Antiviral Properties of Chemical Inhibitors of Cellular Anti-Apoptotic Bcl-2 Proteins

 , , ,
, , ,  , , ,
, , ,  , ,
, ,  add
Show full author list
add
Show full author list
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Viruses
2.3. Cells
2.4. Compound Toxicity and Efficacy Assays
2.5. Scoring Drug Response Profiles
2.6. Virus Titration
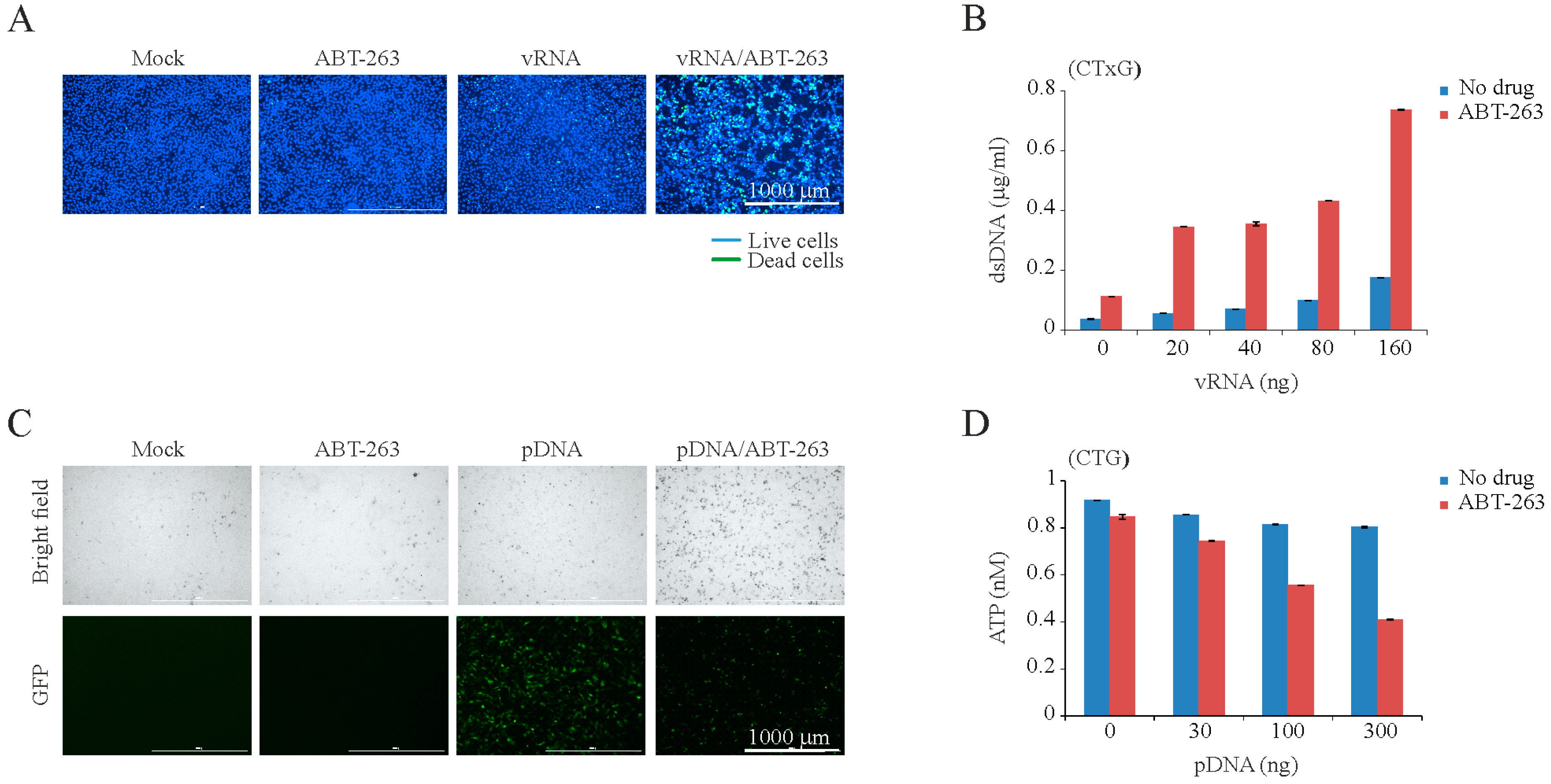
2.7. Transfections of RPE Cells with vRNA or Plasmid DNA
2.8. Live Microscopy of Semliki Forest Virus Infection During Bcl-2-Inhibition
2.9. Dynamic BH3 Peptide Profiling
2.10. Metabolomics
2.11. Immuno-Precipitation and Mass-Spectrometry
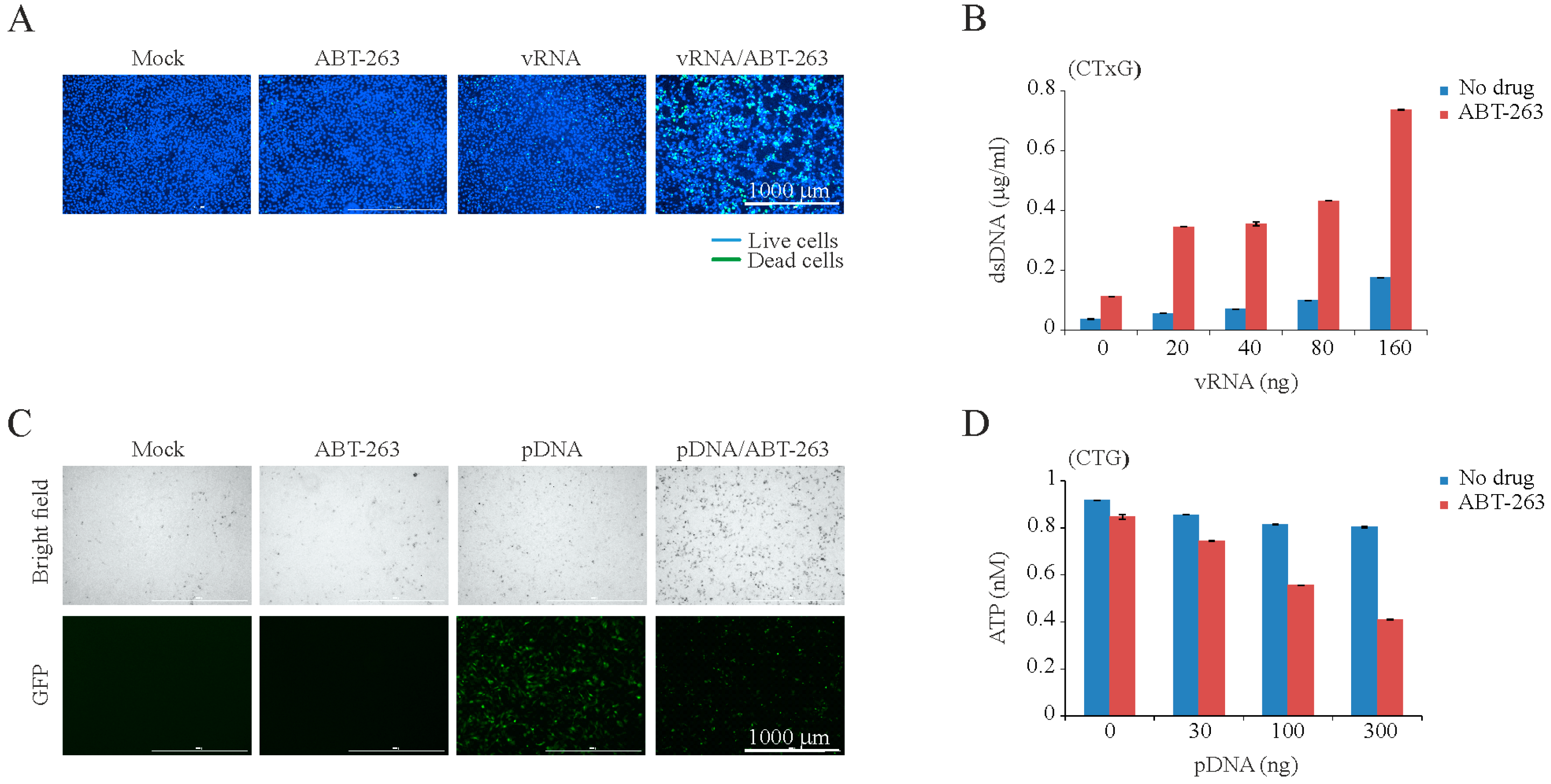
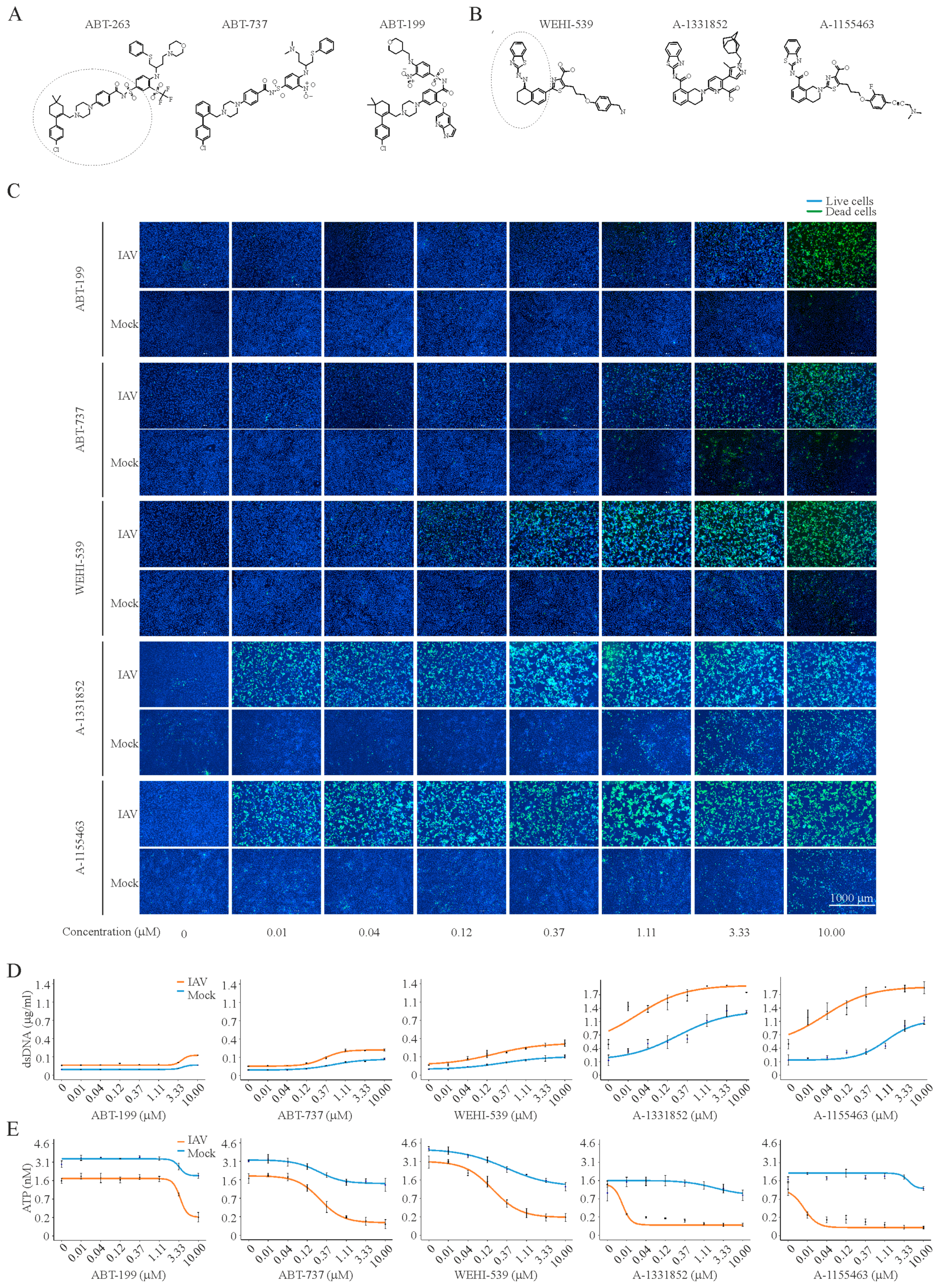
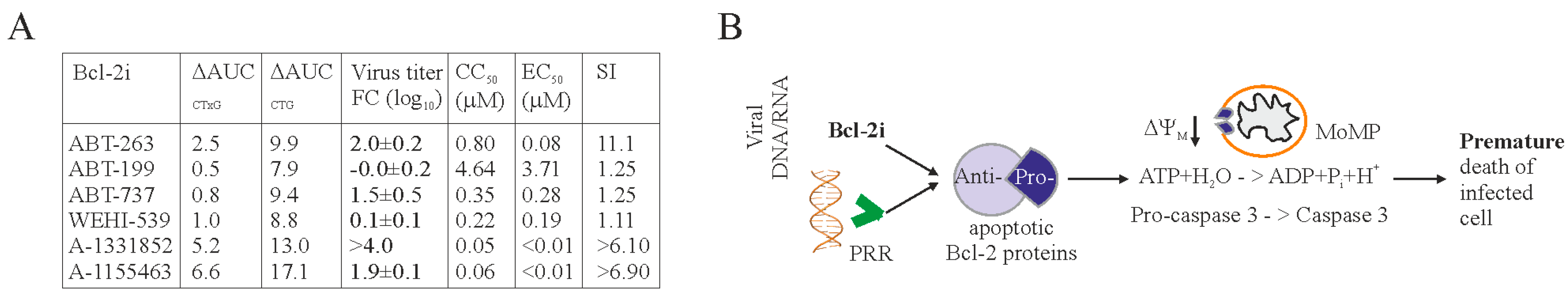
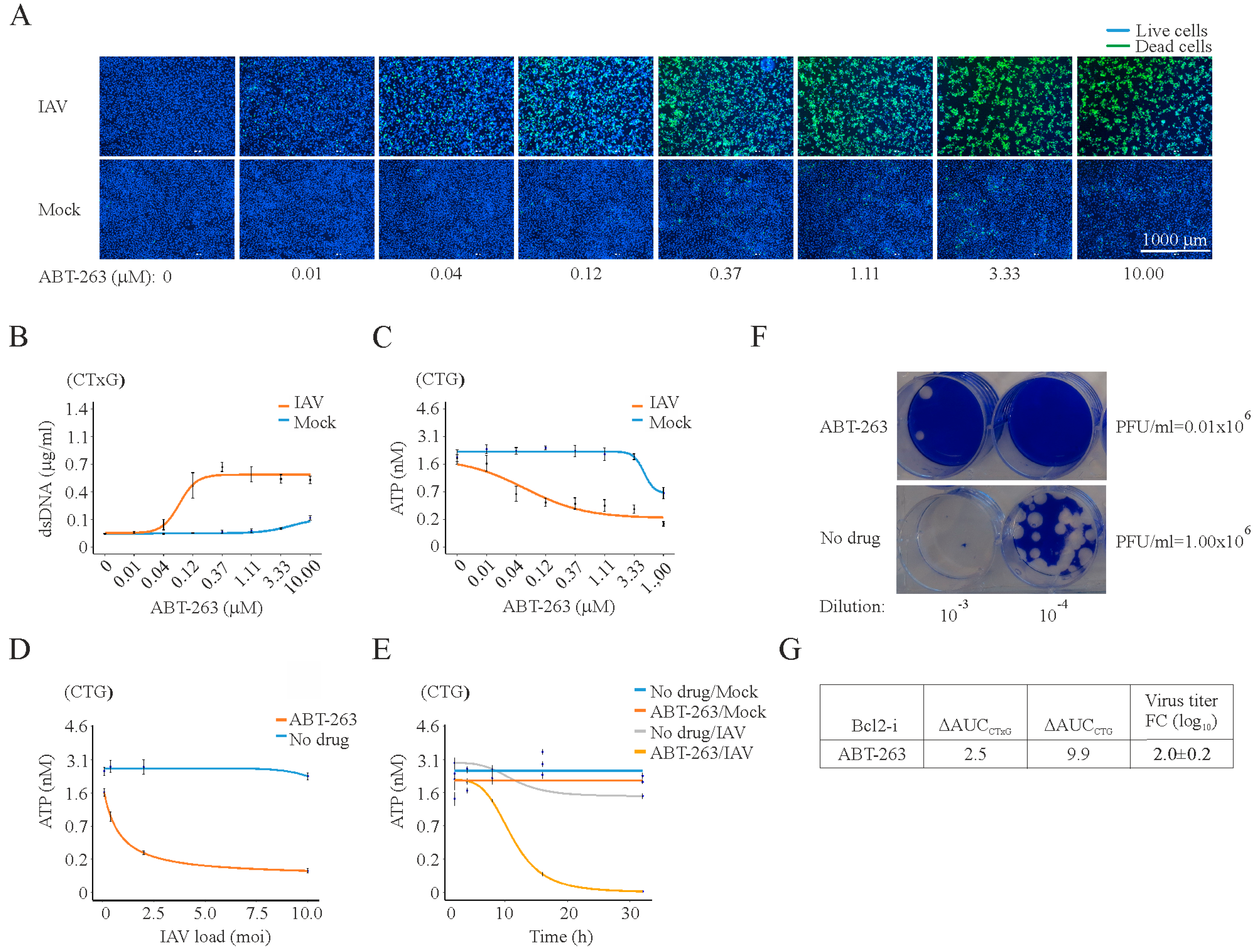
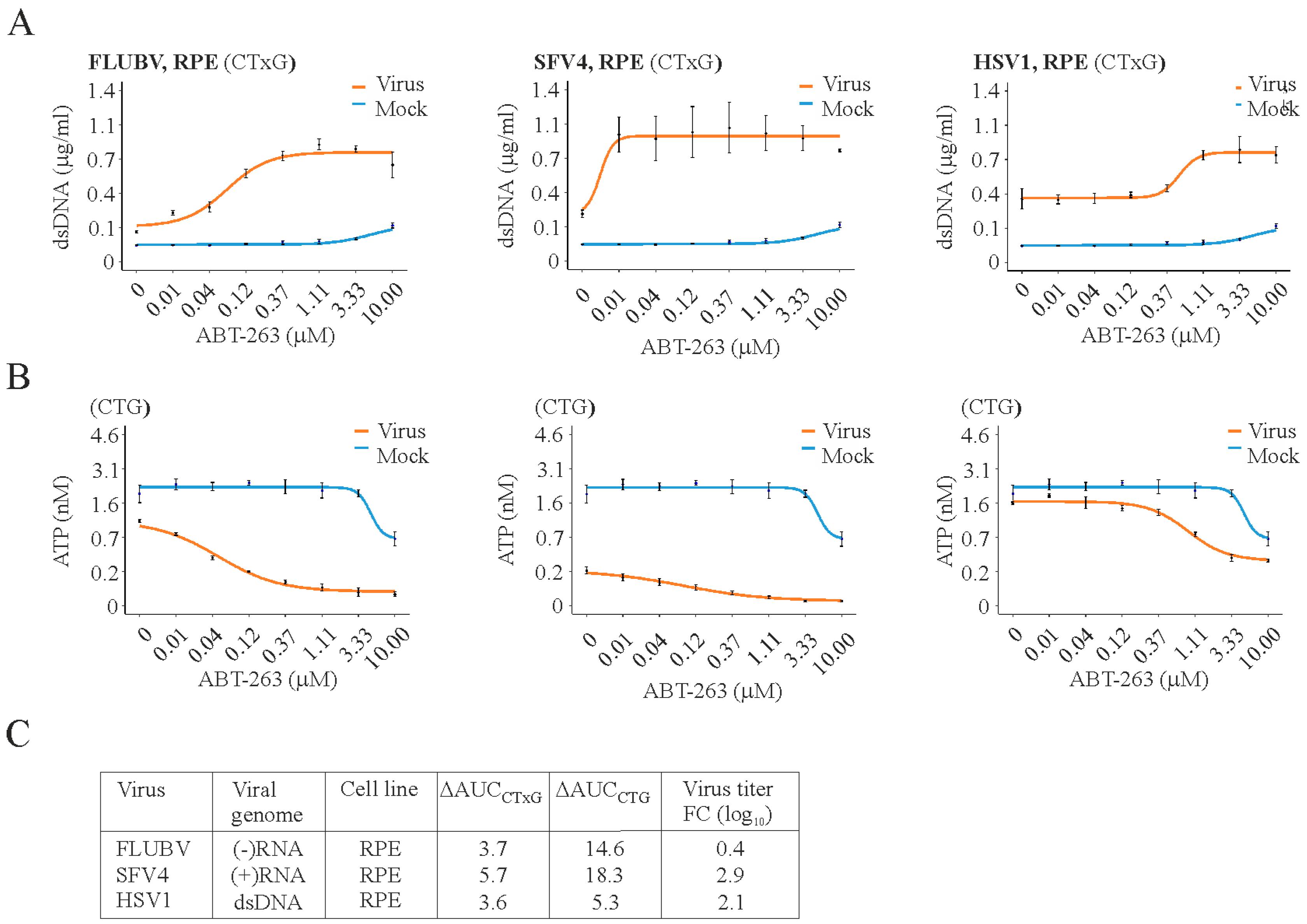
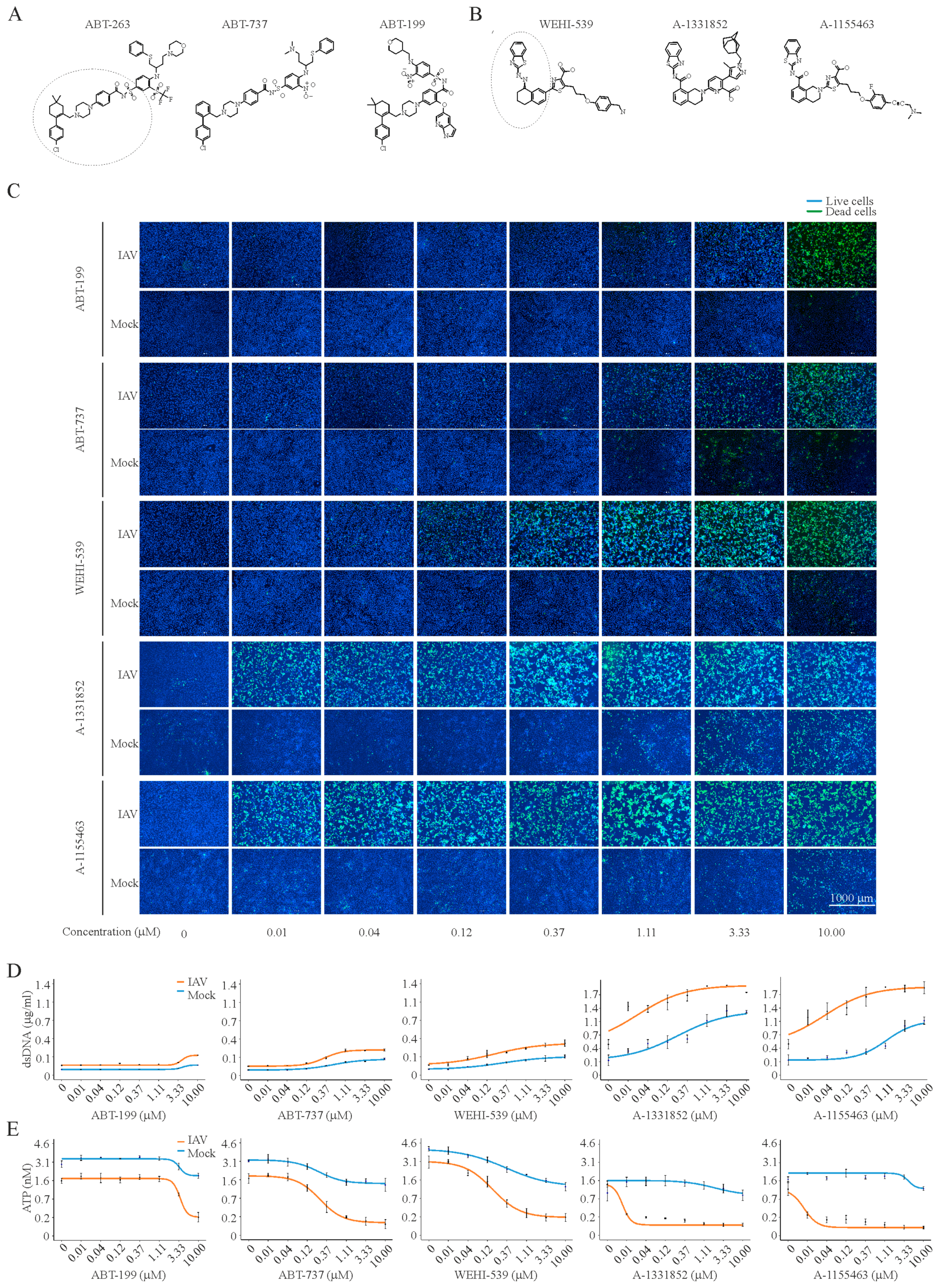
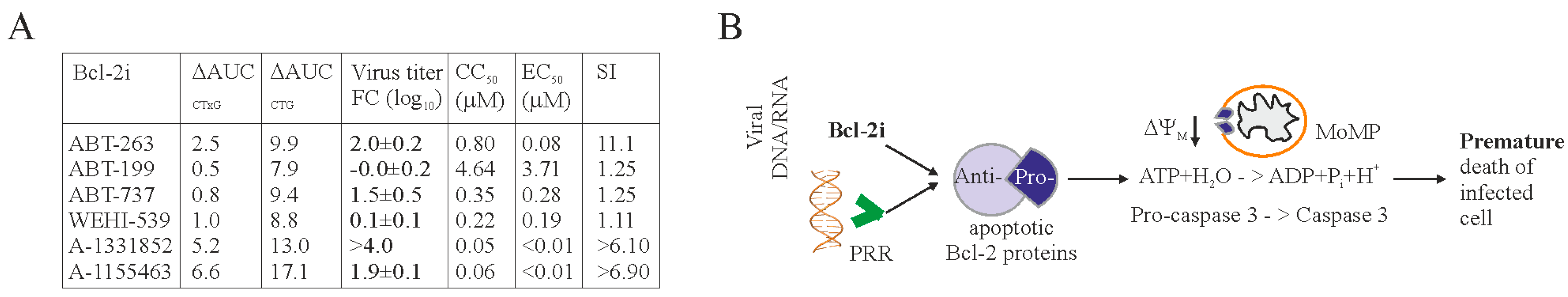
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Global, R. National life expectancy. All-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar]
- Vos, T.; Barber, R.M.; Bell, B.; Bertozzi-Villa, A.; Biryukov, S.; Bolliger, I.; Charlson, F.; Davis, A.; Degenhardt, L.; Dicker, D. Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 743–800. [Google Scholar]
- WHO. Available online: http://www.who.int/medicines/ebola-treatment/WHO-list-of-top-emerging-diseases/en/ (accessed on 1 September 2017).
