1. Introduction
Nowadays, nanotechnology is a multidisciplinary scientific field that is currently undergoing a large technological development. Nano-enhanced medicines are opening new doors for therapeutic delivery and targeting. These new technological advancements are finding new solutions for using efficiently (as for conventional formulations) a number of bioactive molecules having a poor bioavailability, insolubility or drug instability. This results in an unprecedented multidisciplinary convergence of scientists dedicated to the study of a world so small. Nanotechnology is rapidly becoming an interdisciplinary field since biologists, chemists, physicists and engineers are all involved in the study of the properties of substances at the nanoscale regime. The main reason of interest in this field is because the nanoscale is the first point at which molecules can be assembled.
Amongst the different industrial potentials, nanotechnology also has a high impact on the medical industry. With this kind of technology, patients can be administrated with fluids containing nanorobots or nanoprobes programmed to attack and reconstruct the molecular structure of cancer cells and viruses. There is even speculation that nanorobots could slow or reverse the aging process, and life expectancy could increase significantly. Nanorobots could also be programmed to perform delicate surgeries; such nanosurgeons could work at a level which can be a thousand times more precise than the sharpest scalpel [
1].
Today, the main application of nanotechnology lies in the administration of nanoparticles (NPs) containing bioactive molecules as well as contrasting agent, for biomedical applications. NPs for nanomedicine are made from many different materials such as polymeric, inorganic, lipid, but all with the same purpose, which is the controlled delivery of drug or contrasting agent for diagnosis. In comparison with conventional formulations (e.g., macro-formulations like tablets, or micro-formulations like microspheres, microcapsules etc.), NPs present many advantages. The nanoscale allows better penetration of the colloids through biological tissues, through biological barriers and also through cellular membranes. It therefore follows that the efficiency in bringing the active molecules towards the administration sites is significantly increased; thus, for instance, improving drug absorption. The classical administration routes for the NPs are oral, pulmonary, dermal, rectal and injectable. However, still today, the main challenge of nanomedicines lies in the efficient administration of these NPs. The difficulty is the manipulation of NPs in order to bring them to the absorption sites, e.g., pulmonary alveoli, intestinal barriers, mucous membranes, etc. This is particularly true in the case of pulmonary administration. On the one hand, the size range of nanoparticles is too small to insure their retention in the deep lung: actually, NPs can be inhaled, but they are totally expelled during exhalation. Ideal particle sizes for the pulmonary alveoli administration are between 2 and 5 µm, corresponding to the typical size of microparticles. On the other hand, while the best systems for targeting the pulmonary alveoli are such microparticles, the latter are too big to diffuse and allow an efficient administration of the encapsulated drug as NPs can. The result is a real dilemma. Actually, a solution has been found by combining micro and nanoparticles, namely the encapsulation of nanoparticles in microparticles. The microparticles are brought onto the alveoli, and specifically release the NPs, which can diffuse through the barriers. Such multiscale nano-in-microparticles systems are called Trojan particles. Many different other potential applications of these Trojan systems are reported, and tailored in function for the administration routes, e.g., oral, dermal, intraocular. In the case of pulmonary, Trojan particles overcome an important technical challenge that is to bring and concentrate the NPs onto the specific absorption sites above-mentioned. In the present article, we propose a detailed review of the technologies inherent to the Trojan microparticles, by presenting in the first part, the technical details of their fabrication, processes and formulation methods, and, in the second part, by reviewing the potential of Trojan microparticles in terms of biopharmaceutical aspects and applications.
