Inflammation, not Cholesterol, Is a Cause of Chronic Disease




Abstract
1. Introduction
1.1. Biological Significance of Cholesterol—Circulating Blood Cholesterol
1.2. Cholesterol Levels: Demonising a Risk Factor but Not the Causative Mechanisms of Chronic Diseases
1.3. Revisiting the Lipid Hypothesis: Outcomes of the Mediterranean Diet against Inflammation
2. Re-Discovering Chronic Inflammation as the Cause for Chronic Diseases
3. The Role of PAF in Chronic Diseases and the Beneficial Effects of the Mediterranean Diet
3.1. PAF Structure, Activities, and Metabolism: The Role of PAF
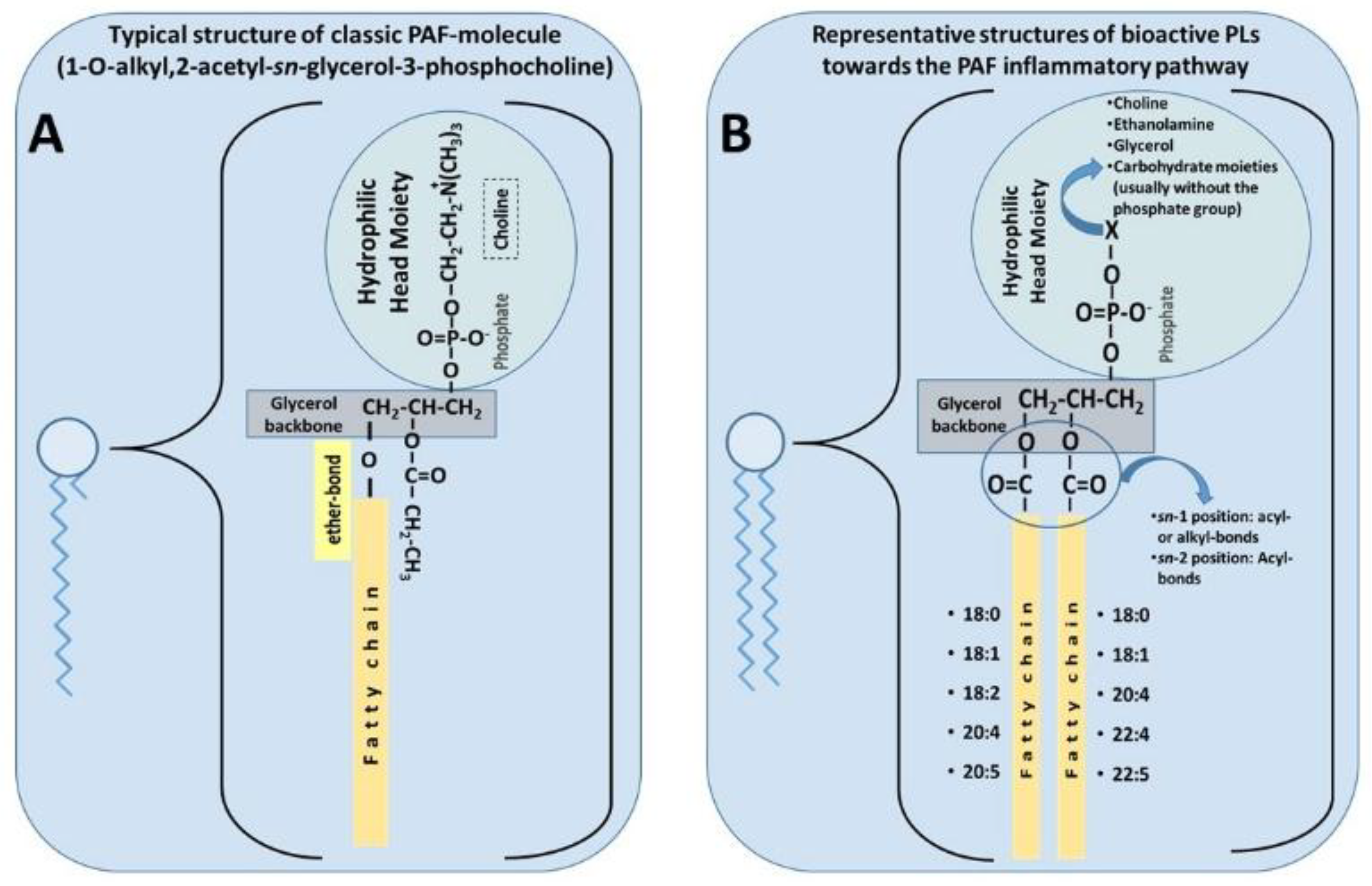
3.1.1. PAF Structure and Physiological Roles
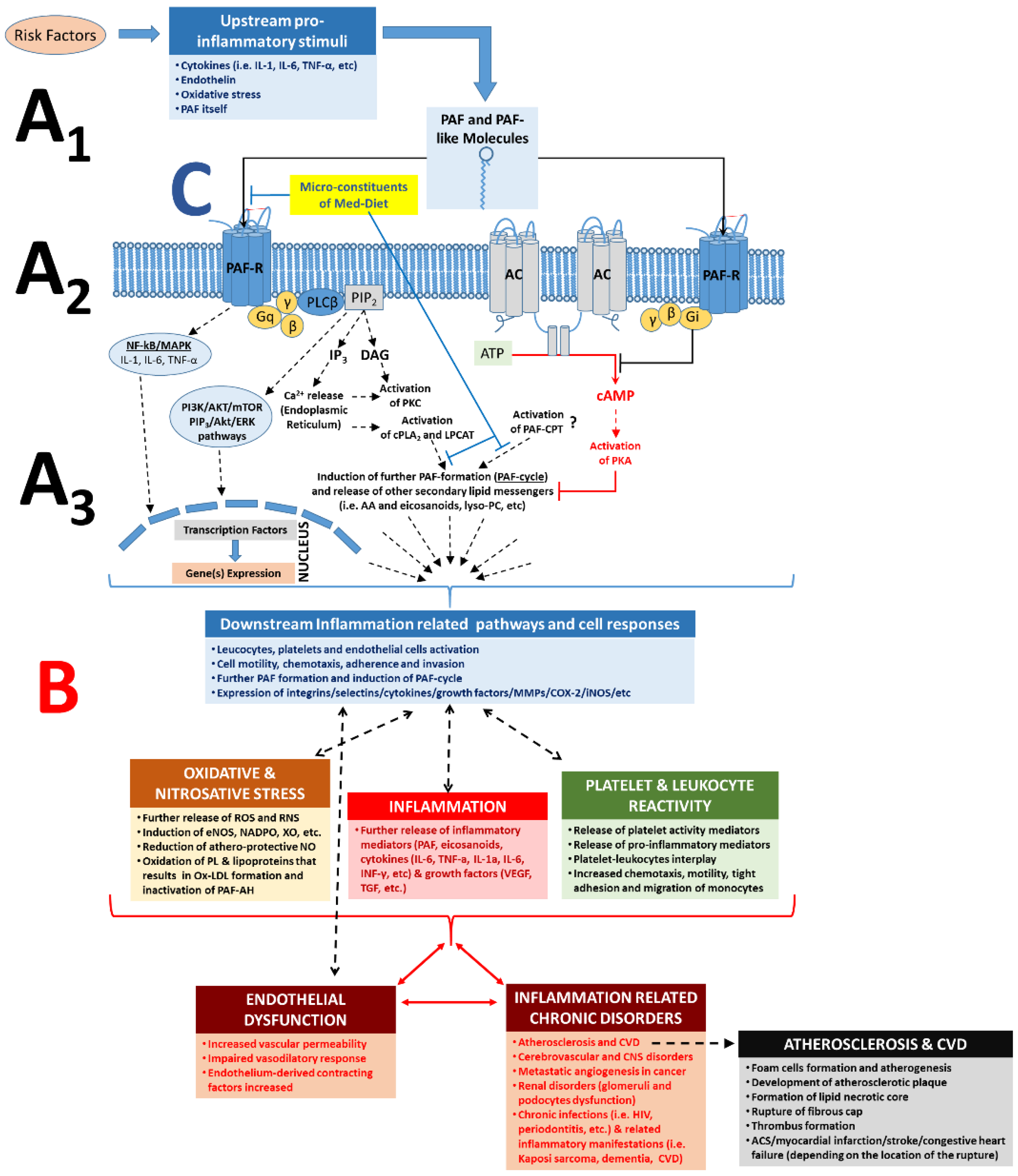
3.1.2. The PAF/PAF-Receptor Signalling Pathways
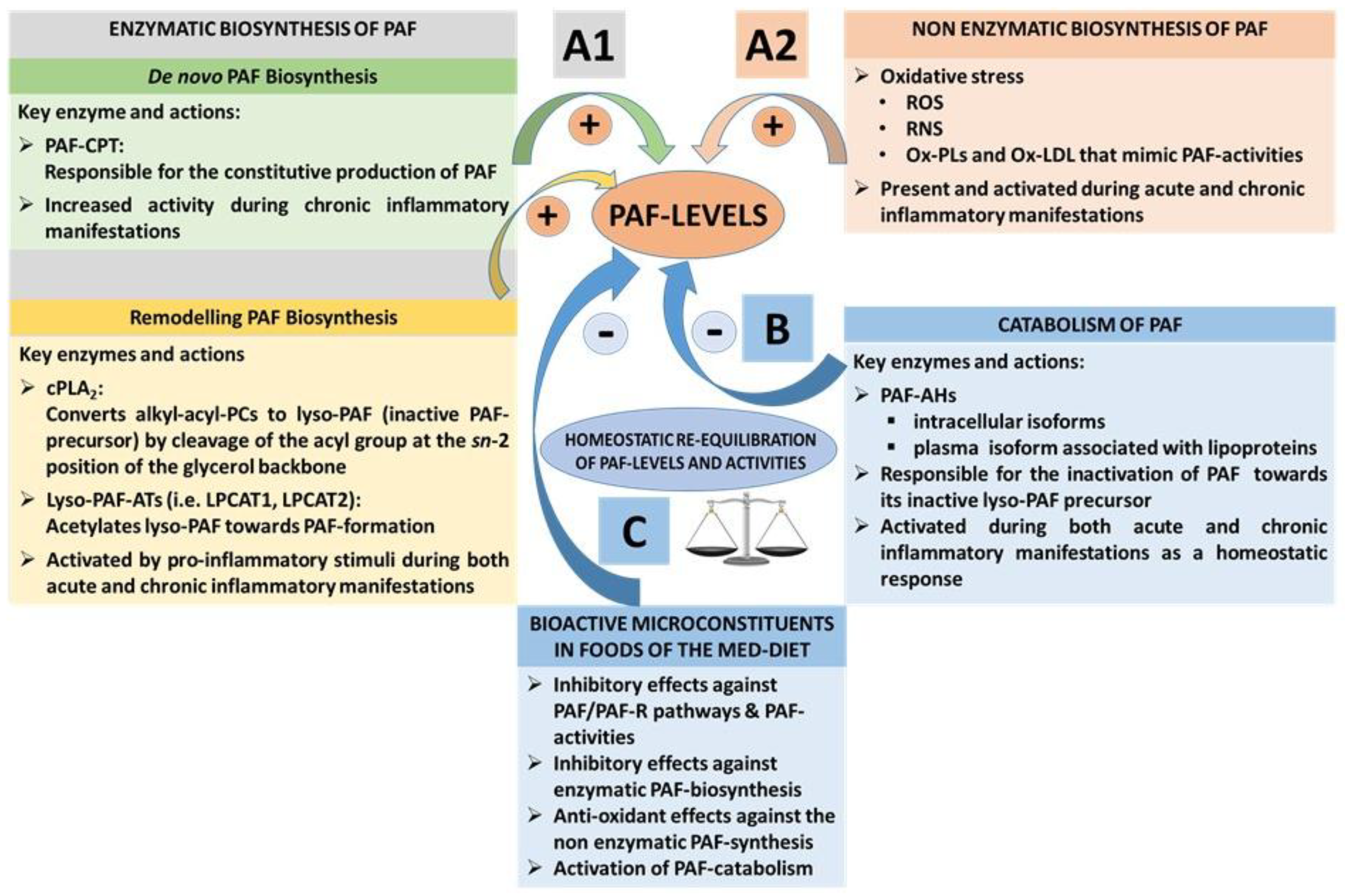
3.1.3. PAF Levels Result from Enzymatic Biosynthesis, Non-Enzymatic Oxidative Synthesis, and Enzymatic Catabolism
3.2. The PAF Pathway and Metabolism in Chronic Diseases
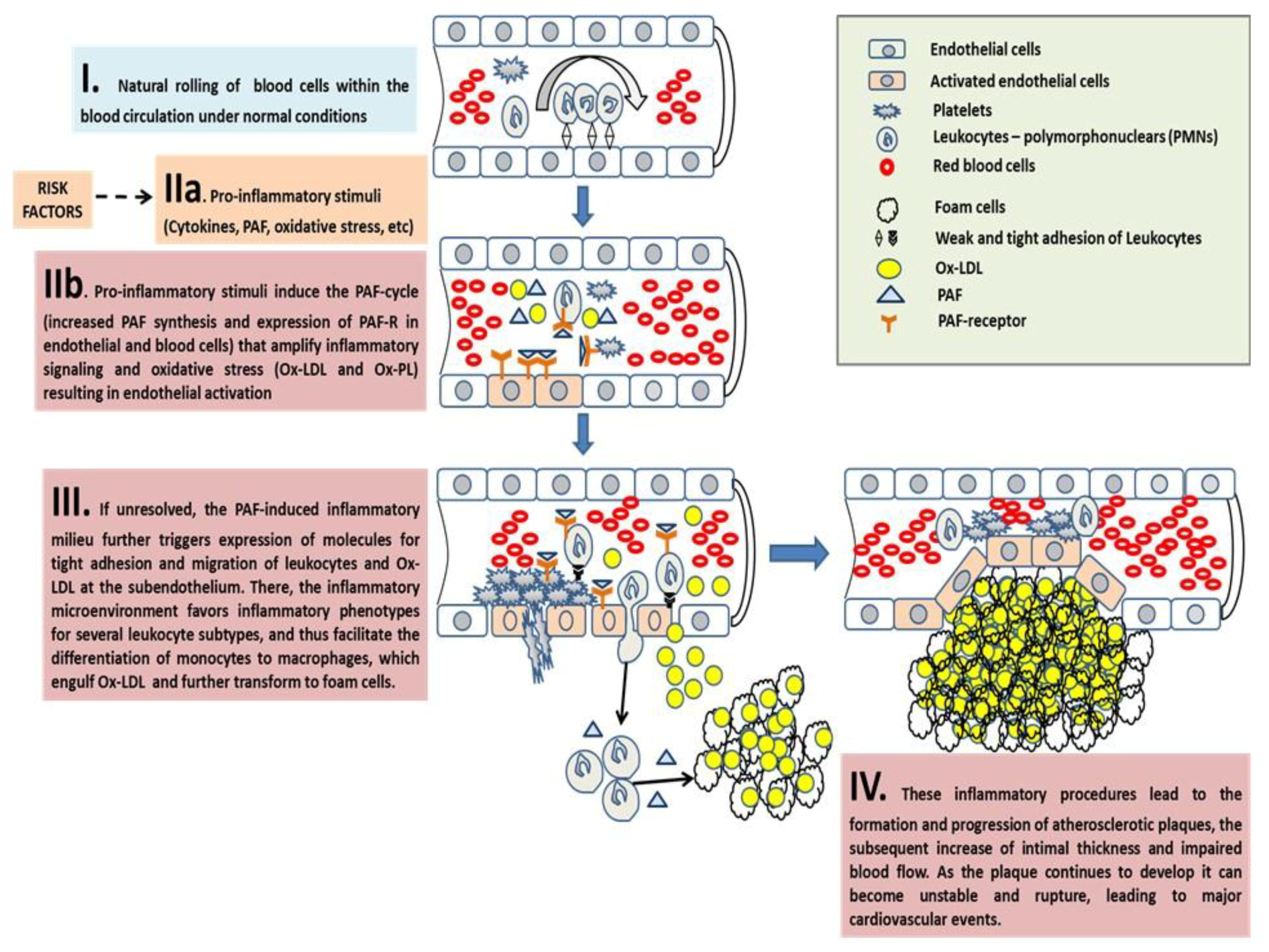
3.2.1. PAF in Atherosclerosis and CVD
The Pro-Inflammatory Crosstalk between PAF with Several Cells and the Endothelium Induces Early Pro-Atherogenic Phases of Endothelial Dysfunction
The Inflammatory Crosstalk Between PAF and Several Cells at the Intima and Subintima Leads to the Induction of Plaque Development and Increased Plaque Growth and Expansion
The Overgrowth and Instability of Plaques and Subsequent Acute Cardiovascular Events
Concluding Remarks on PAF in Atherosclerosis and CVD
3.2.2. The Role of PAF in Cancer and Metastatic Angiogenesis
3.2.3. The Role of PAF in Glomerulosclerosis and Renal Disorders
3.2.4. The Role of PAF in Cerebrovascular and Central Nervous System Disorders
3.2.5. The Role of PAF in Allergies and Asthma
3.2.6. The Role of PAF in Chronic Infections and Inflammation-Associated Comorbidities
3.2.7. The Role of PAF in Various Inflammation-Related Chronic Diseases
3.3. Targeting the PAF Pathways and Metabolism – Beneficial Outcomes of the Mediterranean Diet
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Huff, T.; Jialal, I.I. Physiology, Cholesterol; StatPearls Publishing: Orlando, FL, USA, 2017. [Google Scholar]
- Cox, R.A.; García-Palmieri, M.R. Cholesterol, triglycerides, and associated lipoproteins. In Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Walker, H.K., Hall, W.D., Hurst, J.W., Eds.; Butterworths: Boston, MA, USA, 1990. [Google Scholar]
- Lordan, R.; Tsoupras, A.; Mitra, B.; Zabetakis, I. Dairy fats and cardiovascular disease: Do we really need to be concerned? Foods 2018, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.K. Emerging risk biomarkers in cardiovascular diseases and disorders. J. Lipids 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S. Risk Assessment and Guidelines for the Management of High Blood Cholesterol. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Schaefer, E.J.; Tsunoda, F.; Diffenderfer, M.; Polisecki, E.; Thai, N.; Asztalos, B. The Measurement of Lipids, Lipoproteins, Apolipoproteins, Fatty Acids, and Sterols, and Next Generation Sequencing for the Diagnosis and Treatment of Lipid Disorders. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Iqbal, F.; Baker, W.S.; Khan, M.I.; Thukuntla, S.; McKinney, K.H.; Abate, N.; Tuvdendorj, D. Current and future therapies for addressing the effects of inflammation on HDL cholesterol metabolism. Br. J. Pharmacol. 2017, 174, 3986–4006. [Google Scholar] [CrossRef] [PubMed]
- Spinas, E.; Kritas, S.K.; Saggini, A.; Mobili, A.; Caraffa, A.; Antinolfi, P.; Pantalone, A.; Tei, M.; Speziali, A.; Saggini, R.; et al. Role of mast cells in atherosclerosis: A classical inflammatory disease. Int. J. Immunopathol. Pharmacol. 2014, 27, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Anagnostis, P.; Paschou, S.A.; Goulis, D.G.; Athyros, V.G.; Karagiannis, A. Dietary management of dyslipidaemias. Is there any evidence for cardiovascular benefit? Maturitas 2018, 108, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Pitsavos, C.; Panagiotakos, D.B.; Menotti, A.; Chrysohoou, C.; Skoumas, J.; Stefanadis, C.; Dontas, A.; Toutouzas, P. Forty-Year Follow-Up of Coronary Heart Disease Mortality and Its Predictors: The Corfu Cohort of the Seven Countries Study. Prev. Cardiol. 2003, 6, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Menotti, A.; Lanti, M.; Kromhout, D.; Blackburn, H.; Nissinen, A.; Dontas, A.; Kafatos, A.; Nedeljkovic, S.; Adachi, H. Forty-year coronary mortality trends and changes in major risk factors in the first 10 years of follow-up in the seven countries study. Eur. J. Epidemiol. 2007, 22, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Menotti, A.; Kromhout, D.; Blackburn, H.; Fidanza, F.; Buzina, R.; Nissinen, A. Food intake patterns and 25-year mortality from coronary heart disease: Cross-cultural correlations in the Seven Countries Study. Eur. J. Epidemiol. 1999, 15, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Menotti, A.; Keys, A.; Blackburn, H.; Kromhout, D.; Karvonen, M.; Nissinen, A.; Pekkanen, J.; Punsar, S.; Fidanza, F.; Giampaoli, S.; et al. Comparison of multivariate predictive power of major risk factors for coronary heart diseases in different countries: Results from eight nations of the Seven Countries Study, 25-year follow-up. J. Cardiovasc. Risk 1996, 3, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Grunfeld, C. Cholesterol Lowering Drugs. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Ravnskov, U.; Diamond, D.M.; Hama, R.; Hamazaki, T.; Hammarskjold, B.; Hynes, N.; Kendrick, M.; Langsjoen, P.H.; Malhotra, A.; Mascitelli, L.; et al. Lack of an association or an inverse association between low-density-lipoprotein cholesterol and mortality in the elderly: A systematic review. BMJ Open 2016, 6, e010401. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Hou, L.; Wang, W. Dietary total fat and fatty acids intake, serum fatty acids and risk of breast cancer: A meta-analysis of prospective cohort studies. Int. J. Cancer 2016, 138, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.Q.; Cui, L.H.; Li, C.C.; Yu, Z.; Piao, J.M. Effects of Serum Triglycerides on Prostate Cancer and Breast Cancer Risk: A Meta-Analysis of Prospective Studies. Nutr. Cancer 2016, 68, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Liu, H.; Gao, R. Serum Lipids and Breast Cancer Risk: A Meta-Analysis of Prospective Cohort Studies. PLoS ONE 2015, 10, e0142669. [Google Scholar] [CrossRef] [PubMed]
- Kromhout, D. Serum cholesterol in cross-cultural perspective. The Seven Countries Study. Acta Cardiol. 1999, 54, 155–158. [Google Scholar] [PubMed]
- Papandreou, C.; Tuomilehto, H. Coronary heart disease mortality in relation to dietary, lifestyle and biochemical risk factors in the countries of the Seven Countries Study: A secondary dataset analysis. J. Hum. Nutr. Diet. 2014, 27, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Tektonidis, T.G.; Åkesson, A.; Gigante, B.; Wolk, A.; Larsson, S.C. A Mediterranean diet and risk of myocardial infarction, heart failure and stroke: A population-based cohort study. Atherosclerosis 2015, 243, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; de Lorgeril, M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet 1992, 339, 1523–1526. [Google Scholar] [CrossRef]
