
Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus



{kind=link}
{kind=link}
Abstract
:1. Introduction
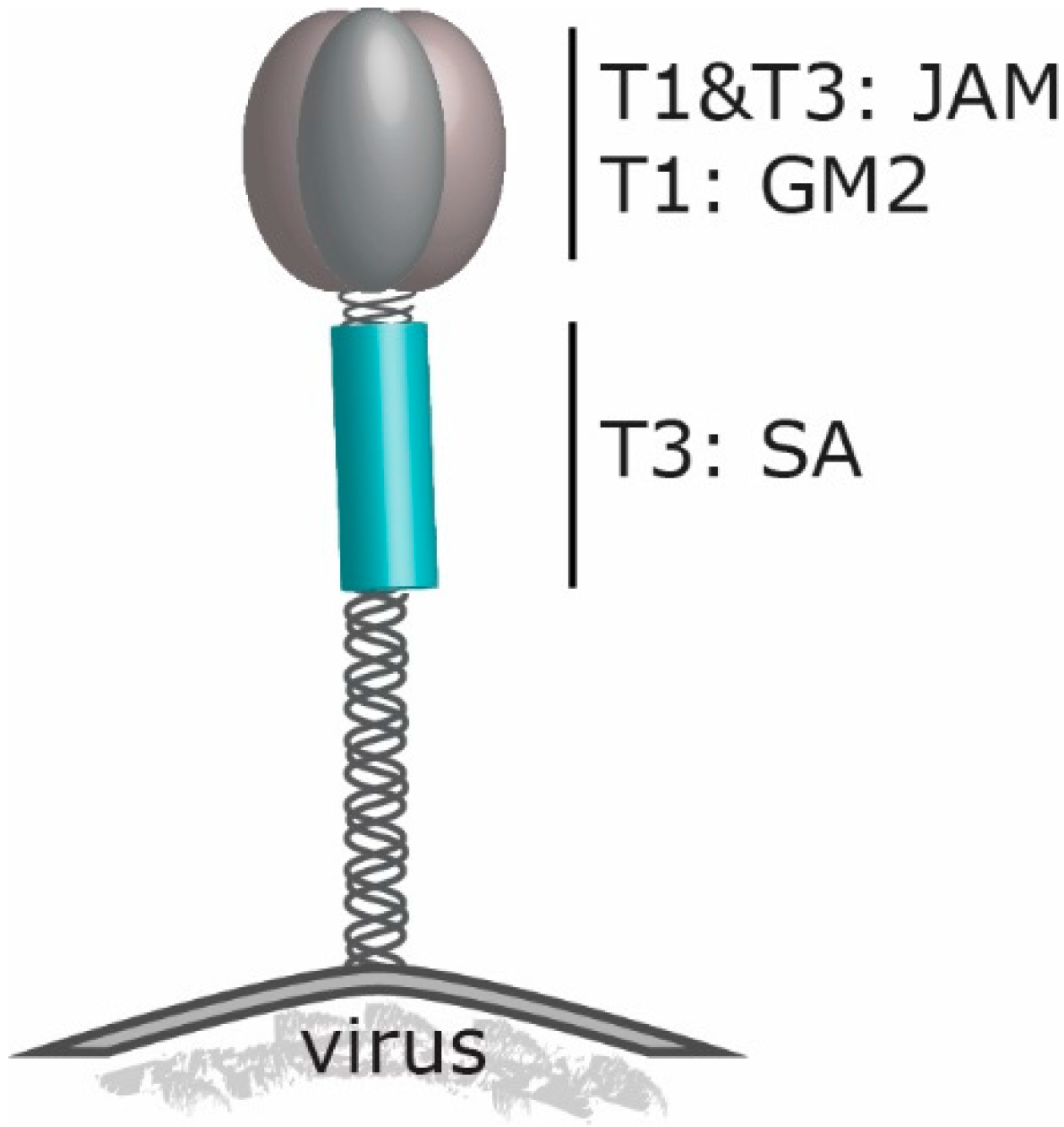
2. Reovirus’ Engagement to Cell Surface Molecules

3. Reovirus and the Great Escape: What Pathways Are Involved in Reovirus Induced Cell Death
4. Reovirus and Its Relation with Neutralizing Antibodies
5. Reovirus and Immune Stimulation
6. Genetic Modification of Reovirus
7. Reovirus and Animal Models
8. Future Directions
Author Contributions
Conflicts of Interest
References
- Eisenstein, S.; Chen, S.H.; Pan, P.Y. Immune cells: More than simple carriers for systemic delivery of oncolytic viruses. Oncolytic. Virother. 2014, 3, 83–91. [Google Scholar] [PubMed]
- Bell, J.; McFadden, G. Viruses for tumor therapy. Cell Host Microbe. 2014, 15, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Hammill, A.M.; Conner, J.; Cripe, T.P. Oncolytic virotherapy reaches adolescence. Pediatric Blood Cancer 2010, 55, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Hashiro, G.; Loh, P.C.; Yau, J.T. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch. Virol. 1977, 54, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Clements, D.; Helson, E.; Gujar, S. Reovirus in cancer therapy: An evidence-based review. Oncolytic. Virother. 2014, 3, 69–82. [Google Scholar] [CrossRef]
- Chakrabarty, R.; Tran, H.; Selvaggi, G.; Hagerman, A.; Thompson, B.; Coffey, M. The oncolytic virus, pelareorep, as a novel anticancer agent: A review. Invest. New Drugs 2015, 33, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L.; Squier, M.K.; Rodgers, S.E.; Schneider, B.E.; Oberhaus, S.M.; Grdina, T.A.; Cohen, J.J.; Dermody, T.S. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J. Virol. 1995, 69, 6972–6979. [Google Scholar] [PubMed]
- Chappell, J.D.; Duong, J.L.; Wright, B.W.; Dermody, T.S. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J. Virol. 2000, 74, 8472–8479. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Danthi, P. Determinants of strain-specific differences in efficiency of reovirus entry. J. Virol. 2010, 84, 12723–12732. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L.; Fields, B.N. Neutralization of reovirus: The gene responsible for the neutralization antigen. J. Exp. Med. 1977, 146, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L.; Ramig, R.F.; Mustoe, T.A.; Fields, B.N. Identification of the gene coding for the hemagglutinin of reovirus. Virology 1978, 86, 581–584. [Google Scholar] [CrossRef]
- Gong, J.; Mita, M.M. Activated ras signaling pathways and reovirus oncolysis: An update on the mechanism of preferential reovirus replication in cancer cells. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Shmulevitz, M.; Lee, P.W. Connecting reovirus oncolysis and ras signaling. Cell Cycle 2005, 4, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Shmulevitz, M.; Pan, D.; Stoltz, D.; Lee, P.W.K. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. 2007, 15, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.L.; Hirasawa, K.; Yang, A.D.; Shields, M.A.; Lee, P.W. Reovirus oncolysis: The Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 11099–11104. [Google Scholar] [CrossRef] [PubMed]
- Smakman, N.; van Den Wollenberg, D.J.M.; Borel Rinkes, I.H.M.; Hoeben, R.C.; Kranenburg, O. Sensitization to apoptosis underlies KrasD12-dependent oncolysis of murine C26 colorectal carcinoma cells by reovirus T3D. J. Virol. 2005, 79, 14981–14985. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Egan, C.; Alain, T.; Urbanski, S.J.; Lee, P.W.; Forsyth, P.A.; Johnston, R.N. Acquired resistance to reoviral oncolysis in ras-transformed fibrosarcoma cells. Oncogene 2007, 26, 4124–4134. [Google Scholar] [CrossRef] [PubMed]
