.L. Endogenous functions of the aryl hydrocarbon receptor (AHR): intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J. Biol. Chem. 2008, 283, 36061–36065. [Google Scholar]
- Schlecht, C.; Klammer, H.; Jarry, H.; Wuttke, W. Effects of estradiol, benzophenone-2 and benzophenone-3 on the expression pattern of the estrogen receptors (ER) alpha and beta, the estrogen receptor-related receptor 1 (ERR1) and the aryl hydrocarbon receptor (AhR) in adult ovariectomized rats. Toxicology 2004, 205, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.J.; Van Overmeire, I.; Goeyens, L.; Denison, M.S.; De Vito, M.J.; Clark, G.C. Analysis of Ah receptor pathway activation by brominated flame retardants. Chemosphere 2004, 55, 1509–1518. [Google Scholar]
- Saeki, K.I.; Kato, T.A.; Yamada, K.; Mizutani, T.; Miyata, N.; Matsuda, T.; Matsui, S.; Fukuhara, K. Activation of the human Ah receptor by aza-polycyclic aromatic hydrocarbons and their halogenated derivatives. Biol. Pharm. Bull. 2003, 26, 448. [Google Scholar]
- Abnet, C.C.; Tanguay, R.L.; Heideman, W.; Peterson, R.E. Transactivation activity of human, zebrafish, and rainbow trout aryl hydrocarbon receptors expressed in COS-7 cells: greater insight into species differences in toxic potency of polychlorinated dibenzo-p-dioxin, dibenzofuran, and biphenyl congeners. Toxicol. Appl. Pharmacol. 1999, 159, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Till, M.; Riebniger, D.; Schmitz, H.J.; Schrenk, D. Potency of various polycyclic aromatic hydrocarbons as inducers of CYP1A1 in rat hepatocyte cultures. Chem. Biol. Interact. 1999, 117, 135–150. [Google Scholar]
- Safe, S.; Wang, F.; Porter, W.; Duan, R.; McDougal, A. Ah receptor agonists as endocrine disruptors: Antiestrogenic activity and mechanisms. Toxicol. Lett. 1998, 102-103, 343. [Google Scholar] [CrossRef] [PubMed]
- Kharat, I.; Saatcioglu, F. Antiestrogenic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin are mediated by direct transcriptional interference with the liganded estrogen receptor: Cross-talk between aryl hydrocarbon- and estrogen-mediated signaling. J. Biol. Chem. 1996, 271, 10533–10537. [Google Scholar]
- Navas, J.M.; Segner, H. Antiestrogenicity of beta-naphthoflavone and PAHs in cultured rainbow trout hepatocytes: evidence for a role of the arylhydrocarbon receptor. Aquat. Toxicol. 2000, 51, 79–92. [Google Scholar]
- Takahashi, O.; Oishi, S.; Yoneyama, M.; Ogata, A.; Kamimura, H. Antiestrogenic effect of paradichlorobenzene in immature mice and rats. Arch. Toxicol. 2007, 81, 505–517. [Google Scholar]
- Swedenborg, E.; Ruegg, J.; Makela, S.; Pongratz, I. Endocrine disruptive chemicals: mechanisms of action and involvement in metabolic disorders. J. Mol. Endocrinol. 2009, 43, 1–10. [Google Scholar]
- McDougal, A.; Gupta, M.S.; Morrow, D.; Ramamoorthy, K.; Lee, J.E.; Safe, S.H. Methyl-substituted diindolylmethanes as inhibitors of estrogen-induced growth of T47D cells and mammary tumors in rats. Breast Cancer Res. Treat. 2001, 66, 147–157. [Google Scholar]