- Martínez-González, M.Á.; Ruiz-Canela, M.; Hruby, A.; Liang, L.; Trichopoulou, A.; Hu, F.B. Intervention Trials with the Mediterranean Diet in Cardiovascular Prevention: Understanding Potential Mechanisms through Metabolomic Profiling. J. Nutr. 2016, 146, 913S–919S. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, H.E.; Koeller, E.; Greer, N.; MacDonald, R.; Kane, R.; Wilt, T.J. Effects on Health Outcomes of a Mediterranean Diet With No Restriction on Fat Intake: A Systematic Review and Meta-analysis. Ann. Intern. Med. 2016, 165, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Sanches Machado d’Almeida, K.; Ronchi Spillere, S.; Zuchinali, P.; Correa Souza, G. Mediterranean Diet and Other Dietary Patterns in Primary Prevention of Heart Failure and Changes in Cardiac Function Markers: A Systematic Review. Nutrients 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Fridman, D. Impact of Mediterranean Diet on Cancer: Focused Literature Review. Cancer Genom. Proteom. 2017, 14, 403–408. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Hoffmann, G. Mediterranean dietary pattern, inflammation and endothelial function: A systematic review and meta-analysis of intervention trials. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.Y.; Chao, X.; Su, T.; Fu, X.; Tse, A.K.; Fong, W.F.; Yu, Z.L. The anticancer and antiobesity effects of Mediterranean diet. Crit. Rev. Food Sci. Nutr. 2017, 57, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Di Daniele, N.; Noce, A.; Vidiri, M.F.; Moriconi, E.; Marrone, G.; Annicchiarico-Petruzzelli, M.; D’Urso, G.; Tesauro, M.; Rovella, V.; De Lorenzo, A. Impact of Mediterranean diet on metabolic syndrome, cancer and longevity. Oncotarget 2017, 8, 8947–8979. [Google Scholar] [CrossRef] [PubMed]
- Godos, J.; Zappala, G.; Bernardini, S.; Giambini, I.; Bes-Rastrollo, M.; Martinez-Gonzalez, M. Adherence to the Mediterranean diet is inversely associated with metabolic syndrome occurrence: A meta-analysis of observational studies. Int. J. Food Sci. Nutr. 2017, 68, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Salas-Salvado, J.; Guasch-Ferre, M.; Lee, C.H.; Estruch, R.; Clish, C.B.; Ros, E. Protective Effects of the Mediterranean Diet on Type 2 Diabetes and Metabolic Syndrome. J. Nutr. 2016. [Google Scholar] [CrossRef]
- Alkhatib, A.; Tsang, C.; Tiss, A.; Bahorun, T.; Arefanian, H.; Barake, R.; Khadir, A.; Tuomilehto, J. Functional Foods and Lifestyle Approaches for Diabetes Prevention and Management. Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Archundia Herrera, M.C.; Subhan, F.B.; Chan, C.B. Dietary Patterns and Cardiovascular Disease Risk in People with Type 2 Diabetes. Curr. Obes. Rep. 2017, 6, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Jannasch, F.; Kroger, J.; Schulze, M.B. Dietary Patterns and Type 2 Diabetes: A Systematic Literature Review and Meta-Analysis of Prospective Studies. J. Nutr. 2017, 147, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Dow, C.; Mancini, F.; Rajaobelina, K.; Boutron-Ruault, M.C.; Balkau, B.; Bonnet, F.; Fagherazzi, G. Diet and risk of diabetic retinopathy: A systematic review. Eur. J. Epidemiol. 2018, 33, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Guilleminault, L.; Williams, E.J.; Scott, H.A.; Berthon, B.S.; Jensen, M.; Wood, L.G. Diet and Asthma: Is It Time to Adapt Our Message? Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.; Kouvari, M.; D’Cunha, N.M.; Georgousopoulou, E.N.; Panagiotakos, D.B.; Mellor, D.D.; Kellett, J.; Naumovski, N. The effects of the Mediterranean diet on rheumatoid arthritis prevention and treatment: A systematic review of human prospective studies. Rheumatol. Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kojima, G.; Avgerinou, C.; Iliffe, S.; Walters, K. Adherence to Mediterranean Diet Reduces Incident Frailty Risk: Systematic Review and Meta-Analysis. J. Am. Geriatr. Soc. 2018. [Google Scholar] [CrossRef] [PubMed]
- Suarez, M.; Boque, N.; Del Bas, J.M.; Mayneris-Perxachs, J.; Arola, L.; Caimari, A. Mediterranean Diet and Multi-Ingredient-Based Interventions for the Management of Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Salomone, F.; Mlynarsky, L. The Mediterranean dietary pattern as the diet of choice for non-alcoholic fatty liver disease: Evidence and plausible mechanisms. Liver Int. 2017, 37, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Shivashankar, R.; Lewis, J.D. The Role of Diet in Inflammatory Bowel Disease. Curr. Gastroenterol. Rep. 2017, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Aridi, Y.S.; Walker, J.L.; Wright, O.R.L. The Association between the Mediterranean Dietary Pattern and Cognitive Health: A Systematic Review. Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Tan, L.; Wang, H.F.; Jiang, T.; Zhu, X.C.; Lu, H.; Tan, M.S.; Yu, J.T. Dietary Patterns and Risk of Dementia: A Systematic Review and Meta-Analysis of Cohort Studies. Mol. Neurobiol. 2016, 53, 6144–6154. [Google Scholar] [CrossRef] [PubMed]
- Petersson, S.D.; Philippou, E. Mediterranean Diet, Cognitive Function, and Dementia: A Systematic Review of the Evidence. Adv. Nutr. 2016, 7, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.; Andrade, J.P. Nutritional and Lifestyle Interventions for Age-Related Macular Degeneration: A Review. Oxid. Med. Cell. Longev. 2017, 2017, 6469138. [Google Scholar] [CrossRef] [PubMed]
- Panagiotakos, D.B.; Notara, V.; Kouvari, M.; Pitsavos, C. The Mediterranean and other Dietary Patterns in Secondary Cardiovascular Disease Prevention: A Review. Curr. Vasc. Pharmacol. 2016, 14, 442–451. [Google Scholar] [CrossRef] [PubMed]
- de Lorgeril, M.; Salen, P.; Martin, J.L.; Monjaud, I.; Delaye, J.; Mamelle, N. Mediterranean diet, traditional risk factors, and the rate of cardiovascular complications after myocardial infarction: Final report of the Lyon Diet Heart Study. Circulation 1999, 99, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.T.; de Groot, L.C.; Kromhout, D.; Perrin, A.E.; Moreiras-Varela, O.; Menotti, A.; van Staveren, W.A. Mediterranean diet, lifestyle factors, and 10-year mortality in elderly European men and women: The HALE project. JAMA 2004, 292, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wilmot, K.A.; Ghasemzadeh, N.; Molloy, D.L.; Burkman, G.; Mekonnen, G.; Gongora, M.C.; Quyyumi, A.A.; Sperling, L.S. Mediterranean Dietary Patterns and Cardiovascular Health. Annu. Rev. Nutr. 2015, 35, 425–449. [Google Scholar] [CrossRef] [PubMed]
- Sofi, F.; Dinu, M.; Pagliai, G.; Cesari, F.; Gori, A.M.; Sereni, A.; Becatti, M.; Fiorillo, C.; Marcucci, R.; Casini, A. Low-Calorie Vegetarian Versus Mediterranean Diets for Reducing Body Weight and Improving Cardiovascular Risk Profile: CARDIVEG Study (Cardiovascular Prevention With Vegetarian Diet). Circulation 2018. [Google Scholar] [CrossRef] [PubMed]
- Ndanuko, R.N.; Tapsell, L.C.; Charlton, K.E.; Neale, E.P.; Batterham, M.J. Dietary Patterns and Blood Pressure in Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Adv. Nutr. 2016, 7, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Martínez-González, M.A.; Corella, D.; Salas-Salvadó, J.; Fitó, M.; Chiva-Blanch, G.; Fiol, M.; Gómez-Gracia, E.; Arós, F.; Lapetra, J. Effect of a high-fat Mediterranean diet on bodyweight and waist circumference: A prespecified secondary outcomes analysis of the PREDIMED randomised controlled trial. Lancet Diabetes Endocrinol. 2016, 4, 666–676. [Google Scholar] [CrossRef]
- Hernáez, Á.; Castañer, O.; Elosua, R.; Pintó, X.; Estruch, R.; Salas-Salvadó, J.; Corella, D.; Arós, F.; Serra-Majem, L.; Fiol, M.; et al. Mediterranean diet improves high-density lipoprotein function in high-cardiovascular-risk individuals: A randomized controlled trial. Circulation 2017, 135, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Psaltopoulou, T.; Naska, A.; Orfanos, P.; Trichopoulos, D.; Mountokalakis, T.; Trichopoulou, A. Olive oil, the Mediterranean diet, and arterial blood pressure: The Greek European Prospective Investigation into Cancer and Nutrition (EPIC) study. Am. J. Clin. Nutr. 2004, 80, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Martinez-Gonzalez, M.A.; Corella, D.; Salas-Salvado, J.; Ruiz-Gutierrez, V.; Covas, M.I.; Fiol, M.; Gomez-Gracia, E.; Lopez-Sabater, M.C.; Vinyoles, E.; et al. Effects of a Mediterranean-style diet on cardiovascular risk factors: A randomized trial. Ann. Intern. Med. 2006, 145, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Phospholipids of Animal and Marine Origin: Structure, Function, and Anti-Inflammatory Properties. Molecules 2017, 22, 1964. [Google Scholar] [CrossRef] [PubMed]