- Alain, T.; Kim, T.S.; Lun, X.; Liacini, A.; Schiff, L.A.; Senger, D.L.; Forsyth, P.A. Proteolytic disassembly is a critical determinant for reovirus oncolysis. Mol. Ther. 2007, 15, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.A.; Gromeier, M. Oncolytic viruses for cancer therapy I. Cell-external factors: Virus entry and receptor interaction. Oncol. Res. Treat. 2005, 28, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P.; Holm, G.H.; Stehle, T.; Dermody, T.S. Reovirus receptors, cell entry, and proapoptotic signaling. Adv. Exp. Med. Biol. 2013, 790, 42–71. [Google Scholar] [PubMed]
- Danthi, P.; Kobayashi, T.; Holm, G.H.; Hansberger, M.W.; Abel, T.W.; Dermody, T.S. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J. Virol. 2008, 82, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, K.M.; Johnson, E.M.; Stehle, T.; Dermody, T.S. Attachment and cell entry of mammalian orthoreovirus. Curr. Top. Microbiol. Immunol. 2006, 309, 1–38. [Google Scholar] [PubMed]
- Morchang, A.; Panaampon, J.; Suttitheptumrong, A.; Yasamut, U.; Noisakran, S.; Yenchitsomanus, P.T.; Limjindaporn, T. Role of cathepsin B in dengue virus-mediated apoptosis. Biochem. Biophys. Res. Commun. 2013, 438, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Di Piazza, M.; Mader, C.; Geletneky, K.; Herrero y Calle, M.; Weber, E.; Schlehofer, J.; Deleu, L.; Rommelaere, J. Cytosolic activation of cathepsins mediates parvovirus h-1-induced killing of cisplatin and trail-resistant glioma cells. J. Virol. 2007, 81, 4186–4198. [Google Scholar] [CrossRef] [PubMed]
- McGuire, K.A.; Barlan, A.U.; Griffin, T.M.; Wiethoff, C.M. Adenovirus type 5 rupture of lysosomes leads to cathepsin B-dependent mitochondrial stress and production of reactive oxygen species. J. Virol. 2011, 85, 10806–10813. [Google Scholar] [CrossRef] [PubMed]
- Van Den Wollenberg, D.J.M.; Dautzenberg, I.J.C.; Van Den Hengel, S.K.; Cramer, S.J.; De Groot, R.J.; Hoeben, R.C. Isolation of reovirus T3D mutants capable of infecting human tumor cells independent of junction adhesion molecule-A. PLoS ONE 2012, 7, e48064. [Google Scholar] [CrossRef] [PubMed]
- Van Den Hengel, S.K.; Balvers, R.K.; Dautzenberg, I.J.; Van Den Wollenberg, D.J.; Kloezeman, J.J.; Lamfers, M.L.; Sillivis-Smit, P.A.; Hoeben, R.C. Heterogeneous reovirus susceptibility in human glioblastoma stem-like cell cultures. Cancer Gene Ther. 2013, 20, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.A.; Schelling, P.; Wetzel, J.D.; Johnson, E.M.; Forrest, J.C.; Wilson, G.A.; Aurrand-Lions, M.; Imhof, B.A.; Stehle, T.; Dermody, T.S. Junctional adhesion molecule A serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J. Virol. 2005, 79, 7967–7978. [Google Scholar] [CrossRef] [PubMed]
- Stettner, E.; Dietrich, M.H.; Reiss, K.; Dermody, T.S.; Stehle, T. Structure of serotype 1 reovirus attachment protein sigma1 in complex with junctional adhesion molecule a reveals a conserved serotype-independent binding epitope. J. Virol. 2015, 89, 6136–6140. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, E.; Guglielmi, K.M.; Strauss, H.M.; Dermody, T.S.; Stehle, T. Structure of reovirus σ1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog. 2008, 4, e1000235. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.; Coffin, R.S.; Anderson, P.N. The Nogo receptor, its ligands and axonal regeneration in the spinal cord; a review. J. Neurocytol. 2002, 31, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Konopka-Anstadt, J.L.; Mainou, B.A.; Sutherland, D.M.; Sekine, Y.; Strittmatter, S.M.; Dermody, T.S. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell. Host Microbe. 2014, 15, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Q.; Cheng, C.Y. A seamless trespass: Germ cell migration across the seminiferous epithelium during spermatogenesis. J. Cell Biol. 2007, 178, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Pesavento, P.A.; Stokol, T.; Liu, H.; van der List, D.A.; Gaffney, P.M.; Parker, J.S. Distribution of the feline calicivirus receptor junctional adhesion molecule a in feline tissues. Vet. Pathol. 2011, 48, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Bhella, D. The role of cellular adhesion molecules in virus attachment and entry. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Dautzenberg, I.J.; van Den Wollenberg, D.J.; van Den Hengel, S.K.; Limpens, R.W.; Barcena, M.; Koster, A.J.; Hoeben, R.C. Mammalian orthoreovirus T3D infects U-118 MG cell spheroids independent of junction adhesion molecule-a. Gene Ther. 2014, 21, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.D.; Joyce, J.A. Proteolytic networks in cancer. Trends Cell Biol. 2011, 21, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Kallunki, T.; Olsen, O.D.; Jaattela, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Barton, E.S.; Connolly, J.L.; Forrest, J.C.; Chappell, J.D.; Dermody, T.S. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 2001, 276, 2200–2211. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Stencel, J.E.; Liu, Y.; Blaum, B.S.; Reiter, D.M.; Feizi, T.; Dermody, T.S.; Stehle, T. The GM2 glycan serves as a functional coreceptor for serotype 1 reovirus. PLoS Pathog. 2012, 8, e1003078. [Google Scholar] [CrossRef] [PubMed]
- Stencel-Baerenwald, J.; Reiss, K.; Blaum, B.S.; Colvin, D.; Li, X.N.; Abel, T.; Boyd, K.; Stehle, T.; Dermody, T.S. Glycan engagement dictates hydrocephalus induction by serotype 1 reovirus. MBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Reiter, D.M.; Frierson, J.M.; Halvorson, E.E.; Kobayashi, T.; Dermody, T.S.; Stehle, T. Crystal structure of reovirus attachment protein σ1 in complex with sialylated oligosaccharides. PLoS Pathog. 2011, 7, e1002166. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.A.; Sidman, R.L.; Fields, B.N. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc. Natl. Acad. Sci. USA 1991, 88, 3852–3856. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L.; Powers, M.L.; Fields, B.N. Absolute linkage of virulence and central nervous system cell tropism of reoviruses to viral hemagglutinin. J. Infect. Dis. 1980, 141, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.A.; Knipe, D.M.; Fischbach, G.D.; Fields, B.N. Type 3 reovirus neuroinvasion after intramuscular inoculation: Direct invasion of nerve terminals and age-dependent pathogenesis. Virology 2002, 303, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Van den Wollenberg, D.J.; Dautzenberg, I.J.; Ros, W.; Lipinska, A.D.; van den Hengel, S.K.; Hoeben, R.C. Replicating reoviruses with a transgene replacing the codons for the head domain of the viral spike. Gene Ther. 2015, 22, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P.; Pruijssers, A.J.; Berger, A.K.; Holm, G.H.; Zinkel, S.S.; Dermody, T.S. Bid regulates the pathogenesis of neurotropic reovirus. PLoS Pathog. 2010, 6, e1000980. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.L.; Dermody, T.S. Virion disassembly is required for apoptosis induced by reovirus. J. Virol. 2002, 76, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, J.J.; Dermody, T.S.; Holm, G.H. Apoptosis induced by mammalian reovirus is β interferon (IFN) independent and enhanced by IFN regulatory factor 3- and NF-λB-dependent expression of Noxa. J.Virol. 