- Safe, S.; Qin, C.; McDougal, A. Development of selective aryl hydrocarbon receptor modulators for treatment of breast cancer. Expert Opin. Investig. Drugs 1999, 8, 1385–1396. [Google Scholar]
- Amakura, Y.; Tsutsumi, T.; Sasaki, K.; Maitani, T.; Nakamura, M.; Kitagawa, H.; Fujino, J.; Toyoda, M.; Yoshida, T. Activation of the aryl hydrocarbon receptor by some vegetable constituents determined using in vitro reporter gene assay. Biol. Pharm. Bull. 2003, 26, 532. [Google Scholar]
- Amakura, Y.; Tsutsumi, T.; Sasaki, K.; Maitani, T.; Yoshida, T. Screening of the inhibitory effect of vegetable constituents on the aryl hydrocarbon receptor-mediated activity induced by 2,3,7,8-tetrachlorodibenzo-p- dioxin. Biol. Pharm. Bull. 2003, 26, 1754. [Google Scholar]
- Amakura, Y.; Tsutsumi, T.; Sasaki, K.; Nakamura, M.; Yoshida, T.; Maitani, T. Influence of food polyphenols on aryl hydrocarbon receptor-signaling pathway estimated by in vitro bioassay. Phytochemistry 2008, 69, 3117–3130. [Google Scholar]
- Ashida, H.; Fukuda, I.; Yamashita, T.; Kanazawa, K. Flavones and flavonols at dietary levels inhibit a transformation of aryl hydrocarbon receptor induced by dioxin. FEBS Lett. 2000, 476, 213–217. [Google Scholar]
- Fukuda, I.; Sakane, I.; Yabushita, Y.; Sawamura, S.; Kanazawa, K.; Ashida, H. Black tea extract suppresses transformation of aryl hydrocarbon receptor induced by dioxin. BioFactors 2004, 21, 367. [Google Scholar]
- Fukuda, I.; Yabushita, Y.; Kodoi, R.; Nishiumi, S.; Kanazawa, K.; Ashida, H.; Sakane, I.; Kakuda, T.; Sawamura, S.I. Pigments in Green Tea Leaves (Camellia sinensis) Suppress Transformation of the Aryl Hydrocarbon Receptor Induced by Dioxin. J. Agric. Food Chem. 2004, 52, 2499. [Google Scholar]
- Nishiumi, S.; Hosokawa, K.; Mukai, R.; Fukuda, I.; Hishida, A.; Iida, O.; Yoshida, K.; Ashida, H. Screening of indigenous plants from Japan for modulating effects on transformation of the aryl hydrocarbon receptor. Asian Pac. J. Cancer Prev. 2006, 7, 208–220. [Google Scholar]
- Fukuda, I.; Mukai, R.; Kawase, M.; Yoshida, K.i.; Ashida, H. Interaction between the aryl hydrocarbon receptor and its antagonists, flavonoids. Biochem. Biophys. Res. Commun. 2007, 359, 822–827. [Google Scholar]
- Hamada, M.; Satsu, H.; Natsume, Y.; Nishiumi, S.; Fukuda, I.; Ashida, H.; Shimizu, M. TCDD-induced CYP1A1 expression, an index of dioxin toxicity, is suppressed by flavonoids permeating the human intestinal Caco-2 cell monolayers. J. Agric. Food Chem. 2006, 54, 8891–8898. [Google Scholar] [CrossRef] [PubMed]
- Shertzer, H.G.; Puga, A.; Chang, C.; Smith, P.; Nebert, D.W.; Setchell, K.D.; Dalton, T.P. Inhibition of CYP1A1 enzyme activity in mouse hepatoma cell culture by soybean isoflavones. Chem. Biol. Interact. 1999, 123, 31–49. [Google Scholar]
- Han, E.H.; Ji, Y.K.; Hye, G.J. Effect of biochanin A on the aryl hydrocarbon receptor and cytochrome P450 1A1 in MCF-7 human breast carcinoma cells. Arch. Pharm. Res. 2006, 29, 570–576. [Google Scholar]
- Chan, H.Y.; Wang, H.; Leung, L.K. The red clover (Trifolium pratense) isoflavone biochanin A modulates the biotransformation pathways of 7,12-dimethylbenz[a]anthracene. Br. J. Nutr. 2003, 90, 87–92. [Google Scholar]