- Tsoupras, A.B.; Iatrou, C.; Frangia, C.; Demopoulos, C.A. The Implication of platelet-activating factor in cancer growth and metastasis: Potent beneficial role of PAF-inhibitors and antioxidants. Infect. Disord. Drug Targets 2009, 9, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Demopoulos, C.A.; Karantonis, H.C.; Antonopoulou, S. Platelet activating factor—A molecular link between atherosclerosis theories. Eur. J. Lipid Sci. Technol. 2003, 105, 705–716. [Google Scholar] [CrossRef]
- Legein, B.; Temmerman, L.; Biessen, E.A.L.; Lutgens, E. Inflammation and immune system interactions in atherosclerosis. Cellular Mol. Life Sci. 2013, 70, 3847–3869. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; True, H.D.; Patel, J. Leukocyte Trafficking in Cardiovascular Disease: Insights from Experimental Models. Mediat. Inflamm. 2017, 2017, 9746169. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and Inflammatory Mechanisms of Atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Hansson, G.K. Inflammation and Immunity in Diseases of the Arterial Tree: Players and Layers. Circ. Res. 2015, 116, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Pirillo, A.; Norata, G.D. Vascular inflammation and low-density lipoproteins: Is cholesterol the link? A lesson from the clinical trials. Br. J. Pharmacol. 2017, 174, 3973–3985. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Feng, Y. Hypercholesterolemia Tunes Hematopoietic Stem/Progenitor Cells for Inflammation and Atherosclerosis. Int. J. Mol. Sci. 2016, 17, 1162. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Grunfeld, C. The Effect of Inflammation and Infection on Lipids and Lipoproteins. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Angelovich, T.A.; Hearps, A.C.; Jaworowski, A. Inflammation-induced foam cell formation in chronic inflammatory disease. Immunol. Cell Biol. 2015, 93, 683. [Google Scholar] [CrossRef] [PubMed]
- Yost, C.C.; Weyrich, A.S.; Zimmerman, G.A. The platelet activating factor (PAF) signaling cascade in systemic inflammatory responses. Biochimie 2010, 92, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Tsoupras, A.B.; Chini, M.; Tsogas, N.; Lioni, A.; Tsekes, G.; Demopoulos, C.A.; Lazanas, M.C. In vitro anti-inflammatory and anti-coagulant effects of antibiotics towards Platelet Activating Factor and thrombin. J. Inflamm. 2011, 8, 17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Papakonstantinou, V.D.; Chini, M.; Mangafas, N.; Stamatakis, G.M.; Tsogas, N.; Tsoupras, A.B.; Psarra, K.; Fragopoulou, E.; Antonopoulou, S.; Gargalianos, P.; et al. In vivo effect of two first-line ART regimens on inflammatory mediators in male HIV patients. Lipids Health Dis. 2014, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Tsoupras, A.B.; Chini, M.; Mangafas, N.; Tsogas, N.; Stamatakis, G.; Tsantila, N.; Fragopoulou, E.; Antonopoulou, S.; Gargalianos, P.; Demopoulos, C.A.; et al. Platelet-Activating Factor and Its Basic Metabolic Enzymes in Blood of Naive HIV-Infected Patients. Angiology 2012, 63, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Tsoupras, A.B.; Chini, M.; Tsogas, N.; Fragopoulou, E.; Nomikos, T.; Lioni, A.; Mangafas, N.; Demopoulos, C.A.; Antonopoulou, S.; Lazanas, M.C. Anti-platelet-activating factor effects of highly active antiretroviral therapy (HAART): A new insight in the drug therapy of HIV infection? AIDS Res. Hum. Retroviruses 2008, 24, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Chini, M.; Tsoupras, A.B.; Mangafas, N.; Tsogas, N.; Papakonstantinou, V.D.; Fragopoulou, E.; Antonopoulou, S.; Gargalianos, P.; Demopoulos, C.A.; Lazanas, M.C. Effects of highly active antiretroviral therapy on platelet activating factor metabolism in naive HIV-infected patients: II) study of the abacavir/lamivudine/efavirenz HAART regimen. Int. J. Immunopathol. Pharmacol. 2012, 25, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Chini, M.; Tsoupras, A.B.; Mangafas, N.; Tsogas, N.; Papakonstantinou, V.D.; Fragopoulou, E.; Antonopoulou, S.; Gargalianos, P.; Demopoulos, C.A.; Lazanas, M.C. Effects of HAART on platelet-activating factor metabolism in naive HIV-infected patients I: Study of the tenofovir-DF/emtricitabine/efavirenz HAART regimen. AIDS Res. Hum. Retroviruses 2012, 28, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Tsoupras, A.B.; Chini, M.; Tsogas, N.; Mangafas, N.; Demopoulos, C.A.; Lazanas, M.C. In vivo effects of a Ginkgo biloba extract on platelet activating factor metabolism in two asymptomatic HIV-infected patients. Eur. J. Inflamm. 2011, 9, 107–116. [Google Scholar] [CrossRef]
- McManus, L.M.; Pinckard, R.N. PAF, a putative mediator of oral inflammation. Crit. Rev. Oral Biol. Med. 2000, 11, 240–258. [Google Scholar] [CrossRef] [PubMed]
- Tsoupras, A.B.; Antonopoulou, S.; Baltas, G.; Samiotaki, M.; Panayotou, G.; Kotsifaki, H.; Mantzavinos, Z.; Demopoulos, C.A. Isolation and identification of hydroxyl–platelet-activating factor from natural sources. Life Sci. 2006, 79, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulou, S.; Tsoupras, A.; Baltas, G.; Kotsifaki, H.; Mantzavinos, Z.; Demopoulos, C.A. Hydroxyl-platelet-activating factor exists in blood of healthy volunteers and periodontal patients. Mediat. Inflamm. 2003, 12, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Reznichenko, A.; Korstanje, R. The Role of Platelet-Activating Factor in Mesangial Pathophysiology. Am. J. Pathol. 2015, 185, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Verouti, S.N.; Tsoupras, A.B.; Alevizopoulou, F.; Demopoulos, C.A.; Iatrou, C. Paricalcitol effects on activities and metabolism of Platelet Activating Factor and on inflammatory cytokines in hemodialysis patients. Int. J. Artif. Organs 2013, 36, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Tsoupras, A.; Fragopoulou, E.; Iatrou, C.; Demopoulos, C. In vitro protective effects of olive pomace polar lipids towards platelet activating factor metabolism in human renal cells. Curr. Top. Nutraceutical Res. 2011, 9, 105. [Google Scholar]
- Tsoupras, A.B.; Fragopoulou, E.; Nomikos, T.; Iatrou, C.; Antonopoulou, S.; Demopoulos, C.A. Characterization of the de novo biosynthetic enzyme of platelet activating factor, DDT-insensitive cholinephosphotransferase, of human mesangial cells. Mediat. Inflamm. 2007, 2007. [Google Scholar] [CrossRef] [PubMed]
- Dalbeni, A.; Giollo, A.; Tagetti, A.; Atanasio, S.; Orsolini, G.; Cioffi, G.; Ognibeni, F.; Rossini, M.; Minuz, P.; Fava, C.; et al. Traditional cardiovascular risk factors or inflammation: Which factors accelerate atherosclerosis in arthritis patients? Int. J. Cardiol. 2017, 236, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Welsh, P.; Grassia, G.; Botha, S.; Sattar, N.; Maffia, P. Targeting inflammation to reduce cardiovascular disease risk: A realistic clinical prospect? Br. J. Pharmacol. 2017, 174, 3898–3913. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O. Multiple Roles for Neutrophils in Atherosclerosis. Circ. Res. 2012, 110, 875. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, T.; Papakonstantinou, V.; Detopoulou, P.; Fragopoulou, E.; Chini, M.; Lazanas, M.C.; Antonopoulou, S. The Role of Platelet-Activating Factor in Chronic Inflammation, Immune Activation, and Comorbidities Associated with HIV Infection. AIDS Rev. 2015, 17, 191–201. [Google Scholar] [PubMed]
- Demopoulos, C.; Pinckard, R.; Hanahan, D.J. Platelet-activating factor. Evidence for 1-O-alkyl-2-acetyl-sn-glyceryl-3-phosphorylcholine as the active component (a new class of lipid chemical mediators). J. Biol. Chem. 1979, 254, 9355–9358. [Google Scholar] [PubMed]
- Ishii, S.; Nagase, T.; Shimizu, T. Platelet-activating factor receptor. Prostaglandins Other Lipid Mediat. 2002, 68–69, 599–609. [Google Scholar] [CrossRef]