3. Biopharmaceutical Applications
In order to optimize the pharmacological activity and the therapeutic action, each drug has to be formulated in a particular dosage form with excipients improving the therapeutic effectiveness of the drug. Indeed, the therapeutic action of extra-vascular (pulmonary, oral, dermal, nasal and rectal) administrations is related to drug bioavailability. This is a quantitative and kinetics concept measuring the rate and the amount of a drug to penetrate the different membrane barriers allowing it to reach the target (organs, tissues, receptors). Therefore, the final goal of a biopharmaceutical development is to design a dosage form which can allow drug release “on demand” and to be able to reach specific targets. This approach also contributes to improve the socio-economic and industrial aspects. The biopharmaceutical developments of accelerated, sustained or prolonged drug release from a dosage form can illustrate this approach. Indeed, in this case, these new medicines can optimize the therapeutic effect of the drug and in some cases lead to a second pharmacological application of an already well-known drug. The ideal drug candidates for a biopharmaceutical development comprise drugs with poor solubility, permeability and stability when there are administered by another route rather than the intravascular one. As an example, one can mention (i) polypeptide antibiotics (capreomycin, teicoplanin, vancomycin, conjugated ampicillin) which are very active towards Gram-positive and Gram-negative microorganisms, and allow loco-regional or site-specific delivery; (ii) paclitaxel, 5-fluorouracil, tamoxifen or antitumor agents of natural origin for oral therapy in order to improve the bioavailability or to favor a lymphatic uptake; (iii) nucleic acids, guanosine derivatives, adenosine derivatives (ciclopentiladenosine) or in combination with dopamine for nasal delivery in order to target the brain or antigenic peptides in order to induce an immune response.
To overcome these problems, some strategies consist of modifying the chemical structures of the active compounds. Obviously, the chemical modifications will change the physicochemical characteristics of the drugs and will generate new chemical entities.
More recently, alternative strategies have been suggested which consist of development of new products rather than chemical modification of the molecule. Pharmaceutical technology, for example, uses particular excipients that can mask the chemical structure, allowing the drug to pass unmodified for biological availability. Thus, pharmaceutical technology does not generate new drugs but new dosage forms. The development of an already known active compound formulated in an innovative dosage form makes available a new product at lower costs, revaluating active compounds well established in clinical practice. Technology indeed is able to optimize the biological availability of drugs.
To reach these aims, new raw materials, new dosage forms and technologies are required. From a biopharmaceutical point of view, the ideal size of the basic particle has to be in nanometric scale (as described above) but from a pharmaceutical technology point of view, the handling (flow, electrostatic problems...) of bigger particles is easier. Indeed, particles having a very small size i.e., in the nanoscale, show difficulty in free flow and are prone to electrostatic charges on the surfaces. For these reasons, and easy handling the ideal size of the particles has to be in a micrometric range. To satisfy the biopharmaceutical and technological requirements, the best compromise consists of building hybrid micrometric particles which can encapsulate particles of nanometric range.
Hybrid carriers having nano/micro dimensions, also named as Trojan microparticles, have been studied and developed as new drug products capable of carrying and release of active substances with unfavorable biopharmaceutical properties. Indeed, the use of particulate systems can affect their biological availability and is capable of improving drug bioavailability. These particulate vectors can be incorporated into classical dosage forms for different administration routes.
The main goal of these pharmaceutical nano/microtechnologies is based on the discovery of new particulate carriers for drugs exhibiting critical biopharmaceutical or kinetics properties. The use of new raw materials or their combinations coupled to innovative physical structures and new manufacturing procedures allow reaching this aim. Therefore, the Trojan microparticles technologies are focused on the preparation of hybrid particles containing drugs having low solubility, poor permeability and short stability by means of new materials, mechanisms, structures, technologies and manufacturing procedures. These new strategies for the formulation of drugs have lead to the alteration of pharmacokinetics/pharmacodynamics of active substances by modifying their absorption and distribution by means of rate and site controlled delivery.
From a biopharmaceutical point of view, several advantages can be attributed to these particulate technologies, like: (i) reduction of the dose administered, (ii) increasing the bioavailability of poorly soluble and absorbed drugs, (iii) prolonged action of the drug after one administration, (iv) improvement of the therapeutic index and therefore reducing the toxicity, (v) improvement of the compliance of the dose intake by the patients, (vi) possibility of use of non-invasive routes of administration for drugs having unsuitable biopharmaceutical and/or pharmaockinetics properties for administrations alternative to parenteral route and then to favor the pharmaco-economical aspects of certain drugs administrable only by injectable administrations (vii) reduction of the general exposition of the host to the active agent. Indeed, a lot of active compounds, although they are very potent, are not able to reach an effective blood concentration, due to absorption, distribution, half-life or stability problems.