- Henry, E.C.; Rucci, G.; Gasiewicz, T.A. Characterization of multiple forms of the Ah receptor: comparison of species and tissues. Biochemistry 1989, 28, 6430–6440. [Google Scholar]
- Bergander, L.; Wincent, E.; Rannug, A.; Foroozesh, M.; Alworth, W.; Rannug, U. Metabolic fate of the Ah receptor ligand 6-formylindolo[3,2-b]carbazole. Chem. Biol. Interact. 2004, 149, 151–164. [Google Scholar]
- Nebert, D.W.; Petersen, D.D.; Fornace, A.J., Jr. Cellular responses to oxidative stress: the [Ah] gene battery as a paradigm. Environ. Health Perspect. 1990, 88, 13–25. [Google Scholar]
- Rivera, S.P.; Wang, F.; Saarikoski, S.T.; Taylor, R.T.; Chapman, B.; Zhang, R.; Hankinson, O. A novel promoter element containing multiple overlapping xenobiotic and hypoxia response elements mediates induction of cytochrome P4502S1 by both dioxin and hypoxia. J. Biol. Chem. 2007, 282, 10881–10893. [Google Scholar]
- Dalton, T.P.; Dieter, M.Z.; Matlib, R.S.; Childs, N.L.; Shertzer, H.G.; Genter, M.B.; Nebert, D.W. Targeted knockout of Cyp1a1 gene does not alter hepatic constitutive expression of other genes in the mouse [Ah] battery. Biochem. Biophys. Res. Commun. 2000, 267, 184–189. [Google Scholar]
- Uno, S.; Dalton, T.P.; Derkenne, S.; Curran, C.P.; Miller, M.L.; Shertzer, H.G.; Nebert, D.W. Oral exposure to benzo[a]pyrene in the mouse: detoxication by inducible cytochrome P450 is more important than metabolic activation. Mol. Pharmacol. 2004, 65, 1225–1237. [Google Scholar]
- Nowack, R. Review article: cytochrome P450 enzyme, and transport protein mediated herb-drug interactions in renal transplant patients: grapefruit juice, St John's Wort - and beyond! Nephrology (Carlton) 2008, 13, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Backlund, M.; Johansson, I.; Mkrtchian, S.; Ingelman-Sundberg, M. Signal transduction-mediated activation of the aryl hydrocarbon receptor in rat hepatoma H4IIE cells. J. Biol. Chem. 1997, 272, 31755–31763. [Google Scholar]
- Chan, H.Y.; Leung, L.K. A potential protective mechanism of soya isoflavones against 7,12-dimethylbenz[a]anthracene tumour initiation. Br. J. Nutr. 2003, 90, 457–465. [Google Scholar]
- Kasai, A.; Hiramatsu, N.; Hayakawa, K.; Yao, J.; Kitamura, M. Blockade of the dioxin pathway by herbal medicine Formula Bupleuri Minor: identification of active entities for suppression of AhR activation. Biol. Pharm. Bull. 2008, 31, 838–846. [Google Scholar]
- Kikuchi, H.; Hossain, A. Signal transduction-mediated CYP1A1 induction by omeprazole in human HepG2 cells. Exp. Toxicol. Pathol. 1999, 51, 342–346. [Google Scholar]
- Kumar, A.; Upadhyay, G.; Modi, D.R.; Singh, M.P. The involvement of secondary signaling molecules in cytochrome P-450 1A1-mediated inducible nitric oxide synthase expression in benzo(a)pyrene-treated rat polymorphonuclear leukocytes. Life Sci. 2007, 81, 1575–1584. [Google Scholar]
- Lemaire, G.; Delescluse, C.; Pralavorio, M.; Ledirac, N.; Lesca, P.; Rahmani, R. The role of protein tyrosine kinases in CYP1A1 induction by omeprazole and thiabendazole in rat hepatocytes. Life Sci. 2004, 74, 2265–2278. [Google Scholar]