2012, 86, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Pan, L.Z.; Hill, R.; Marcato, P.; Shmulevitz, M.; Vassilev, L.T.; Lee, P.W. Stabilisation of p53 enhances reovirus-induced apoptosis and virus spread through p53-dependent NF-λB activation. Br. J. Cancer 2011, 105, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, D.J.; Bickel, R.J.; Tyler, K.L. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J. Virol. 2002, 76, 11414–11424. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.L.; Rodgers, S.E.; Clarke, P.; Ballard, D.W.; Kerr, L.D.; Tyler, K.L.; Dermody, T.S. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J. Virol. 2000, 74, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, G.J.; Garofalo, M.; Valeri, N.; Roulstone, V.; Volinia, S.; Cohn, D.E.; Phelps, M.; Harrington, K.J.; Vile, R.; Melcher, A.; et al. Reovirus-associated reduction of microRNA-let-7d is related to the increased apoptotic death of cancer cells in clinical samples. Modern Pathol. 2012, 25, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.; Meintzer, S.M.; Gibson, S.; Widmann, C.; Garrington, T.P.; Johnson, G.L.; Tyler, K.L. Reovirus-induced apoptosis is mediated by trail. J. Virol. 2000, 74, 8135–8139. [Google Scholar] [CrossRef] [PubMed]
- Garant, K.A.; Shmulevitz, M.; Pan, L.; Daigle, R.M.; Ahn, D.G.; Gujar, S.A.; Lee, P.W. Oncolytic reovirus induces intracellular redistribution of Ras to promote apoptosis and progeny virus release. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.L.; Barton, E.S.; Dermody, T.S. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J. Virol. 2001, 75, 4029–4039. [Google Scholar] [CrossRef] [PubMed]
- Coffey, C.M.; Sheh, A.; Kim, I.S.; Chandran, K.; Nibert, M.L.; Parker, J.S. Reovirus outer capsid protein micro1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J. Virol. 2006, 80, 8422–8438. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, M.L.; Werner, B.G.; Hom, L.G.; Anguish, L.J.; Coffey, C.M.; Parker, J.S. Reovirus infection or ectopic expression of outer capsid protein micro1 induces apoptosis independently of the cellular proapoptotic proteins Bax and Bak. J. Virol. 2011, 85, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Hammer, K.; Tollefson, W.C.; Konopka-Anstadt, J.L.; Kobayashi, T.; Dermody, T.S. Nonstructural protein sigma1s mediates reovirus-induced cell cycle arrest and apoptosis. J. Virol. 2013, 87, 12967–12979. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.K.; Danthi, P. Reovirus activates a caspase-independent cell death pathway. MBio 2013, 4, e00178–e00213. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus modulates autophagy during oncolysis of multiple myeloma. Autophagy 2013, 9, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin. Cancer Res. 2012, 18, 4962–4972. [Google Scholar] [CrossRef] [PubMed]
- Hiller, B.E.; Berger, A.K.; Danthi, P. Viral gene expression potentiates reovirus-induced necrosis. Virology 2015, 484, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Christofferson, D.E.; Yuan, J.Y. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Levine, B. Viruses and autophagy. Rev. Med. Virol. 2009, 19, 359–378. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.I.; Huang, W.R.; Lai, I.H.; Cheng, C.Y.; Liu, H.J. The p17 nonstructural protein of avian reovirus triggers autophagy enhancing virus replication via activation of phosphatase and tensin deleted on chromosome 10 (PTEN) and AMP-activated protein kinase (AMPK), as well as dsRNA-dependent protein kinase (PKR)/EIF2α signaling pathways. J. Biol. Chem. 2013, 288, 3571–3584. [Google Scholar] [PubMed]