- Honda, Z.; Ishii, S.; Shimizu, T. Platelet-activating factor receptor. J. Biochem. 2002, 131, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Palur Ramakrishnan, A.V.K.; Varghese, T.P.; Vanapalli, S.; Nair, N.K.; Mingate, M.D. Platelet activating factor: A potential biomarker in acute coronary syndrome? Cardiovasc. Ther. 2017, 35, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.D.; Navab, M.; Hama, S.Y.; Sevanian, A.; Prescott, S.M.; Stafforini, D.M.; McIntyre, T.M.; Du, B.N.; Fogelman, A.M.; Berliner, J.A. Effect of platelet activating factor-acetylhydrolase on the formation and action of minimally oxidized low density lipoprotein. J. Clin. Investig. 1995, 95, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, W.; Laird, J.; Hazen, S.L.; Salomon, R.G. Polyunsaturated phospholipids promote the oxidation and fragmentation of gamma-hydroxyalkenals: Formation and reactions of oxidatively truncated ether phospholipids. J. Lipid Res. 2008, 49, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Venable, M.E.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Platelet-activating factor: A phospholipid autacoid with diverse actions. J. Lipid Res. 1993, 34, 691–702. [Google Scholar] [PubMed]
- Damiani, E.; Ullrich, S.E. Understanding the connection between platelet-activating factor, a UV-induced lipid mediator of inflammation, immune suppression and skin cancer. Prog. Lipid Res. 2016, 63, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.; Zhu, W.; Chen, G.; Luo, X.; Liew, O.W.; Puah, C.M.; Chen, K.; Jiang, H. Understanding the regulation mechanisms of PAF receptor by agonists and antagonists: Molecular modeling and molecular dynamics simulation studies. Proteins 2007, 67, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.D.; Harris, C.S.; Carswell, C.L.; Baenziger, J.E.; Bennett, S.A. Heterogeneity in the sn-1 carbon chain of platelet-activating factor glycerophospholipids determines pro- or anti-apoptotic signaling in primary neurons. J. Lipid Res. 2008, 49, 2250–2258. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M. Bioactive oxidatively truncated phospholipids in inflammation and apoptosis: Formation, targets, and inactivation. Biochim. Biophys. Acta 2012, 1818, 2456–2464. [Google Scholar] [CrossRef] [PubMed]
- Montrucchio, G.; Alloatti, G.; Camussi, G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol. Rev. 2000, 80, 1669–1699. [Google Scholar] [CrossRef] [PubMed]
- Castro Faria Neto, H.C.; Stafforini, D.M.; Prescott, S.M.; Zimmerman, G.A. Regulating inflammation through the anti-inflammatory enzyme platelet-activating factor-acetylhydrolase. Mem. Inst. Oswaldo Cruz 2005, 100, 83–91. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Triggiani, M.; Schleimer, R.; Warner, J.; Chilton, F. Differential synthesis of 1-acyl-2-acetyl-sn-glycero-3-phosphocholine and platelet-activating factor by human inflammatory cells. J. Immunol. 1991, 147, 660–666. [Google Scholar] [PubMed]
- Francescangeli, E.; Freysz, L.; Goracci, G. PAF-Synthesizing Enzymes in Neural Cells during Differentiation and in Gerbil Brain during Ischemia. In Platelet-Activating Factor and Related Lipid Mediators 2; Springer: Berlin/Heidelberg, Germany, 1996; pp. 21–27. [Google Scholar]
- Tarui, M.; Shindou, H.; Kumagai, K.; Morimoto, R.; Harayama, T.; Hashidate, T.; Kojima, H.; Okabe, T.; Nagano, T.; Nagase, T.; et al. Selective inhibitors of a PAF biosynthetic enzyme lysophosphatidylcholine acyltransferase 2. J. Lipid Res. 2014, 55, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Shindou, H.; Hishikawa, D.; Harayama, T.; Eto, M.; Shimizu, T. Generation of membrane diversity by lysophospholipid acyltransferases. J. Biochem. 2013, 154, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Nasopoulou, C.; Tsoupras, A.B.; Karantonis, H.C.; Demopoulos, C.A.; Zabetakis, I. Fish polar lipids retard atherosclerosis in rabbits by down-regulating PAF biosynthesis and up-regulating PAF catabolism. Lipids Health Dis. 2011, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Detopoulou, P.; Nomikos, T.; Fragopoulou, E.; Antonopoulou, S.; Kotroyiannis, I.; Vassiliadou, C.; Panagiotakos, D.B.; Chrysohoou, C.; Pitsavos, C.; Stefanadis, C. Platelet activating factor (PAF) and activity of its biosynthetic and catabolic enzymes in blood and leukocytes of male patients with newly diagnosed heart failure. Clin. Biochem. 2009, 42, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Marathe, G.K.; Zimmerman, G.A.; Prescott, S.M.; McIntyre, T.M. Activation of vascular cells by PAF-like lipids in oxidized LDL. Vasc. Pharmacol. 2002, 38, 193–200. [Google Scholar] [CrossRef]
- Marathe, G.K.; Prescott, S.M.; Zimmerman, G.A.; McIntyre, T.M. Oxidized LDL Contains Inflammatory PAF-Like Phospholipids. Trends Cardiovasc. Med. 2001, 11, 139–142. [Google Scholar] [CrossRef]
- Stafforini, D.M. Biology of Platelet-activating Factor Acetylhydrolase (PAF-AH, Lipoprotein Associated Phospholipase A2). Cardiovasc. Drugs Ther. 2009, 23, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, K.; Inoue, K. Overview of PAF-degrading enzymes. The Enzymes 2015, 38, 1–22. [Google Scholar] [PubMed]
- Stafforini, D.M.; Zimmerman, G.A. Unraveling the PAF-AH/Lp-PLA(2) controversy. J. Lipid Res. 2014, 55, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Tellis, C.C.; Tselepis, A. Pathophysiological role and clinical significance of lipoprotein-associated phospholipase A2 (Lp-PLA2) bound to LDL and HDL. Curr. Pharm. Des. 2014, 20, 6256–6269. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Arai, H. Intracellular PAF-acetylhydrolase type I. The Enzymes 2015, 38, 23–35. [Google Scholar] [PubMed]
- Kono, N.; Arai, H. Intracellular platelet-activating factor acetylhydrolase, type II: A unique cellular phospholipase A2 that hydrolyzes oxidatively modified phospholipids. The Enzymes 2015, 38, 43–54. [Google Scholar] [PubMed]
- Mazereeuw, G.; Herrmann, N.; Bennett, S.A.; Swardfager, W.; Xu, H.; Valenzuela, N.; Fai, S.; Lanctot, K.L. Platelet activating factors in depression and coronary artery disease: A potential biomarker related to inflammatory mechanisms and neurodegeneration. Neurosci. Biobehav. Rev. 2013, 37, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, V.; Bar-Eli, M. Inflammation and melanoma growth and metastasis: The role of platelet-activating factor (PAF) and its receptor. Cancer Metastasis Rev. 2007, 26, 359. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics—2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Stokes, K.Y.; Granger, D.N. Platelets: A critical link between inflammation and microvascular dysfunction. J. Physiol. 2012, 590, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Ed Rainger, G.; Chimen, M.; Harrison, M.J.; Yates, C.M.; Harrison, P.; Watson, S.P.; Lordkipanidzé, M.; Nash, G.B. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets 2015, 26, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, A.A.; McLeod, E.; Wahle, K.W.J.; Arthur, J.R. Cytokine-induced monocyte adhesion to endothelial cells involves platelet-activating factor: Suppression by conjugated linoleic acid. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2006, 1761, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, Z.S.; Jackson, S.P. The Role of Platelets in Atherothrombosis. ASH Educ. Program Book 2011, 2011, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Ghasemzadeh, M.; Hosseini, E. Intravascular leukocyte migration through platelet thrombi: Directing leukocytes to sites of vascular injury. Thromb. Haemost. 2015, 113, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Franks, Z.G.; Campbell, R.A.; Weyrich, A.S.; Rondina, M.T. Platelet–leukocyte interactions link inflammatory and thromboembolic events in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Sager, H.B.; Nahrendorf, M. Inflammation: A trigger for acute coronary syndrome. Q. J. Nucl. Med. Mol. Imaging 2016, 60, 185–193. [Google Scholar] [PubMed]
- Cochain, C.; Zernecke, A. Macrophages in vascular inflammation and atherosclerosis. Pflügers Archiv Eur. J. Physiol. 2017, 469, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Shimokado, A.; Sun, Y.; Akasaka, T.; Muragaki, Y. Diverse Roles of Macrophages in Atherosclerosis: From Inflammatory Biology to Biomarker Discovery. Mediat. Inflamm. 2012, 2012, 693083. [Google Scholar] [CrossRef] [PubMed]