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.M.; Kim, J.; Tenson, T.; Min, J.-Y.; Kainov, D.E. Influenza virus infection, interferon response, viral counter-response, and apoptosis. Viruses 2017, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Ebert, G.; Allison, C.; Preston, S.; Cooney, J.; Toe, J.G.; Stutz, M.D.; Ojaimi, S.; Baschuk, N.; Nachbur, U.; Torresi, J.; et al. Eliminating hepatitis B by antagonizing cellular inhibitors of apoptosis. Proc. Natl. Acad. Sci. USA 2015, 112, 5803–5808. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.T.; Cortens, J.P.; Du, Q.; Wilkins, J.A.; Coombs, K.M. Influenza virus induces apoptosis via BAD-mediated mitochondrial dysregulation. J. Virol. 2013, 87, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.E.; Datan, E.; Matassov, D.; Zakeri, Z.F. Lack of Bax prevents influenza A virus-induced apoptosis and causes diminished viral replication. J. Virol. 2009, 83, 8233–8246. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, V.S.; Olsen, C.W.; Dybdahl-Sissoko, N.; Evans, D. Apoptosis: A mechanism of cell killing by influenza A and B viruses. J. Virol. 1994, 68, 3667–3673. [Google Scholar] [PubMed]
- Kakkola, L.; Denisova, O.V.; Tynell, J.; Viiliainen, J.; Ysenbaert, T.; Matos, R.C.; Nagaraj, A.; Ohman, T.; Kuivanen, S.; Paavilainen, H.; et al. Anticancer compound ABT-263 accelerates apoptosis in virus-infected cells and imbalances cytokine production and lowers survival rates of infected mice. Cell Death Dis. 2013, 4, e742. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Sainski, A.M.; Dai, H.; Natesampillai, S.; Pang, Y.P.; Bren, G.D.; de Araujo Correia, M.C.; Sampath, R.; Rizza, S.A.; O'Brien, D.; et al. Prime, shock, and kill: Priming CD4 T cells from HIV patients with a BCL-2 antagonist before HIV reactivation reduces HIV reservoir size. J. Virol. 2016, 90, 4032–4048. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.; Beljanski, V.; van Grevenynghe, J.; Richards, S.; Ben Yebdri, F.; He, Z.; Nichols, C.; Belgnaoui, S.M.; Steel, C.; Goulet, M.L.; et al. BCL-2 inhibitors sensitize therapy-resistant chronic lymphocytic leukemia cells to VSV oncolysis. Mol. Ther. 2013, 21, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Sainski-Nguyen, A.M.; Natesampillai, S.; Aboulnasr, F.; Kaufmann, S.; Badley, A.D. Maintenance of the HIV reservoir is antagonized by selective BCL-2 inhibition. J. Virol. 2017, 91, e00012-17. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, C.J.; Cory, S. ABT-199, a new Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood 2013, 121, 2285–2288. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Huang, D. Targeting BCL-2 with BH3 mimetics: Basic science and clinical application of venetoclax in chronic lymphocytic leukemia and related B cell malignancies. Clin. Pharmacol. Ther. 