Two classes of raw materials, lipids and polymers, can be used to build these colloidal particles using nano/microtechnologies based techniques. Besides the choice of the nature of the raw materials, the synthesis protocol is also a key factor to be taken into account since this can drastically modify the particle size, surface charge and the physicochemical characteristics (hydrophilic/hydrophobic). Many biocompatible raw materials have been suggested for this purpose which may include phospholipids, medium- and long-chain-triglyceride lipid-based systems, polylactide and copolymers, polysaccharides such as hyaluronic acids, chitosan, alginate etc. As mentioned above, these particles can be prepared by various methods using preformed polymers or polymerizing a monomer during the preparation process.
The Trojan microparticles, due to their architecture, can improve the mucosal absorption of drugs either by means of prolonged retention and preferred transport at mucosal site or by protection of carrier substance from degradation. The optimizing of the biopharmaceutical properties of drugs by means of particulate carriers is a real challenge leading to innovation.
Puerarin is an isoflavone isolated from a Chinese herbal medicine, Radix Puerariae Lobatae, and has been reported to have a comprehensive pharmacological action in the treatment of diabetes and cardiovascular diseases. Puerarin is a water-soluble drug which has a short half-life in the body. One way to increase its therapeutic efficacy is to prepare nanoparticles of this active compound and then to microencapsulate using a supercritical CO
2 process or to use a co-precipitation process using biodegradable polymers to produce a sustained release effect.
In vitro, Chen
et al. [
10] found that the puerarin nanoparticles were released very quickly through the dialysis bag, and the drug dissolved completely in 6 h. After microencapsulation of puerarin by poly(