- Rios, F.J.O.; Gidlund, M.; Jancar, S. Pivotal Role for Platelet-Activating Factor Receptor in CD36 Expression and oxLDL Uptake by Human Monocytes/Macrophages. Cell. Physiol. Biochem. 2011, 27, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Sumita, C.; Yamane, M.; Matsuda, T.; Maeda, M.; Nariai, T.; Fujio, Y.; Azuma, J. Platelet activating factor induces cytoskeletal reorganization through Rho family pathway in THP-1 macrophages. FEBS Lett. 2005, 579, 4038–4042. [Google Scholar] [CrossRef] [PubMed]
- Angeli, V.; Llodrá, J.; Rong, J.X.; Satoh, K.; Ishii, S.; Shimizu, T.; Fisher, E.A.; Randolph, G.J. Dyslipidemia Associated with Atherosclerotic Disease Systemically Alters Dendritic Cell Mobilization. Immunity 2004, 21, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.M.; Bizzarro, B.; Sá-Nunes, A.; Rios, F.J.O.; Jancar, S. Activation of PAF-receptor induces regulatory dendritic cells through PGE2 and IL-10. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Li, N. CD4+ T cells in atherosclerosis: Regulation by platelets. Thromb. Haemost. 2013, 109, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, K.; Murata, T.; Tanaka, H.; Matsuoka, T.; Sakata, D.; Yoshida, N.; Katagiri, K.; Kinashi, T.; Tanaka, T.; Miyasaka, M.; et al. Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat. Immunol. 2003, 4, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Prescott, S.M.; Zimmerman, G.A.; Stafforini, D.M.; McIntyre, T.M. Platelet-activating factor and related lipid mediators. Annu. Rev. Biochem. 2000, 69, 419–445. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J.; Huang, Y.H.; Rönnelid, J.; Schäfer-Elinder, L. Platelet-Activating Factor and Oxidized LDL Induce Immune Activation by a Common Mechanism. Arterioscl. Thromb. Vasc. Biol. 1997, 17, 963. [Google Scholar] [CrossRef] [PubMed]
- Koltai, M.; Hosford, D.; Guinot, P.; Esanu, A.; Braquet, P. Platelet activating factor (PAF). A review of its effects, antagonists and possible future clinical implications (Part I). Drugs 1991, 42, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Koltai, M.; Hosford, D.; Guinot, P.; Esanu, A.; Braquet, P. PAF. A review of its effects, antagonists and possible future clinical implications (Part II). Drugs 1991, 42, 174–204. [Google Scholar] [CrossRef] [PubMed]
- Negro Alvarez, J.M.; Miralles Lopez, J.C.; Ortiz Martinez, J.L.; Abellan Aleman, A.; Rubio del Barrio, R. Platelet-activating factor antagonists. Allergol. Immunopathol. 1997, 25, 249–258. [Google Scholar]
- Singh, P.; Singh, I.N.; Mondal, S.C.; Singh, L.; Garg, V.K. Platelet-activating factor (PAF)-antagonists of natural origin. Fitoterapia 2013, 84, 180–201. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, G.; Rabinovici, R.; Leor, J.; Winkler, J.D.; Vonhof, S. Platelet-activating factor and cardiac diseases: Therapeutic potential for PAF inhibitors. J. Lipid Mediat. Cell Signal. 1997, 15, 255–284. [Google Scholar] [CrossRef]
- Loucks, E.B.; Symersky, P.; Qayumi, A.K. Platelet-activating factor antagonism: A new concept in the management of regional myocardial ischemia-reperfusion injury. J. Investig. Surg. 1997, 10, 321–338. [Google Scholar] [CrossRef]
- Papakonstantinou, V.D.; Lagopati, N.; Tsilibary, E.C.; Demopoulos, C.A.; Philippopoulos, A.I. A Review on Platelet Activating Factor Inhibitors: Could a New Class of Potent Metal-Based Anti-Inflammatory Drugs Induce Anticancer Properties? Bioinorg. Chem. Appl. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Fragopoulou, E.; Choleva, M.; Antonopoulou, S.; Demopoulos, C.A. Wine and its metabolic effects. A comprehensive review of Clinical Trials. Metabolism 2018. [Google Scholar] [CrossRef] [PubMed]
- Nasopoulou, C.; Karantonis, H.C.; Perrea, D.N.; Theocharis, S.E.; Iliopoulos, D.G.; Demopoulos, C.A.; Zabetakis, I. In vivo anti-atherogenic properties of cultured gilthead sea bream (Sparus aurata) polar lipid extracts in hypercholesterolaemic rabbits. Food Chem. 2010, 120, 831–836. [Google Scholar] [CrossRef]
- Tsantila, N.; Karantonis, H.C.; Perrea, D.N.; Theocharis, S.E.; Iliopoulos, D.G.; Iatrou, C.; Antonopoulou, S.; Demopoulos, C.A. Atherosclerosis regression study in rabbits upon olive pomace polar lipid extract administration. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Tsantila, N.; Karantonis, H.C.; Perrea, D.N.; Theocharis, S.E.; Iliopoulos, D.G.; Antonopoulou, S.; Demopoulos, C.A. Antithrombotic and antiatherosclerotic properties of olive oil and olive pomace polar extracts in rabbits. Mediat. Inflamm. 2007, 2007. [Google Scholar] [CrossRef] [PubMed]
- Karantonis, H.C.; Antonopoulou, S.; Perrea, D.N.; Sokolis, D.P.; Theocharis, S.E.; Kavantzas, N.; Iliopoulos, D.G.; Demopoulos, C.A. In vivo antiatherogenic properties of olive oil and its constituent lipid classes in hyperlipidemic rabbits. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Argyrou, C.; Vlachogianni, I.; Stamatakis, G.; Demopoulos, C.A.; Antonopoulou, S.; Fragopoulou, E. Postprandial effects of wine consumption on Platelet Activating Factor metabolic enzymes. Prostaglandins Other Lipid Mediat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Xanthopoulou, M.N.; Kalathara, K.; Melachroinou, S.; Arampatzi-Menenakou, K.; Antonopoulou, S.; Yannakoulia, M.; Fragopoulou, E. Wine consumption reduced postprandial platelet sensitivity against platelet activating factor in healthy men. Eur. J. Nutr. 2017, 56, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Xanthopoulou, M.N.; Asimakopoulos, D.; Antonopoulou, S.; Demopoulos, C.A.; Fragopoulou, E. Effect of Robola and Cabernet Sauvignon extracts on platelet activating factor enzymes activity on U937 cells. Food Chem. 2014, 165, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Fragopoulou, E.; Antonopoulou, S.; Tsoupras, A.; Tsantila, N.; Grypioti, A.; Gribilas, G.; Gritzapi, H.; Konsta, E.; Skandalou, E.; Papadopoulou, A. Antiatherogenic properties of red/white wine, musts, grape-skins, and yeast. In Proceedings of the 45th International Conference on the Bioscience of Lipids, University of Ioannina, Ioannina, Greece, 25–29 May 2004; p. 66. [Google Scholar]
- Fragopoulou, E.; Nomikos, T.; Tsantila, N.; Mitropoulou, A.; Zabetakis, I.; Demopoulos, C.A. Biological activity of total lipids from red and white wine/must. J. Agric. Food Chem. 2001, 49, 5186–5193. [Google Scholar] [CrossRef] [PubMed]
- Fragopoulou, E.; Nomikos, T.; Antonopoulou, S.; Mitsopoulou, C.A.; Demopoulos, C.A. Separation of Biologically Active Lipids from Red Wine. J. Agric. Food. Chem. 2000, 48, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Panayiotou, A.; Samartzis, D.; Nomikos, T.; Fragopoulou, E.; Karantonis, H.C.; Demopoulos, C.A.; Zabetakis, I. Lipid fractions with aggregatory and antiaggregatory activity toward platelets in fresh and fried cod (Gadus morhua): correlation with platelet-activating factor and atherogenesis. J. Agric. Food. Chem. 2000, 48, 6372–6379. [Google Scholar] [CrossRef] [PubMed]
- Sioriki, E.; Smith, T.K.; Demopoulos, C.A.; Zabetakis, I. Structure and cardioprotective activities of polar lipids of olive pomace, olive pomace-enriched fish feed and olive pomace fed gilthead sea bream (Sparus aurata). Food Res. Int. 2016, 83, 143–151. [Google Scholar] [CrossRef]
- Sioriki, E.; Nasopoulou, C.; Demopoulos, C.A.; Zabetakis, I. Comparison of sensory and cardioprotective properties of olive-pomace enriched and conventional gilthead sea bream (Sparus aurata): The effect of grilling. J. Aquat. Food Prod. Technol. 2015, 24, 782–795. [Google Scholar] [CrossRef]
- Nasopoulou, C.; Smith, T.; Detopoulou, M.; Tsikrika, C.; Papaharisis, L.; Barkas, D.; Zabetakis, I. Structural elucidation of olive pomace fed sea bass (Dicentrarchus labrax) polar lipids with cardioprotective activities. Food Chem. 2014, 145, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Nasopoulou, C.; Gogaki, V.; Stamatakis, G.; Papaharisis, L.; Demopoulos, C.; Zabetakis, I. Evaluation of the in Vitro Anti-Atherogenic Properties of Lipid Fractions of Olive Pomace, Olive Pomace Enriched Fish Feed and Gilthead Sea Bream (Sparus aurata) Fed with Olive Pomace Enriched Fish Feed. Mar. Drugs 2013, 11, 3676. [Google Scholar] [CrossRef] [PubMed]