- Helsby, N.A.; Williams, J.; Kerr, D.; Gescher, A.; Chipman, J.K. The isoflavones equol and genistein do not induce xenobiotic-metabolizing enzymes in mouse and in human cells. Xenobiotica 1997, 27, 587–596. [Google Scholar]
- Kishida, T.; Nagamoto, M.; Ohtsu, Y.; Watakabe, M.; Ohshima, D.; Nashiki, K.; Mizushige, T.; Izumi, T.; Obata, A.; Ebihara, K. Lack of an inducible effect of dietary soy isoflavones on the mRNA abundance of hepatic cytochrome P-450 isozymes in rats. Biosci. Biotechnol. Biochem. 2004, 68, 508–515. [Google Scholar]
- Helsby, N.A.; Chipman, J.K.; Gescher, A.; Kerr, D. Inhibition of mouse and human CYP 1A- and 2E1-dependent substrate metabolism by the isoflavonoids genistein and equol. Food Chem. Toxicol. 1998, 36, 375–382. [Google Scholar]
- Shon, Y.H.; Park, S.D.; Nam, K.S. Effective chemopreventive activity of genistein against human breast cancer cells. J. Biochem. Mol. Biol. 2006, 39, 448–451. [Google Scholar]
- Choi, E.J.; Kim, T. Daidzein modulates induction of hepatic CYP1A1, 1B1, and AhR by 7,12-dimethylbenz[a]anthracene in mice. Arch. Pharm. Res. 2008, 31, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Durant, P.; Leone-Kabler, S.; Wood, C.E.; Register, T.C.; Townsend, A.; Cline, J.M. Effects of prior oral contraceptive use and soy isoflavonoids on estrogen-metabolizing cytochrome P450 enzymes. J. Steroid. Biochem. Mol. Biol. 2008, 112, 179–185. [Google Scholar]
- Spink, D.C.; Spink, B.C.; Cao, J.Q.; DePasquale, J.A.; Pentecost, B.T.; Fasco, M.J.; Li, Y.; Sutter, T.R. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis 1998, 19, 291–298. [Google Scholar]
- Pang, P.H.; Lin, Y.H.; Lee, Y.H.; Hou, H.H.; Hsu, S.P.; Juan, S.H. Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. J. Cell Physiol. 2008, 215, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zhang, L.; Hoagland, M.S.; Swanson, H.I. Lack of the aryl hydrocarbon receptor leads to impaired activation of AKT/protein kinase B and enhanced sensitivity to apoptosis induced via the intrinsic pathway. J. Pharmacol. Exp. Ther. 2007, 320, 448–457. [Google Scholar]
- Chung, J.Y.; Kim, J.Y.; Kim, W.R.; Lee, S.G.; Kim, Y.J.; Park, J.E.; Hong, Y.P.; Chun, Y.J.; Park, Y.C.; Oh, S.; Yoo, K.S.; Yoo, Y.H.; Kim, J.M. Abundance of aryl hydrocarbon receptor potentiates benzo[a]pyrene-induced apoptosis in Hepa1c1c7 cells via CYP1A1 activation. Toxicology 2007, 235, 62–72. [Google Scholar]
- Mathieu, M.C.; Lapierre, I.; Brault, K.; Raymond, M. Aromatic hydrocarbon receptor (AhR).AhR nuclear translocator- and p53-mediated induction of the murine multidrug resistance mdr1 gene by 3-methylcholanthrene and benzo(a)pyrene in hepatoma cells. J. Biol. Chem. 2001, 276, 4819–4827. [Google Scholar] [PubMed]
- Schreck, I.; Chudziak, D.; Schneider, S.; Seidel, A.; Platt, K.L.; Oesch, F.; Weiss, C. Influence of aryl hydrocarbon- (Ah) receptor and genotoxins on DNA repair gene expression and cell survival of mouse hepatoma cells. Toxicology 2009, 259, 91–96. [Google Scholar]