- Huang, W.R.; Chiu, H.C.; Liao, T.L.; Chuang, K.P.; Shih, W.L.; Liu, H.J. Avian reovirus protein p17 functions as a nucleoporin tpr suppressor leading to activation of p53, p21 and PTEN and inactivation of P13K/AKT/mTOR and ERK signaling pathways. PLoS ONE 2015, 10, e0133699. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Jiang, K.; Zhang, X.; Zhang, M.; Zhou, Z.; Hu, M.; Yang, R.; Sun, C.; Wu, Y. Avian reovirus triggers autophagy in primary chicken fibroblast cells and vero cells to promote virus production. Arch. Virol. 2012, 157, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wang, Z.; Tao, L.; Wang, Y. Er stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010, 6, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Roulstone, V.; Pedersen, M.; Kyula, J.; Mansfield, D.; Khan, A.A.; McEntee, G.; Wilkinson, M.; Karapanagiotou, E.; Coffey, M.; Marais, R.; et al. BRAF- and MEK-targeted small molecule inhibitors exert enhanced antimelanoma effects in combination with oncolytic reovirus through ER stress. Mol. Ther. 2015, 23, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Kelly, K.R.; Coffey, M.; Freeman, J.W.; Nawrocki, S.T. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Espitia, C.M.; Mahalingam, D.; Oyajobi, B.O.; Coffey, M.; Giles, F.J.; Carew, J.S.; Nawrocki, S.T. Reovirus therapy stimulates endoplasmic reticular stress, Noxa induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene 2012, 31, 3023–3038. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.L.; Coffey, M.C.; Hirasawa, K.; Demetrick, D.J.; Nishikawa, S.G.; DiFrancesco, L.M.; Strong, J.E.; Lee, P.W. Reovirus oncolysis of human breast cancer. Hum. Gene Ther. 2002, 13, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, K.; Nishikawa, S.G.; Norman, K.L.; Alain, T.; Kossakowska, A.; Lee, P.W.K. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002, 62, 1696–1701. [Google Scholar] [PubMed]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Oncolytic reovirus effectively targets breast cancer stem cells. Mol. Ther. 2009, 17, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Lech, P.J.; Pappoe, R.; Nakamura, T.; Tobin, G.J.; Nara, P.L.; Russell, S.J. Antibody neutralization of retargeted measles viruses. Virology 2014, 454, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Calcedo, R.; Franco, J.; Qin, Q.; Richardson, D.W.; Mason, J.B.; Boyd, S.; Wilson, J.M. Preexisting neutralizing antibodies to adeno-associated virus capsids in large animals other than monkeys may confound in vivo gene therapy studies. Hum. Gene Ther. Methods 2015, 26, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.R.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus serotype 5 neutralizing antibodies target both hexon and fiber following vaccination and natural infection. J. Virol. 2012, 86, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Selb, B.; Weber, B. A study of human reovirus IgG and IgA antibodies by elisa and western blot. J. Virol. Methods 1994, 47, 15–25. [Google Scholar] [CrossRef]
- Minuk, G.Y.; Paul, R.W.; Lee, P.W. The prevalence of antibodies to reovirus type 3 in adults with idiopathic cholestatic liver disease. J. Med. Virol. 