- Nasopoulou, C.; Stamatakis, G.; Demopoulos, C.A.; Zabetakis, I. Effects of olive pomace and olive pomace oil on growth performance, fatty acid composition and cardio protective properties of gilthead sea bream (Sparus aurata) and sea bass (Dicentrarchus labrax). Food Chem. 2011, 129, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Nasopoulou, C.; Karantonis, H.C.; Andriotis, M.; Demopoulos, C.A.; Zabetakis, I. Antibacterial and anti-PAF activity of lipid extracts from sea bass (Dicentrarchus labrax) and gilthead sea bream (Sparus aurata). Food Chem. 2008, 111, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Nasopoulou, C.; Nomikos, T.; Demopoulos, C.; Zabetakis, I. Comparison of antiatherogenic properties of lipids obtained from wild and cultured sea bass (Dicentrarchus labrax) and gilthead sea bream (Sparus aurata). Food Chem. 2007, 100, 560–567. [Google Scholar] [CrossRef]
- Rementzis, J.; Antonopoulou, S.; Argyropoulos, D.; Demopoulos, C.A. Biologically active lipids from S. scombrus. In Platelet-Activating Factor and Related Lipid Mediators 2; Springer: Berlin/Heidelberg, Germany, 1996; pp. 65–72. [Google Scholar]
- Karantonis, H.C.; Antonopoulou, S.; Demopoulos, C.A. Antithrombotic lipid minor constituents from vegetable oils. Comparison between olive oils and others. J. Agric. Food Chem. 2002, 50, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Nasopoulou, C.; Gogaki, V.; Panagopoulou, E.; Demopoulos, C.; Zabetakis, I. Hen egg yolk lipid fractions with antiatherogenic properties. Anim. Sci. J. 2013, 84, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Poutzalis, S.; Anastasiadou, A.; Nasopoulou, C.; Megalemou, K.; Sioriki, E.; Zabetakis, I. Evaluation of the in vitro anti-atherogenic activities of goat milk and goat dairy products. Dairy Sci. Technol. 2016, 96, 317–327. [Google Scholar] [CrossRef]
- Tsorotioti, S.E.; Nasopoulou, C.; Detopoulou, M.; Sioriki, E.; Demopoulos, C.A.; Zabetakis, I. In vitro anti-atherogenic properties of traditional Greek cheese lipid fractions. Dairy Sci. Technol. 2014, 94, 269–281. [Google Scholar] [CrossRef]
- Megalemou, K.; Sioriki, E.; Lordan, R.; Dermiki, M.; Nasopoulou, C.; Zabetakis, I. Evaluation of sensory and in vitro anti-thrombotic properties of traditional Greek yogurts derived from different types of milk. Heliyon 2017, 3, e00227. [Google Scholar] [CrossRef] [PubMed]
- Apitz-Castro, R.; Cabrera, S.; Cruz, M.R.; Ledezma, E.; Jain, M.K. Effects of garlic extract and of three pure components isolated from it on human platelet aggregation, arachidonate metabolism, release reaction and platelet ultrastructure. Thromb. Res. 1983, 32, 155–169. [Google Scholar] [CrossRef]
- Violi, F.; Pratico, D.; Ghiselli, A.; Alessandri, C.; Iuliano, L.; Cordova, C.; Balsano, F. Inhibition of cyclooxygenase-independent platelet aggregation by low vitamin E concentration. Atherosclerosis 1990, 82, 247–252. [Google Scholar] [CrossRef]
- Kakishita, E.; Suehiro, A.; Oura, Y.; Nagai, K. Inhibitory effect of vitamin E (alpha-tocopherol) on spontaneous platelet aggregation in whole blood. Thromb. Res. 1990, 60, 489–499. [Google Scholar] [CrossRef]
- Ji, W.; Chen, J.; Mi, Y.; Wang, G.; Xu, X.; Wang, W. Platelet-activating factor receptor activation promotes prostate cancer cell growth, invasion and metastasis via ERK1/2 pathway. Int. J. Oncol. 2016, 49, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Karandish, F.; Mallik, S. Biomarkers and Targeted Therapy in Pancreatic Cancer. Biomarkers Cancer 2016, 8, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Anandi, V.L.; Ashiq, K.A.; Nitheesh, K.; Lahiri, M. Platelet-activating factor promotes motility in breast cancer cells and disrupts non-transformed breast acinar structures. Oncol. Rep. 2016, 35, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Saito Rde, F.; Tortelli, T.C., Jr.; Jacomassi, M.D.; Otake, A.H.; Chammas, R. Emerging targets for combination therapy in melanomas. FEBS Lett. 2015, 589, 3438–3448. [Google Scholar] [CrossRef] [PubMed]
- Jancar, S.; Chammas, R. PAF receptor and tumor growth. Curr. Drug Targets 2014, 15, 982–987. [Google Scholar] [PubMed]
- Hackler, P.C.; Reuss, S.; Konger, R.L.; Travers, J.B.; Sahu, R.P. Systemic Platelet-activating Factor Receptor Activation Augments Experimental Lung Tumor Growth and Metastasis. Cancer Growth Metastasis 2014, 7, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, M.; Zhang, X.; Cai, Q.; Zhu, Z.; Jiang, W.; Xu, C. Transactivation of epidermal growth factor receptor through platelet-activating factor/receptor in ovarian cancer cells. J. Exp. Clin. Cancer Res. 2014, 33, 85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shida-Sakazume, T.; Endo-Sakamoto, Y.; Unozawa, M.; Fukumoto, C.; Shimada, K.; Kasamatsu, A.; Ogawara, K.; Yokoe, H.; Shiiba, M.; Tanzawa, H.; et al. Lysophosphatidylcholine acyltransferase1 overexpression promotes oral squamous cell carcinoma progression via enhanced biosynthesis of platelet-activating factor. PLoS ONE 2015, 10, e0120143. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Gao, L.; Wang, L.; Tang, G.; He, M.; Yu, Y.; Ni, X.; Sun, Y. Effects of platelet-activating factor and its differential regulation by androgens and steroid hormones in prostate cancers. Br. J. Cancer 2013, 109, 1279–1286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kispert, S.E.; Marentette, J.O.; McHowat, J. Enhanced breast cancer cell adherence to the lung endothelium via PAF acetylhydrolase inhibition: A potential mechanism for enhanced metastasis in smokers. Am. J. Physiol. Cell Physiol. 2014, 307, C951–C956. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kispert, S.; Marentette, J.; McHowat, J. Cigarette smoke induces cell motility via platelet-activating factor accumulation in breast cancer cells: A potential mechanism for metastatic disease. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- da Silva-Junior, I.A.; Dalmaso, B.; Herbster, S.; Lepique, A.P.; Jancar, S. Platelet-Activating Factor Receptor Ligands Protect Tumor Cells from Radiation-Induced Cell Death. Front. Oncol. 2018, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; He, Z.; Ke, J.; Li, S.; Wu, X.; Lian, L.; He, X.; He, X.; Hu, J.; Zou, Y.; et al. PAF receptor antagonist Ginkgolide B inhibits tumourigenesis and angiogenesis in colitis-associated cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 432–440. [Google Scholar] [PubMed]
- Morita, K.; Shiraishi, S.; Motoyama, N.; Kitayama, T.; Kanematsu, T.; Uezono, Y.; Dohi, T. Palliation of bone cancer pain by antagonists of platelet-activating factor receptors. PLoS ONE 2014, 9, e91746. [Google Scholar] [CrossRef] [PubMed]
- Semini, G.; Hildmann, A.; von Haefen, C.; Danker, K. Glycosidated phospholipids—A promising group of anti-tumour lipids. Anticancer Agents Med. Chem. 2014, 14, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G. Potential role of platelet-activating factor in renal pathophysiology. Kidney Int. 1986, 29, 469–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fragopoulou, E.; Iatrou, C.; Demopoulos, C.A. Characterization of acetyl-CoA: Lyso-PAF acetyltransferase of human mesangial cells. Mediat. Inflamm. 2005, 2005, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M. Potential role of platelet activating factor in acute renal failure. Kidney Int. 1999, 55, 1672–1682. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ortiz, A.; Gomez-Chiarri, M.; Lerma, J.L.; Gonzalez, E.; Egido, J. The role of platelet-activating factor (PAF) in experimental glomerular injury. Lipids 1991, 26, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Fragopoulou, E.; Iatrou, C.; Antonopoulou, S.; Ruan, X.Z.; Fernando, R.L.; Powis, S.H.; Moorhead, J.F.; Varghese, Z. Platelet-activating factor (PAF) increase intracellular lipid accumulation by increasing both LDL and scavenger receptors in human mesangial cells. J. Lab. Clin. Med. 2006, 147, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Correa-Costa, M.; Andrade-Oliveira, V.; Braga, T.T.; Castoldi, A.; Aguiar, C.F.; Origassa, C.S.; Rodas, A.C.; Hiyane, M.I.; Malheiros, D.M.; Rios, F.J.; et al. Activation of platelet-activating factor receptor exacerbates renal inflammation and promotes fibrosis. Lab. Investig. 2014, 94, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shields, L.B.E.; Gao, Z.; Wang, Y.; Zhang, Y.P.; Chu, T.; Zhu, Q.; Shields, C.B.; Cai, J. Current Understanding of Platelet-Activating Factor Signaling in Central Nervous System Diseases. Mol. Neurobiol. 2017, 54, 5563–5572. [Google Scholar] [CrossRef] [PubMed]