L-lactide) (PLLA), the puerarin PLLA microparticles showed a burst of release, and about 60.2% of the puerarin was released in the first 4 h; this result demonstrated that this puerarin fraction can be easily removed, which was similar to the results of the encapsulation efficiency measurements. After that, the microparticles released the drug in a sustained process, about 89.6% of the drug was released within 24 h and was finally completely released in 36 h. After incubation in PBS (pH 6.8) for 24 and 48 h, it was seen that after 24 hours the surface became cleaner and some micropores were observed due to the removal of puerarin nanoparticles while after 48 h more micropores were generated by the continuous removal of puerarin nanoparticles.
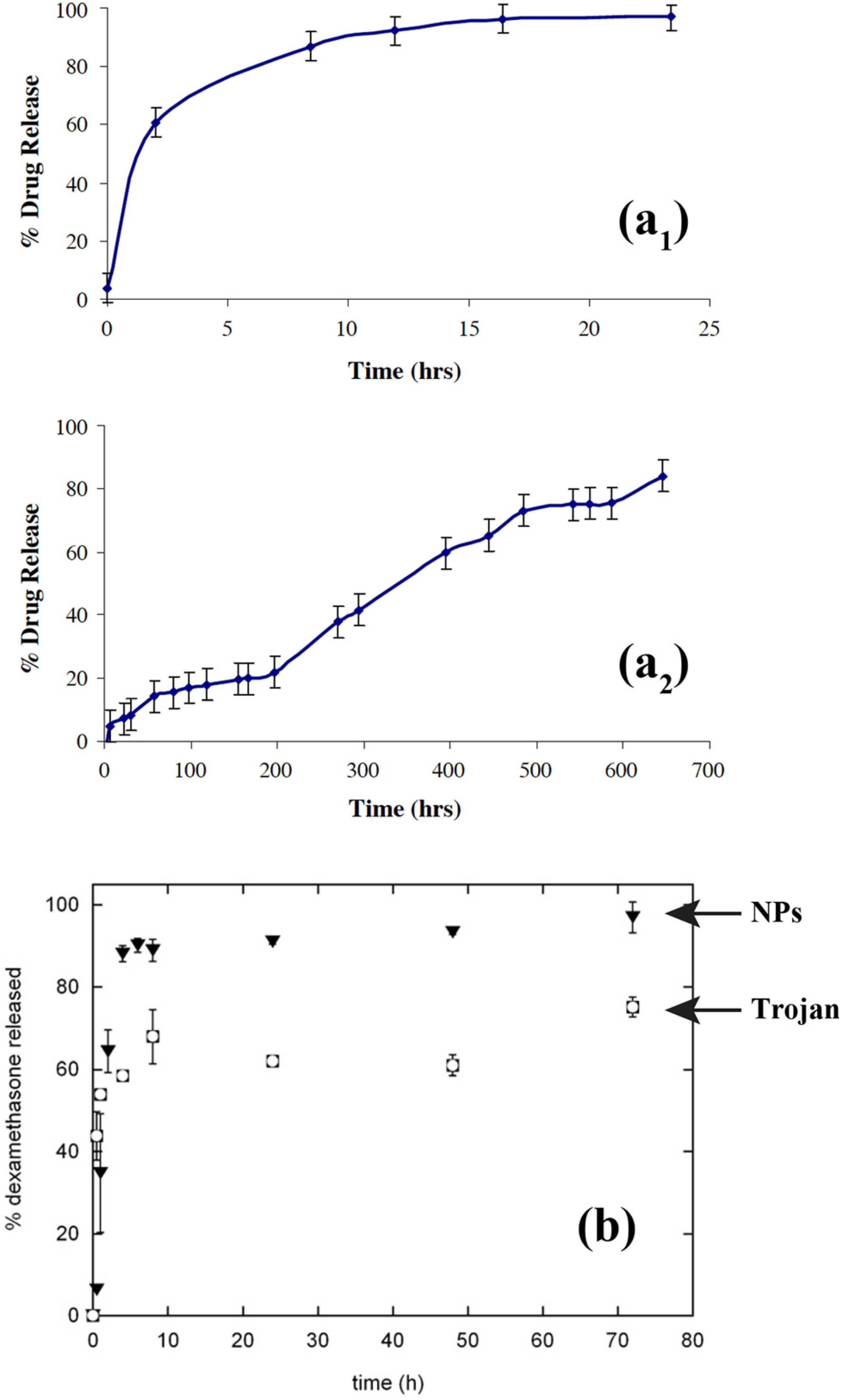
Thote and Gupta also used supercritical carbon dioxide to microencapsulate nanoparticles of dexamethasone phosphate, a hydrophilic drug, in order to sustain its release [
8].
In vitro drug release of dexamethasone phosphate nanoparticles encapsulated in PLGA microparticles was compared to the unprocessed drug particles as provided by the supplier also encapsulated in PLGA microspheres. The
in vitro drug release of the unprocessed drug particles (but microencapsulated) showed an initial burst release of about 5% at time zero, and the entire drug was released within 24 hours. This kinetic release is not advisable for sustained drug release. On the other hand, the
in vitro drug release of the nanoparticles (also microencapsulated) obtained using supercritical antisolvent technique with enhanced mass transfer (SAS-EM) showed continuous release for 700 hours. The release profile showed almost no burst release, however ~15% was released in the first 60 hours followed by a stationary state of about 200 hours, during which only 22% of the encapsulated drug was released. This was followed with continuous increase in the drug release rate up to 500 hours, during which about 75% of the drug was released followed by continued release of the drug over time. From the sustained drug release profile, the authors clearly inferred that the drug was well dispersed in the polymer matrix, which allowed gradual release of the drug with polymer degradation, as opposed to initial burst release observed in the case where the w/o/o/o phase separation technique was used for encapsulation of dexamethasone phosphate. As the complete process was anhydrous, Thote and Gupta suggested that similar sustained release formulations could also be produced for other hydrophilic drugs.