2017, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.F.; Czabotar, P.E.; Smith, B.J.; Deshayes, K.; Zobel, K.; Colman, P.M.; Fairlie, W.D. Crystal structure of ABT-737 complexed with Bcl-xL: Implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 2007, 14, 1711–1713. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Hinds, M.G. The Bcl-2 family: Structures, interactions and targets for drug discovery. Apoptosis 2015, 20, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Lessene, G.; Czabotar, P.E.; Sleebs, B.E.; Zobel, K.; Lowes, K.N.; Adams, J.M.; Baell, J.B.; Colman, P.M.; Deshayes, K.; Fairbrother, W.J.; et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.F.; Hasvold, L.; Wang, L.; Wang, X.; Petros, A.M.; Park, C.H.; Boghaert, E.R.; Catron, N.D.; Chen, J.; Colman, P.M.; et al. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med. Chem. Lett. 2014, 5, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra40. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, S.; Jaunbergs, J.; Roth, M.G.; Ferguson, D.A.; de Brabander, J.K. Evaluating the potential of vacuolar ATPase inhibitors as anticancer agents and multigram synthesis of the potent salicylihalamide analog saliphenylhalamide. Bioorg. Med. Chem. Lett. 2008, 18, 5879–5883. [Google Scholar] [CrossRef] [PubMed]