- Pru, J.K.; Kaneko-Tarui, T.; Jurisicova, A.; Kashiwagi, A.; Selesniemi, K.; Tilly, J.L. Induction of proapoptotic gene expression and recruitment of p53 herald ovarian follicle loss caused by polycyclic aromatic hydrocarbons. Reprod. Sci. 2009, 16, 347–356. [Google Scholar]
- Patel, R.D.; Murray, I.A.; Flaveny, C.A.; Kusnadi, A.; Perdew, G.H. Ah receptor represses acute-phase response gene expression without binding to its cognate response element. Lab. Invest. 2009, 89, 695–707. [Google Scholar]
- Teng, J.; Wang, Z.Y.; Jarrard, D.F.; Bjorling, D.E. Roles of estrogen receptor alpha and beta in modulating urothelial cell proliferation. Endocr. Relat. Cancer 2008, 15, 351–364. [Google Scholar]
- Wang, C.; Fu, M.; D'Amico, M.; Albanese, C.; Zhou, J.N.; Brownlee, M.; Lisanti, M.P.; Chatterjee, V.K.; Lazar, M.A.; Pestell, R.G. Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Mol. Cell Biol. 2001, 21, 3057–3070. [Google Scholar]
- Chen, A.C.; Donovan, S.M. Genistein at a concentration present in soy infant formula inhibits Caco-2BBe cell proliferation by causing G2/M cell cycle arrest. J Nutr 2004, 134, 1303–1308. [Google Scholar]
- Wang, B.F.; Wang, J.S.; Lu, J.F.; Kao, T.H.; Chen, B.H. Antiproliferation effect and mechanism of prostate cancer cell lines as affected by isoflavones from soybean cake. J. Agric. Food Chem. 2009, 57, 2221–2232. [Google Scholar]
- Su, S.J.; Yeh, T.M.; Lei, H.Y.; Chow, N.H. The potential of soybean foods as a chemoprevention approach for human urinary tract cancer. Clin. Cancer Res. 2000, 6, 230–236. [Google Scholar]
- Sakamoto, K. Synergistic effects of thearubigin and genistein on human prostate tumor cell (PC-3) growth via cell cycle arrest. Cancer Lett. 2000, 151, 103–109. [Google Scholar]
- Tophkhane, C.; Yang, S.; Bales, W.; Archer, L.; Osunkoya, A.; Thor, A.D.; Yang, X. Bcl-2 overexpression sensitizes MCF-7 cells to genistein by multiple mechanisms. Int. J. Oncol. 2007, 31, 867–874. [Google Scholar]
- Zhou, J.R.; Mukherjee, P.; Gugger, E.T.; Tanaka, T.; Blackburn, G.L.; Clinton, S.K. Inhibition of murine bladder tumorigenesis by soy isoflavones via alterations in the cell cycle, apoptosis, and angiogenesis. Cancer Res. 1998, 58, 5231–5238. [Google Scholar]
- Majid, S.; Kikuno, N.; Nelles, J.; Noonan, E.; Tanaka, Y.; Kawamoto, K.; Hirata, H.; Li, L.C.; Zhao, H.; Okino, S.T.; Place, R.F.; Pookot, D.; Dahiya, R. Genistein induces the p21WAF1/CIP1 and p16INK4a tumor suppressor genes in prostate cancer cells by epigenetic mechanisms involving active chromatin modification. Cancer Res. 2008, 68, 2736–2744. [Google Scholar]
- Chang, K.L.; Kung, M.L.; Chow, N.H.; Su, S.J. Genistein arrests hepatoma cells at G2/M phase: involvement of ATM activation and upregulation of p21waf1/cip1 and Wee1. Biochem. Pharmacol. 2004, 67, 717–726. [Google Scholar]
- Sanchez, Y.; Amran, D.; de Blas, E.; Aller, P. Regulation of genistein-induced differentiation in human acute myeloid leukaemia cells (HL60, NB4) Protein kinase modulation and reactive oxygen species generation. Biochem. Pharmacol. 2009, 77, 384–396. [Google Scholar]