In order to decrease the initial burst release of low molecular weight drugs from formulations including drug-loaded micro- and nanoparticles, Hasan
et al. [
48] prepared composite microparticles by using w/o/w emulsion process. They encapsulated polymeric nanoparticles into polymeric microparticles by using non-water soluble polymers and appropriate organic solvents. They compared the release kinetics with nanoparticles and classical microparticles prepared by the same method. Poly-ε-caprolactone (PCL) dissolved in methylene chloride was used to make nanoparticles, whereas ethylcellulose and Eudragit RS dissolved in ethyl acetate, a non-solvent of poly-ε-caprolactone, were used for the preparation of microparticles. Ibuprofen and triptorelin acetate were chosen as lipophilic and hydrophilic model drugs, respectively. High entrapment efficiencies were obtained with ibuprofen whereas lower amounts of triptorelin acetate were encapsulated, mainly with formulations prepared with poly-ε-caprolactone and Eudragit RS used alone or blended with ethylcellulose. The authors showed that the burst was significantly lower with composite microparticles and may be explained by the slower diffusion of the drugs through the double polymeric wall formed by the nanoparticle matrix followed by another diffusion step through the microparticle polymeric wall.
The solutions of optimized drug releases given by the technology of Trojan microparticles can be extended to the ocular route. Gómez-Gaete
et al. [
7] combined the therapeutic potential of nanoparticles systems with the ease of manipulation of microparticles by developing Trojan particles. The aim of their new delivery vehicle was to be used for intravitreal administration of dexamethasone (DXM). Indeed, the presence of crystalline drug along with the nanoparticles considerably reduces its ability to load the polymeric matrix. However, Gómez
et al. demonstrated that 1.3 mg DXM (=1.44 mg dexamethasone acetate (DXA)) could be encapsulated into 100 mg nanoparticles which was about 6 fold more than what could be obtained by using dexamethasone. Initially, they optimized dexamethasone acetate (DXA) encapsulation into biodegradable poly(
D,
L-lactide-co-glycolide) (PLGA) nanoparticles, then Trojan particles were formulated by spray drying 1,2-Dipalmitoyl-sn-Glycero-3-Phosphocholine (DPPC), hyaluronic acid (HA) and different concentrations of nanoparticle suspensions. The effects of nanoparticles concentration on the physical characteristics of Trojan particles as well as the effect of the spray drying process on nanoparticles size were investigated. Finally, DXA
in vitro release from nanoparticles and Trojan particles was evaluated under sink condition. Although a burst effect can be observed with both systems, the extent of the burst was lower for Trojan particles. SEM and confocal microscopy confirmed that most of Trojan particles were spherical, hollow and possessed an irregular surface due to the presence of nanoparticles. The authors observed that neither Trojan particle tap density nor size distribution was significantly modified as a function of nanoparticles concentration. They mean size distribution of nanoparticles was also found to increase significantly after spray drying. Finally, based on
in vitro release of DXA they showed that the excipient matrix provided protection to encapsulated nanoparticles by slowing down drug release. Gómez
et al. concluded that even if the active principle was released rather quickly as compared with other implants, the
in situ release of drug loaded nanoparticles should favor their internalization within retinal pigment epithelial cells and might therefore increase the drug efficacy for treating retinal affections.