- Lietzen, N.; Ohman, T.; Rintahaka, J.; Julkunen, I.; Aittokallio, T.; Matikainen, S.; Nyman, T.A. Quantitative subcellular proteome and secretome profiling of influenza A virus-infected human primary macrophages. PLoS Pathog. 2011, 7, e1001340. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Dauber, B.; Melen, K.; Julkunen, I.; Wolff, T. Analysis of influenza B Virus NS1 protein trafficking reveals a novel interaction with nuclear speckle domains. J. Virol. 2009, 83, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Ruokola, P.; Dadu, E.; Kazmertsuk, A.; Hakkanen, H.; Marjomaki, V.; Ihalainen, J.A. Raman spectroscopic signatures of echovirus 1 uncoating. J. Virol. 2014, 88, 8504–8513. [Google Scholar] [CrossRef] [PubMed]
- Smura, T.; Kakkola, L.; Blomqvist, S.; Klemola, P.; Parsons, A.; Kallio-Kokko, H.; Savolainen-Kopra, C.; Kainov, D.E.; Roivainen, M. Molecular evolution and epidemiology of echovirus 6 in Finland. Infect. Genet. Evol. 2013, 16, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Nygardas, M.; Paavilainen, H.; Muther, N.; Nagel, C.H.; Roytta, M.; Sodeik, B.; Hukkanen, V. A herpes simplex virus-derived replicative vector expressing LIF limits experimental demyelinating disease and modulates autoimmunity. PLoS ONE 2013, 8, e64200. [Google Scholar] [CrossRef] [PubMed]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jaaskelainen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Vaha-Koskela, M.J.; Tuittila, M.T.; Nygardas, P.T.; Nyman, J.K.; Ehrengruber, M.U.; Renggli, M.; Hinkkanen, A.E. A novel neurotropic expression vector based on the avirulent A7(74) strain of Semliki Forest virus. J. Neurovirol. 