- Maclennan, K.M.; Smith, P.F.; Darlington, C.L. Platelet-activating factor in the CNS. Prog. Neurobiol. 1996, 50, 585–596. [Google Scholar] [CrossRef]
- Tsuda, M.; Tozaki-Saitoh, H.; Inoue, K. Platelet-activating factor and pain. Biol. Pharm. Bull. 2011, 34, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Barlow, C.L.; Ramirez, S.H.; Abood, M.E.; Brailoiu, G.C. Effects of Platelet-Activating Factor on Brain Microvascular Endothelial Cells. Neuroscience 2018, 377, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Logan, A.; Cocks, B.G.; Rochfort, S. Seasonal variation of polar lipid content in bovine milk. Food Chem. 2017, 237, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Maclennan, K.M.; Darlington, C.L.; Smith, P.F. The CNS effects of Ginkgo biloba extracts and ginkgolide B. Prog. Neurobiol. 2002, 67, 235–257. [Google Scholar] [CrossRef]
- Tomasiak-Lozowska, M.M.; Klimek, M.; Lis, A.; Moniuszko, M.; Bodzenta-Lukaszyk, A. Markers of anaphylaxis—A systematic review. Adv. Med. Sci. 2018, 63, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Triggiani, M.; Patella, V.; Staiano, R.I.; Granata, F.; Marone, G. Allergy and the cardiovascular system. Clin. Exp. Immunol. 2008, 153 (Suppl. 1), 7–11. [Google Scholar] [CrossRef] [PubMed]
- Kasperska-Zajac, A.; Brzoza, Z.; Rogala, B. Platelet-activating factor (PAF): A review of its role in asthma and clinical efficacy of PAF antagonists in the disease therapy. Rec. Patents on Inflammation Allergy Drug Discovery 2008, 2, 72–76. [Google Scholar]
- Kasperska-Zajac, A.; Brzoza, Z.; Rogala, B. Platelet activating factor as a mediator and therapeutic approach in bronchial asthma. Inflammation 2008, 31, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Palgan, K.; Bartuzi, Z. Platelet activating factor in allergies. Int. J. Immunopathol. Pharmacol. 2015, 28, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Kulinski, J.M.; Munoz-Cano, R.; Olivera, A. Sphingosine-1-phosphate and other lipid mediators generated by mast cells as critical players in allergy and mast cell function. Eur. J. Pharmacol. 2016, 778, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, V. Role of histamine and platelet-activating factor in allergic rhinitis. J. Physiol. Biochem. 2004, 60, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Varricchi, G.; Seaf, M.; Marone, G.; Levi-Schaffer, F.; Marone, G. Bidirectional Mast Cell-Eosinophil Interactions in Inflammatory Disorders and Cancer. Front. Med. 2017, 4, 103. [Google Scholar] [CrossRef] [PubMed]
- Mullol, J.; Bousquet, J.; Bachert, C.; Canonica, W.G.; Gimenez-Arnau, A.; Kowalski, M.L.; Marti-Guadano, E.; Maurer, M.; Picado, C.; Scadding, G.; et al. Rupatadine in allergic rhinitis and chronic urticaria. Allergy 2008, 63 (Suppl. 87), 5–28. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Nunez, V.; Bachert, C.; Mullol, J. Rupatadine: Global safety evaluation in allergic rhinitis and urticaria. Expert Opin. Drug. Saf. 2016, 15, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.C.; Teixeira, M.M.; Souza, D.G. Opportunities for the development of novel therapies based on host-microbial interactions. Pharmacol. Res. 2016, 112, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J. The platelet activating factor receptor: A new anti-infective target in respiratory disease? Thorax 2012, 67, 840–841. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chatzovoulos, P.; Tsoupras, A.B.; Samiotaki, M.; Panayotou, G.; Demopoulos, C.A.; Dotsika, E. PAF-metabolic enzymes and PAF-like activity in L. infantum and L. major promastigotes. Eur. J. Inflamm. 2011, 9, 231–239. [Google Scholar] [CrossRef]
- Mathiak, G.; Szewczyk, D.; Abdullah, F.; Ovadia, P.; Rabinovici, R. Platelet-activating factor (PAF) in experimental and clinical sepsis. Shock 1997, 7, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Akinosoglou, K.; Alexopoulos, D. Use of antiplatelet agents in sepsis: A glimpse into the future. Thromb. Res. 2014, 133, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.P. Therapeutic options directed against platelet activating factor, eicosanoids and bradykinin in sepsis. J. Antimicrob. Chemother. 1998, 41 (Suppl. A), 81–94. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ersoy, B.; Huseyinov, A.; Darcan, S. The role of platelet-activating factor in pathogenesis of type 1 diabetes. Diabetes Care 2005, 28, 980. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sfredel, V.; Moţa, M.; Trăilă, A.; Dănoiu, S.; Matcaş, H. Disturbances of the coagulating equilibrium of blood in diabetes mellitus. Rev. Roum. Med. Intern. 1999, 37, 251–260. [Google Scholar]
- Kudolo, G.B.; DeFronzo, R.A. Urinary platelet-activating factor excretion is elevated in non-insulin dependent diabetes mellitus. Prostaglandins Other Lipid Mediat. 1999, 57, 87–98. [Google Scholar] [CrossRef]
- Liu, L.R.; Xia, S.H. Role of platelet-activating factor in the pathogenesis of acute pancreatitis. World J. Gastroenterol. 2006, 12, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xia, S.H.; Chen, H.; Li, X.H. Therapy for acute pancreatitis with platelet-activating factor receptor antagonists. World J. Gastroenterol. 2008, 14, 4735–4738. [Google Scholar] [CrossRef] [PubMed]
- Karidis, N.P.; Kouraklis, G.; Theocharis, S.E. Platelet-activating factor in liver injury: A relational scope. World J. Gastroenterol. 2006, 12, 3695–3706. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.L.; Caplan, M.S. Necrotizing enterocolitis: Pathophysiology, platelet-activating factor, and probiotics. Semin. Pediatr. Surg. 2013, 22, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Ewer, A.K. Role of platelet-activating factor in the pathophysiology of necrotizing enterocolitis. Acta Paediatr. Suppl. 2002, 91, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Nitoda, E.; Moschos, M.M.; Mavragani, C.P.; Koutsilieris, M. Ocular actions of platelet-activating factor: Clinical implications. Expert Opin. Ther. Targets 2012, 16, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.T.; Gozal, E.; Roberts, A.M. Platelet-mediated vascular dysfunction during acute lung injury. Arch. Physiol. Biochem. 2012, 118, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Edwards, L.J.; Constantinescu, C.S. Platelet activating factor/platelet activating factor receptor pathway as a potential therapeutic target in autoimmune diseases. Inflamm. Allergy Drug Targets 2009, 8, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Rollino, C.; Mariano, F.; Quarello, F.; Camussi, G. IL-10 stimulates production of platelet-activating factor by monocytes of patients with active systemic lupus erythematosus (SLE). Clin. Exp. Immunol. 2000, 122, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Thornton, M.; Solomon, M.J. Crohn’s disease: In defense of a microvascular aetiology. Int. J. Colorectal Dis. 2002, 17, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Macconi, D.; Riccardi, E.; Boccardo, P.; Zilio, P.; Bertani, T.; Remuzzi, G. Platelet-activating factor receptor blocking reduces proteinuria and improves survival in lupus autoimmune mice. J. Pharmacol. Exp. Ther. 1991, 258, 601–606. [Google Scholar] [PubMed]