New transmucosal administration routes, like the pulmonary, could also be exploited for systemic absorption of drugs. Edwards
et al. [
51] were the first to show that very light particles (
ρ <~ 0.4 g/cm
3) with d > 5 μm can be deposited in the lungs. As a consequence of their large size and low mass density, porous particles can aerosolize from a DPI more efficiently than smaller nonporous particles, resulting in higher respirable fractions of inhaled therapeutics. Also by virtue of their size, large particles can avoid phagocytic clearance from the lungs until the particles have delivered their therapeutic dose; this attribute can be particularly useful for controlled-release inhalation therapies. To optimize physiologically the kinetic release of the drug incorporated into these Large Porous Particles (LPPs) and to allow producing them easily, Tsapis
et al. [
22] combined the drug release and delivery potential of nanoparticle systems with the ease of flow, processing, and aerosolization potential of Large Porous Particle (LPP) systems by spray drying solutions of polymeric and nonpolymeric NPs into extremely thin-walled macroscale structures. These hybrid LPPs exhibited better flow and aerosolization properties than the NPs; yet, unlike the LPPs, which dissolved in physiological conditions to produce molecular constituents, the hybrid LPPs dissolved to produce NPs, with the drug release and delivery advantages associated with NP delivery systems. Formation of the large porous NP (LPNP) aggregates occurred via a spray-drying process that ensured that the drying time of the sprayed droplet was sufficiently shorter than the characteristic time for redistribution of NPs by diffusion within the drying droplet, implying a local Peclet number much greater than unity. Additional control over LPNPs physical characteristics were achieved by adding other components to the spray-dried solutions, including sugars, lipids, polymers, and proteins. The ability to produce LPNPs appeared to be largely independent of molecular component type as well as the size or chemical nature of the NPs. Hadinoto
et al. [
42,
52] developed similar drug delivery systems based on large hollow nanoparticulate aggregates as therapeutic carrier particles in dry powder inhaler delivery of nanoparticulate drugs. The large hollow carrier particles were manufactured by spray drying of nanoparticulate suspensions of biocompatible acrylic polymer with loaded drugs. The size and concentration of the nanoparticles, as well as the phospholipids inclusion, have been known to influence the resulting morphology (
i.e., size and degree of hollowness) of the spray-dried carrier particles. The effects of the resulting morphology of the carrier particles on the drug release rate were therefore investigated by varying the above three variables. To study these different factors, the authors used aspirin and salbutamol sulfate since these two model drugs have a varying degree of solubility in water. The results indicated that the drug release rate is mostly governed by the degree of hollowness of the carrier particles, and also to a lesser extent by the nanoparticles size, where variation in the drug loading capacity of nanoparticles was observed for different particle size.
Design of appropriate inhaled carriers with adequate aerodynamic properties, drug release, biodegradation and evasion of macrophage uptake is a major challenge for controlled release pulmonary drug delivery [
53,
54]. In order to reach this aim, Al-Qadi
et al. [
55] and El-Sherbiny
et al. [
56] prepared hybrid PEG graft copolymerized onto N-phthaloyl chitosan and chitosan/hyaluronic acid nanoparticles, by ionotropic gelation, and microencapsulated them respectively in sodium alginate and mannitol microspheres, resulting in a dry powder that showed adequate aerodynamic properties for deep pulmonary deposition. Following the encapsulation process, structural analysis of the dry powder was analyzed by confocal laser scanning microscopy, which elucidated that the nanoparticles were homogeneously distributed within the mannitol microsphere. The evidence that nanoparticles were completely encapsulated within the carrier by means of the spray drying process was achieved by application of the sensitive surface analysis techniques, X-ray photoelectron spectroscopy and in combination with time-of-flight secondary ion mass spectroscopy. These outcomes confirmed the success of nanoparticles microencapsulation by spray drying and the authors concluded that the developed delivery system holds great potential for lung delivery of macromolecules. Grenha
et al. [
43,
57] concluded similarly by studying chitosan nanoparticle-loaded mannitol microspheres. The mannitol microspheres contained chitosan/tripolyphosphate nanoparticles and were also prepared by spray-drying. These microspheres were proposed as valuable candidates to transport therapeutic protein-loaded nanoparticles to the lungs owing to their favorable aerodynamic properties. Similar formulations were developed by other researchers in order to open the way for new drug-targeting strategies using nanoparticles for proteins [
58] and pulmonary delivery of drugs and diagnostics. Indeed, some of them [
59] developed a platform for aerosol delivery of nanoparticles. In this case, lactose was used as the excipient and spray-dried with two different types of nanoparticles: gelatin and polybutylcyanoacrylate nanoparticles. The results of these studies showed that some carrier particles were hollow while others had a continuous matrix. Gelatin nanoparticles were incorporated throughout the matrix and sometimes accumulated at one end of the lactose. Polycyanoacrylate nanoparticles mostly clustered in different spots within the lactose carriers. The mean sizes of both nanoparticle types were characterized at two different times: one just before they were spray-dried and other after they were redissolved from the spray-dried powders. Both nanoparticle types remained in the nano-range size after spray-drying. The mean nanoparticle sizes were increased by approximately 30% after spray-drying, though this increase was statistically significant only for the gelatin nanoparticles. Dispersion of the powder with an in-house passive dry powder inhaler and subsequent cascade impaction measurements showed that incorporation of the nanoparticles did not affect the fine particle fraction (FPF) or mass median aerodynamic diameter (MMAD) of the powders. FPF was approximately 40% while MMAD was 3.0 ± 0.2 μm, indicating these formulations yielded aerosols of a suitable particle size for efficient lung delivery of nanoparticles. As for the other delivery systems intended for this administration route, the researchers demonstrated that nanoparticles can be delivered to the lungs via carrier particles that dissolve after coming in contact with the aqueous environment of the lung epithelium.