2003, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Park, W.J.; Park, S.; Kim, S.; Windisch, M.P.; Ryu, W.S. The FDA-approved drug irbesartan inhibits HBV-infection in HepG2 cells stably expressing sodium taurocholate co-transporting polypeptide. Antivir. Ther. 2015, 20, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Chang, S.Y.; Sung, M.; Park, J.H.; Bin Kim, H.; Lee, H.; Choi, J.P.; Choi, W.S.; Min, J.Y. Extensive viable middle east respiratory syndrome (MERS) coronavirus contamination in air and surrounding environment in MERS isolation wards. Clin. Infect. Dis. 2016, 63, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.M. The relationship between variable selection and data augmentation and a method for prediction. Technometrics 1976, 16, 125–127. [Google Scholar] [CrossRef]
- Saint-Marcoux, F.; Royer, B.; Debord, J.; Larosa, F.; Legrand, F.; Deconinck, E.; Kantelip, J.P.; Marquet, P. Pharmacokinetic modelling and development of Bayesian estimators for therapeutic drug monitoring of mycophenolate mofetil in reduced-intensity haematopoietic stem cell transplantation. Clin. Pharmacokinet. 2009, 48, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Prasad, S.; Singer, D.R.; MacAllister, R.J. Ageing is associated with impairment of nitric oxide and prostanoid dilator pathways in the human forearm. Clin. Sci. 2002, 102, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.A.; Skoner, D.P. The relationship between airway hyperreactivity (AHR) and sodium, potassium adenosine triphosphatase (Na+, K+ ATPase) enzyme inhibition. J. Allergy Clin. Immunol. 1997, 99, 367–373. [Google Scholar] [CrossRef]
- Muller, K.H.; Kainov, D.E.; El Bakkouri, K.; Saelens, X.; de Brabander, J.K.; Kittel, C.; Samm, E.; Muller, C.P. The proton translocation domain of cellular vacuolar ATPase provides a target for the treatment of influenza A virus infections. Br. J. Pharmacol. 2011, 164, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Biacchesi, S.; Skiadopoulos, M.H.; Yang, L.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Rapid human metapneumovirus microneutralization assay based on green fluorescent protein expression. J. Virol. Methods 2005, 128, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.; Letai, A. BH3 profiling in whole cells by fluorimeter or FACS. Methods 2013, 61, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Gaelings, L.; Soderholm, S.; Bugai, A.; Fu, Y.; Nandania, J.; Schepens, B.; Lorey, M.B.; Tynell, J.; Vande Ginste, L.; Le Goffic, R.; et al. Regulation of kynurenine biosynthesis during influenza virus infection. FEBS J. 2017, 284, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- MSEA. MetaboAnalyst 3.0—A Comprehensive Tool Suite for Metabolic Data Analysis. Available online: www.msea.ca (accessed on 1 September 2017).