- Raffoul, J.J.; Wang, Y.; Kucuk, O.; Forman, J.D.; Sarkar, F.H.; Hillman, G.G. Genistein inhibits radiation-induced activation of NF-kappaB in prostate cancer cells promoting apoptosis and G2/M cell cycle arrest. BMC Cancer 2006, 6, 107. [Google Scholar]
- Shen, J.C.; Klein, R.D.; Wei, Q.; Guan, Y.; Contois, J.H.; Wang, T.T.; Chang, S.; Hursting, S.D. Low-dose genistein induces cyclin-dependent kinase inhibitors and G(1) cell-cycle arrest in human prostate cancer cells. Mol. Carcinog. 2000, 29, 92–102. [Google Scholar]
- Frey, R.S.; Li, J.; Singletary, K.W. Effects of genistein on cell proliferation and cell cycle arrest in nonneoplastic human mammary epithelial cells: involvement of Cdc2, p21(waf/cip1), p27(kip1), and Cdc25C expression. Biochem. Pharmacol. 2001, 61, 979–989. [Google Scholar] [PubMed]
- Oki, T.; Sowa, Y.; Hirose, T.; Takagaki, N.; Horinaka, M.; Nakanishi, R.; Yasuda, C.; Yoshida, T.; Kanazawa, M.; Satomi, Y.; Nishino, H.; Miki, T.; Sakai, T. Genistein induces Gadd45 gene and G2/M cell cycle arrest in the DU145 human prostate cancer cell line. FEBS Lett. 2004, 577, 55–59. [Google Scholar]
- Mansour, A.; McCarthy, B.; Schwander, S.K.; Chang, V.; Kotenko, S.; Donepudi, S.; Lee, J.; Raveche, E. Genistein induces G2 arrest in malignant B cells by decreasing IL-10 secretion. Cell Cycle 2004, 3, 1597–1605. [Google Scholar]
- Cappelletti, V.; Fioravanti, L.; Miodini, P.; Di Fronzo, G. Genistein blocks breast cancer cells in the G(2)M phase of the cell cycle. J. Cell Biochem. 2000, 79, 594–600. [Google Scholar]
- Casagrande, F.; Darbon, J.M. p21CIP1 is dispensable for the G2 arrest caused by genistein in human melanoma cells. Exp. Cell Res. 2000, 258, 101–108. [Google Scholar]
- Casagrande, F.; Darbon, J.M. Effects of structurally related flavonoids on cell cycle progression of human melanoma cells: regulation of cyclin-dependent kinases CDK2 and CDK1. Biochem. Pharmacol. 2001, 61, 1205–1215. [Google Scholar]
- Darbon, J.M.; Penary, M.; Escalas, N.; Casagrande, F.; Goubin-Gramatica, F.; Baudouin, C.; Ducommun, B. Distinct Chk2 activation pathways are triggered by genistein and DNA-damaging agents in human melanoma cells. J. Biol. Chem. 2000, 275, 15363–15369. [Google Scholar]
- Rauth, S.; Kichina, J.; Green, A. Inhibition of growth and induction of differentiation of metastatic melanoma cells in vitro by genistein: chemosensitivity is regulated by cellular p53. Br. J. Cancer 1997, 75, 1559–1566. [Google Scholar]
- Matsukawa, Y.; Marui, N.; Sakai, T.; Satomi, Y.; Yoshida, M.; Matsumoto, K.; Nishino, H.; Aoike, A. Genistein arrests cell cycle progression at G2-M. Cancer Res. 1993, 53, 1328–1331. [Google Scholar]
- Wang, H.Z.; Zhang, Y.; Xie, L.P.; Yu, X.Y.; Zhang, R.Q. Effects of genistein and daidzein on the cell growth, cell cycle, and differentiation of human and murine melanoma cells(1). J. Nutr. Biochem. 2002, 13, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.M.; Xiao, B.X.; Liu, D.H.; Grant, M.; Zhang, S.; Lai, Y.F.; Guo, Y.B.; Liu, Q. Biphasic effect of daidzein on cell growth of human colon cancer cells. Food Chem. Toxicol. 2004, 42, 1641–1646. [Google Scholar]