Besides the specific uses of Trojan microspheres for pulmonary delivery, the particles have also found applications for oral administration, to reach desirable sustained release profiles. El-Sherbiny
et al. [
60] developed a series of nano/micro hydrogel matrices for oral delivery of silymarin. This drug was chosen as a model of hydrophobic natural therapeutics. Constituted of sodium alginate-based pH responsive hydrogel microspheres encapsulating poly(
D,
L-lactic-co-glycolic acid) (PLGA) nanoparticles (NPs), this new design of particles allowed enhancing the dissolution and oral bioavailability of this model drug via formulation in polymeric matrices. From these studies, the authors showed that alginate-based hydrogel microparticles incorporating silymarin-loaded PLGA NPs are pH-sensitive. Also, the obtained data revealed a considerable effect of the alginate content and the drying method onto the characteristics of the prepared alginate-PLGA particles.
In the field of gene delivery by oral route, Bhavsar
et al. [
49,
61] investigated the development and the evaluation of a novel nanoparticles-in-microsphere oral system (NiMOS) for gene delivery and transfection in specific regions of the gastrointestinal (GI) tract. To achieve this, plasmid DNA, encoding either for β-galactosidase (CMV-βgal) or enhanced green fluorescent protein (EFGP-N1), was encapsulated in type B gelatin nanoparticles. NiMOS were prepared by further protecting the DNA-loaded nanoparticles in a poly(epsilon-caprolactone) (PCL) matrix to form microspheres of less than 5.0 μm in diameter. In order to evaluate the biodistribution following oral administration, radiolabeled (
111In-labeled) gelatin nanoparticles and NiMOS were administered orally to fasted Wistar rats. The results of biodistribution studies showed that, while gelatin nanoparticles traversed through the GI tract rather quickly with more than 85% of the administered dose per gram localizing in the large intestine within the first hour, NiMOS resided in the stomach and small intestine for relatively longer duration. Following oral administration of CMV-βgal or EFGP-N1 plasmid DNA at 100 μg dose in the control and test formulations, the qualitative results presented in this study provide the proof-of-concept for the transfection capability of NiMOS upon oral administration. After 5 days post-administration, the authors observed transgene expression in the small and large intestine of rats. Based on these preliminary results, NiMOS showed significant potential as novel gene delivery vehicle for therapeutic and vaccination purposes.
Several other peculiar applications have been investigated for the Trojan microparticles. These ones comprise the possibility to incorporate iron oxide. In this context, superparamagnetic iron oxide nanoparticles embedded chitosan microspheres were developed for magnetic resonance (MR)-traceable embolotherapy with high resolution and sensitivity through magnetic resonance imaging (MRI) [
62]. Superparamagnetic iron oxide nanoparticles loaded chitosan microspheres were prepared by emulsion and cross-linking technique and 100–200 μm sized spherical microsparticles were obtained. Within 30 days, about 60% of the incorporated superparamagnetic iron oxide nanoparticles were released from low cross-linked microspheres, whereas only about 40% of superparamagnetic iron oxide nanoparticles were released from highly cross-linked microspheres. Highly cross-linked microspheres were more efficient for lower degree of swelling leading to secure entrapment of superparamagnetic iron oxide nanoparticles in matrix.