- Ohman, T.; Lietzen, N.; Valimaki, E.; Melchjorsen, J.; Matikainen, S.; Nyman, T.A. Cytosolic RNA recognition pathway activates 14–3-3 protein mediated signaling and caspase-dependent disruption of cytokeratin network in human keratinocytes. J. Proteome Res. 2010, 9, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Anastasina, M.; Le May, N.; Bugai, A.; Fu, Y.; Soderholm, S.; Gaelings, L.; Ohman, T.; Tynell, J.; Kyttanen, S.; Barboric, M.; et al. Influenza virus NS1 protein binds cellular DNA to block transcription of antiviral genes. Biochim. Biophys. Acta 2016, 1859, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Denisova, O.V.; Kakkola, L.; Feng, L.; Stenman, J.; Nagaraj, A.; Lampe, J.; Yadav, B.; Aittokallio, T.; Kaukinen, P.; Ahola, T.; et al. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza a virus infection. J. Biol. Chem. 2012, 287, 35324–35332. [Google Scholar] [CrossRef] [PubMed]
- Kuivanen, S.; Bespalov, M.M.; Nandania, J.; Ianevski, A.; Velagapudi, V.; De Brabander, J.K.; Kainov, D.E.; Vapalahti, O. Obatoclax, saliphenylhalamide and gemcitabine inhibit Zika virus infection in vitro and differentially affect cellular signaling, transcription and metabolism. Antivir. Res. 2017, 139, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.L.; Steele, C. Innate receptors and cellular defense against pulmonary infections. J. Immunol. 2014, 193, 3842–3850. [Google Scholar] [CrossRef] [PubMed]
- Mansour, D.E.; El-Shazly, A.A.; Elawamry, A.I.; Ismail, A.T. Comparison of ocular findings in patients with H1N1 influenza infection versus patients receiving influenza vaccine during a pandemic. Ophthalmic Res. 2012, 48, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Geiler, J.; Klassert, D.; Doerr, H.W.; Cinatl, J. Infection of human retinal pigment epithelial cells with influenza A viruses. Invest. Ophthalmol. Vis. Sci. 2009, 50, 5419–5425. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Gaelings, L.; Soderholm, S.; Belanov, S.; Nandania, J.; Nyman, T.A.; Matikainen, S.; Anders, S.; Velagapudi, V.; Kainov, D.E. JNJ872 inhibits influenza A virus replication without altering cellular antiviral responses. Antivir. Res. 2016, 133, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.D.; Mansell, A.; Tate, M.D. Hero turned villain: NLRP3 inflammasome-induced inflammation during influenza A virus infection. J. Leukoc. Biol. 2017, 101, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Ludwig, S.; Pleschka, S.; Wolff, T. Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J. Leukoc. Biol. 2012, 92, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, T.; Vijayalingam, S.; Kuppuswamy, M.; Chinnadurai, G. Interaction of cellular proteins with BCL-xL targeted to cytoplasmic inclusion bodies in adenovirus infected cells. Virology 2015, 483, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Blaho, J.A. Cellular players in the herpes simplex virus dependent apoptosis balancing act. Viruses 2009, 1, 965–978. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulanova, D.; Ianevski, A.; Bugai, A.; Akimov, Y.; Kuivanen, S.; Paavilainen, H.; Kakkola, L.; Nandania, J.; Turunen, L.; Ohman, T.; et al. Antiviral Properties of Chemical Inhibitors of Cellular Anti-Apoptotic Bcl-2 Proteins. Viruses 2017, 9, 271. https://doi.org/10.3390/v9100271
Bulanova D, Ianevski A, Bugai A, Akimov Y, Kuivanen S, Paavilainen H, Kakkola L, Nandania J, Turunen L, Ohman T, et al. Antiviral Properties of Chemical Inhibitors of Cellular Anti-Apoptotic Bcl-2 Proteins. Viruses. 2017; 9(10):271. https://doi.org/10.3390/v9100271
Chicago/Turabian StyleBulanova, Daria, Aleksandr Ianevski, Andrii Bugai, Yevhen Akimov, Suvi Kuivanen, Henrik Paavilainen, Laura Kakkola, Jatin Nandania, Laura Turunen, Tiina Ohman, and et al. 2017. "Antiviral Properties of Chemical Inhibitors of Cellular Anti-Apoptotic Bcl-2 Proteins" Viruses 9, no. 10: 271. https://doi.org/10.3390/v9100271