- Tijet, N.; Boutros, P.C.; Moffat, I.D.; Okey, A.B.; Tuomisto, J.; Pohjanvirta, R. Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries. Mol. Pharmacol. 2006, 69, 140–153. [Google Scholar]
- Yoon, C.Y.; Park, M.; Kim, B.H.; Park, J.Y.; Park, M.S.; Jeong, Y.K.; Kwon, H.; Jung, H.K.; Kang, H.; Lee, Y.S.; Lee, B.J. Gene expression profile by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the liver of wild-type (AhR+/+) and aryl hydrocarbon receptor-deficient (AhR-/-) mice. J. Vet. Med. Sci. 2006, 68, 663–668. [Google Scholar]
- Bock, K.W.; Köhle, C. Ah receptor: Dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochem. Pharmacol. 2006. [Google Scholar]
- Miniero, R.; De Felip, E.; Ferri, F.; di Domenico, A. An overview of TCDD half-life in mammals and its correlation to body weight. Chemosphere 2001, 43, 839–844. [Google Scholar]
- Kerger, B.D.; Leung, H.W.; Scott, P.; Paustenbach, D.J.; Needham, L.L.; Patterson, D.G., Jr.; Gerthoux, P.M.; Mocarelli, P. Age- and concentration-dependent elimination half-life of 2,3,7,8-tetrachlorodibenzo-p-dioxin in Seveso children. Environ. Health Perspect. 2006, 114, 1596–1602. [Google Scholar]
- Mitchell, K.A.; Lockhart, C.A.; Huang, G.; Elferink, C.J. Sustained aryl hydrocarbon receptor activity attenuates liver regeneration. Mol. Pharmacol. 2006, 70, 163–170. [Google Scholar]
- Kuznetsov, N.V.; Andersson, P.; Gradin, K.; Stein, P.; Dieckmann, A.; Pettersson, S.; Hanberg, A.; Poellinger, L. The dioxin/aryl hydrocarbon receptor mediates downregulation of osteopontin gene expression in a mouse model of gastric tumourigenesis. Oncogene 2005, 24, 3216–3222. [Google Scholar]
- Moennikes, O.; Loeppen, S.; Buchmann, A.; Andersson, P.; Ittrich, C.; Poellinger, L.; Schwarz, M. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004, 64, 4707–4710. [Google Scholar]
- Andersson, P.; McGuire, J.; Rubio, C.; Gradin, K.; Whitelaw, M.L.; Pettersson, S.; Hanberg, A.; Poellinger, L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 9990–9995. [Google Scholar]
- Tauchi, M.; Hida, A.; Negishi, T.; Katsuoka, F.; Noda, S.; Mimura, J.; Hosoya, T.; Yanaka, A.; Aburatani, H.; Fujii-Kuriyama, Y.; Motohashi, H.; Yamamoto, M. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol. Cell Biol. 2005, 25, 9360–9368. [Google Scholar]
- Hu, W.; Sorrentino, C.; Denison, M.S.; Kolaja, K.; Fielden, M.R. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol. Pharmacol. 2007, 71, 1475–1486. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Medjakovic, S.; Mueller, M.; Jungbauer, A. Potential Health-modulating Effects of Isoflavones and Metabolites via Activation of PPAR and AhR. Nutrients 2010, 2, 241-279. https://doi.org/10.3390/nu2030241
Medjakovic S, Mueller M, Jungbauer A. Potential Health-modulating Effects of Isoflavones and Metabolites via Activation of PPAR and AhR. Nutrients. 2010; 2(3):241-279. https://doi.org/10.3390/nu2030241
Chicago/Turabian StyleMedjakovic, Svjetlana, Monika Mueller, and Alois Jungbauer. 2010. "Potential Health-modulating Effects of Isoflavones and Metabolites via Activation of PPAR and AhR" Nutrients 2, no. 3: 241-279. https://doi.org/10.3390/nu2030241