- Baldi, E.; Emancipator, S.N.; Hassan, M.O.; Dunn, M.J. Platelet activating factor receptor blockade ameliorates murine systemic lupus erythematosus. Kidney Int. 1990, 38, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Karantonis, H.C.; Fragopoulou, E.; Antonopoulou, S.; Rementzis, J.; Phenekos, C.; Demopoulos, C.A. Effect of fast-food Mediterranean-type diet on type 2 diabetics and healthy human subjects’ platelet aggregation. Diabetes Res. Clin. Pract. 2006, 72, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulou, S.; Fragopoulou, E.; Karantonis, H.C.; Mitsou, E.; Sitara, M.; Rementzis, J.; Mourelatos, A.; Ginis, A.; Phenekos, C. Effect of traditional Greek Mediterranean meals on platelet aggregation in normal subjects and in patients with type 2 diabetes mellitus. J. Med. Food 2006, 9, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Fragopoulou, E.; Detopoulou, P.; Nomikos, T.; Pliakis, E.; Panagiotakos, D.B.; Antonopoulou, S. Mediterranean wild plants reduce postprandial platelet aggregation in patients with metabolic syndrome. Metabolism 2012, 61, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.W.; Ramji, D.P. Cytokines: Roles in atherosclerosis disease progression and potential therapeutic targets. Fut. Med. Chem. 2016, 8, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Capra, V.; Bäck, M.; Barbieri, S.S.; Camera, M.; Tremoli, E.; Rovati, G.E. Eicosanoids and Their Drugs in Cardiovascular Diseases: Focus on Atherosclerosis and Stroke. Med. Res. Rev. 2013, 33, 364–438. [Google Scholar] [CrossRef] [PubMed]
- Feige, E.; Mendel, I.; George, J.; Yacov, N.; Harats, D. Modified phospholipids as anti-inflammatory compounds. Curr. Opin. Lipidol. 2010, 21, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Lordan, R.; Zabetakis, I. Invited review: The anti-inflammatory properties of dairy lipids. J. Dairy Sci. 2017, 100, 4197–4212. [Google Scholar] [CrossRef] [PubMed]
- Lordan, R.; Zabetakis, I. Ovine and caprine lipids promoting cardiovascular health in milk and its derivatives. Adv. Dairy Res. 2017, 5. [Google Scholar] [CrossRef]
- Rizzo, M.; Otvos, J.; Nikolic, D.; Montalto, G.; Toth, P.; Banach, M. Subfractions and subpopulations of HDL: An update. Curr. Med. Chem. 2014, 21, 2881–2891. [Google Scholar] [CrossRef] [PubMed]
- Marathe, G.K.; Pandit, C.; Lakshmikanth, C.L.; Chaithra, V.H.; Jacob, S.P.; D’Souza, C.J.M. To hydrolyze or not to hydrolyze: The dilemma of platelet-activating factor acetylhydrolase. J. Lipid Res. 2014, 55, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Goszcz, K.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Bioactive polyphenols and cardiovascular disease: Chemical antagonists, pharmacological agents or xenobiotics that drive an adaptive response? Br. J. Pharmacol. 2017, 174, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Sesso, H.D.; Christen, W.G.; Bubes, V.; Smith, J.P.; MacFadyen, J.; Schvartz, M.; Manson, J.E.; Glynn, R.J.; Buring, J.E.; Gaziano, J.M. Multivitamins in the prevention of cardiovascular disease in men: The physicians’ health study II randomized controlled trial. JAMA 2012, 308, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.; Yusuf, S.; Hoogwerf, B.; Pogue, J.; Yi, Q.; Zinman, B.; Bosch, J.; Dagenais, G.; Mann, J.F.E.; Gerstein, H.C. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: Results of the HOPE study and MICRO-HOPE substudy. Diabetes Care 2002, 25, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- The Heart Outcomes Prevention Evaluation Study Investigators. Vitamin E Supplementation and Cardiovascular Events in High-Risk Patients. NEMJ 2000, 342, 154–160. [Google Scholar] [CrossRef]
- Goszcz, K.; Deakin, S.J.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Antioxidants in cardiovascular therapy: Panacea or false hope? Front. Cardiovas. Med. 2015, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Sargeant, L.A.; Khaw, K.T.; Bingham, S.; Day, N.E.; Luben, R.N.; Oakes, S.; Welch, A.; Wareham, N.J. Fruit and vegetable intake and population glycosylated haemoglobin levels: The EPIC-Norfolk Study. Eur. J. Clin. Nutr. 2001, 55, 342. [Google Scholar] [CrossRef] [PubMed]
- Myung, S.-K.; Ju, W.; Cho, B.; Oh, S.-W.; Park, S.M.; Koo, B.-K.; Park, B.-J. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. Br. Med. J. 2013, 346. [Google Scholar] [CrossRef] [PubMed]
- Gale, C.R.; Martyn, C.N.; Winter, P.D.; Cooper, C. Vitamin C and risk of death from stroke and coronary heart disease in cohort of elderly people. BMJ 1995, 310, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Detopoulou, P.; Demopoulos, C.; Karantonis, H.; Antonopoulou, S. Mediterranean diet and its protective mechanisms against cardiovascular disease: An insight into Platelet Activating Factor (PAF) and diet interplay. Ann. Nutr. Disord. Ther. 2015, 2, 1–10. [Google Scholar]
- Stoner, G.D.; Sardo, C.; Apseloff, G.; Mullet, D.; Wargo, W.; Pound, V.; Singh, A.; Sanders, J.; Aziz, R.; Casto, B.; et al. Pharmacokinetics of Anthocyanins and Ellagic Acid in Healthy Volunteers Fed Freeze-Dried Black Raspberries Daily for 7 Days. J. Clin. Pharmacol. 2005, 45, 1153–1164. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studied Food and Components | Type of Study | Results |
|---|---|---|
| PL of red and white wine, musts, grape-skins, and yeast | In vitro studies in WRP and in U937 macrophages, In vivo postprandial dietary interventions studies in humans | Inhibition of PAF-induced platelet aggregation and modulation of PAF metabolism towards reduced PAF levels [141,146,147,148,149,150,151] |
| PL of Fish (sea bass, sea bream, salmon, etc.) | In vitro studies in WRP, and in HMC, Ex vivo studies in hPRP In vivo studies in hyperlipidaemic rabbits | Inhibition of PAF-induced platelet aggregation, modulation of PAF metabolism towards reduced PAF levels, and reduction of the thickness of atherosclerotic lesions in hypercholestrolaemic rabbits [81,103,142,152,153,154,155,156,157,158,159,160], unpublished data for salmon PL |
| PL of olive oil and olive pomace | In vitro studies in WRP and in HMC, In vivo study in hyperlipidaemic rabbits | Inhibition of PAF-induced platelet aggregation and modulation of PAF metabolism towards reduced PAF levels, and reduction of the thickness of atherosclerotic lesions in hypercholestrolaemic rabbits and regression of the already formed atherosclerotic lesions [80,144,145,161] |
| PL of seed oils (soybean, corn, sunflower, and sesame oil) | In vitro studies in WRP | Inhibition of PAF induced platelet aggregation [161] |
| PL of Hen egg | In vitro studies in WRP | Inhibition of PAF-induced platelet aggregation [162] |
| PL of dairy products (milk, yoghurt, cheese, etc.) | In vitro studies in WRP and ex vivo studies in hPRP | Inhibition of PAF-induced platelet aggregation [3,163,164,165] unpublished data for bovine, ovine, and caprine milk, and yogurt and cheese in hPRP |
| Lipid extracts from garlic | Ex vivo studies in hPRP | Inhibition of PAF-induced platelet aggregation and de-aggregation of aggregated platelets [166] |
| Vitamin D and its analogues | In vitro studies in WRP and human leukocytes, ex vivo studies in hPRP and in vivo studies in haemodialysis patients | Inhibition of PAF-induced platelet aggregation and modulation of PAF metabolism towards reduced PAF levels and reduction of the inflammatory milieu (reduced levels of several cytokines) [79] |
| Vitamin E | Ex vivo studies in hPRP and whole blood | Inhibition of PAF-induced platelet aggregation [167,168] |
| Mediterranean-based meals and diets, rich in PL with anti-PAF effects | In vivo studies in humans | Reduction of PAF-induced platelet activity in patients with diabetes-II, metabolic syndrome, and healthy subjects |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsoupras, A.; Lordan, R.; Zabetakis, I. Inflammation, not Cholesterol, Is a Cause of Chronic Disease. Nutrients 2018, 10, 604. https://doi.org/10.3390/nu10050604
Tsoupras A, Lordan R, Zabetakis I. Inflammation, not Cholesterol, Is a Cause of Chronic Disease. Nutrients. 2018; 10(5):604. https://doi.org/10.3390/nu10050604
Chicago/Turabian StyleTsoupras, Alexandros, Ronan Lordan, and Ioannis Zabetakis. 2018. "Inflammation, not Cholesterol, Is a Cause of Chronic Disease" Nutrients 10, no. 5: 604. https://doi.org/10.3390/nu10050604
APA StyleTsoupras, A., Lordan, R., & Zabetakis, I. (2018). Inflammation, not Cholesterol, Is a Cause of Chronic Disease. Nutrients, 10(5), 604. https://doi.org/10.3390/nu10050604