Drug-loaded microspheres using the proof-of-concept of Trojan particles, a kind of target-orientation drug, were also investigated by several researchers. Drug delivery systems constituted of a magnetic polymeric carrier, composed of biodegradable poly(D,L-lactide) microspheres, maghemite nanoparticles and anti-cancer drug (paclitaxel) was successfully prepared in dichloromethane using highspeed homogenization [
63]. Maghemite nanoparticles were well dispersed in poly(
D,
L-lactide) matrix. The carrier was magnetically responsive and release of loaded paclitaxel was enhanced by applying an oscillating magnetic field. The thermal energy generated by maghemite nanoparticles due to magnetic hysteresis loss was very low and had a negligible effect in influencing the release behavior. The authors showed that alternating movement of the nanoparticles, stimulated by magnetic force, resulting in deterioration of the mechanical properties of polymer matrix was likely to be the main reason for the enhancement in drug release. Similar systems containing paclitaxel or interferon alpha-2b were respectively formulated by Cui
et al. [
64] and Zhou
et al. [
65].
In an anecdotic way, one can also mention the use of hybrid particles for the enzyme immobilization. Sadjadi
et al. [
66] investigated the assembly of the silver nanoparticles on the surface of the amine-functionalized zeolite microspheres or formation of zeolitesilver nanoparticle “core-shell” structure and thereafter, using this nanosystem in immobilization of fungal protease. The assembly of silver nanoparticles on zeolite surface occurs through the amine groups present in 3-aminopropyltrimethoxysilane. The fungal proteases bound to the massive “core-shell” structures can be easily separated from the reaction medium by mild centrifugation and exhibited excellent reuse characteristics. The biocatalytic activity of fungal protease in the bioconjugate was marginally enhanced relative to the free enzyme in solution.
Using the technology of the encapsulation of nanoparticles into microspheres, Lee
et al. [
67] reported the antibacterial properties of magnetically directed microparticles containing Ag nanoparticle loaded multilayers. Original development of Trojan microspheres was realized in tissue-regeneration matrix and drug delivery systems. Park
et al. [
68] applied these biomaterials as novel cell supporting matrix for stem cell delivery. They devised a novel method for the fabrication of nanostructured 3D scaffolds. The growth factor loaded heparin/poly(
L-lysine) nanoparticles were physically attached on the positively charged surface of PLGA microspheres precoated with low molecular weight poly(ethyleneimmine) via a layer-by-layer system. Growth factor loaded heparin/poly(
L-lysine) nanoparticles, which were simply produced as polyion complex micelles with diameters of 50–150 nm, were fabricated in the first step. Microsphere matrix (size, 20~80 nm) containing TGF-β 3 showed a significantly higher number of specific lacunae phenotypes at the end of the 4 week study
in vitro culture of mesenchymal stem cells. Thus, the researchers concluded that growth factor delivery of polylactide-co-glycolide microsphere can be used to engineer synthetic extracellular matrix. This polymeric microsphere matrix containing TGF-
β 3 showed promise as coatings for implantable biomedical devices to improve biocompatibility and ensure
in vivo performance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
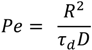
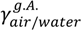 = (71.35 ± 0.01) mN·m−1 and
= (71.35 ± 0.01) mN·m−1 and 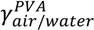 = (42.67 ± 0.10) mN·m−1. This result also explains the presence of “wire-like” objects formed during the droplet formation, which was due to an interfacial phenomenon. Here the surface tension measurements were performed using a drop tensiometer (Teclis, Longessaigne, France).
= (42.67 ± 0.10) mN·m−1. This result also explains the presence of “wire-like” objects formed during the droplet formation, which was due to an interfacial phenomenon. Here the surface tension measurements were performed using a drop tensiometer (Teclis, Longessaigne, France).