1985, 16, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Adair, R.A.; Roulstone, V.; Scott, K.J.; Morgan, R.; Nuovo, G.J.; Fuller, M.; Beirne, D.; West, E.J.; Jennings, V.A.; Rose, A.; et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.J.; Prestwich, R.J.; Kottke, T.; Errington, F.; Thompson, J.M.; Harrington, K.J.; Pandha, H.S.; Coffey, M.; Selby, P.J.; Vile, R.G.; et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009, 16, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.; Kottke, T.; Donnelly, O.; Thompson, J.; Willmon, C.; Diaz, R.; Zaidi, S.; Coffey, M.; Selby, P.; Harrington, K.; et al. Cytokine conditioning enhances systemic delivery and therapy of an oncolytic virus. Mol. Ther. 2014, 22, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Khalil, D.N.; Budhu, S.; Gasmi, B.; Zappasodi, R.; Hirschhorn-Cymerman, D.; Plitt, T.; de Henau, O.; Zamarin, D.; Holmgaard, R.B.; Murphy, J.T.; et al. The new era of cancer immunotherapy: Manipulating T-cell activity to overcome malignancy. Adv. Cancer Res. 2015, 128, 1–68. [Google Scholar] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef] [PubMed]
- Devaud, C.; John, L.B.; Westwood, J.A.; Darcy, P.K.; Kershaw, M.H. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory t cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, S.P.; Restifo, N.P. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012, 72, 3125–3130. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Ilett, E.J.; Errington, F.; Diaz, R.M.; Steele, L.P.; Kottke, T.; Thompson, J.; Galivo, F.; Harrington, K.J.; Pandha, H.S.; et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin. Cancer Res. 2009, 15, 4374–4381. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Lee, P.W. Oncolytic virus-mediated reversal of impaired tumor antigen presentation. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of oncovexgm-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. First oncolytic virus edges towards approval in surprise vote. Nat. Biotech. 2015, 33, 569–570. [Google Scholar] [CrossRef] [PubMed]
- The angeles clinic and research institute. Available online: http://www.theangelesclinic.org (accessed on 18 December 2015).
- Egen, J.G.; Kuhns, M.S.; Allison, J.P. CTLA-4: New insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 2002, 3, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Bauzon, M.; Hermiston, T. Armed therapeutic viruses—A disruptive therapy on the horizon of cancer immunotherapy. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Rajani, K.; Parrish, C.; Shim, K.; Ilett, L.; Thompson, J.; Kottke, T.; Pulido, J.; Errington-Mais, F.; Selby, P.; Pandha, H.; et al. Combination therapy with reovirus and PD-1 blockade effectively establishes tumor control via innate and adaptive immune responses. In Proceedings of AACR Tumor Immunology and Immunotherapy Conference, Orlando, FL, USA, 2014.
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Clements, D.R.; Sterea, A.M.; Kim, Y.; Helson, E.; Dean, C.A.; Nunokawa, A.; Coyle, K.M.; Sharif, T.; Marcato, P.; Gujar, S.A.; et al. Newly recruited cd11b+, gr-1+, ly6chigh myeloid cells augment tumor-associated immunosuppression immediately following the therapeutic administration of oncolytic reovirus. J. Immunol. 2015, 194, 4397–4412. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, L.; Peranzoni, E.; Ugel, S.; Marigo, I.; Fernandez Gomez, A.; Mesa, C.; Geilich, M.; Winkels, G.; Traggiai, E.; Casati, A. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by gm-csf. Eur. J. Immunol. 2010, 40, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.K.; Kmieciak, M.; Knutson, K.L.; Bear, H.D.; Manjili, M.H. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1-bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res. Trans. 2010, 123, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.; Sampath, P.; Hou, W.; Thorne, S.H. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin. Cancer Res. 2015, 21. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Antar, A.A.R.; Boehme, K.W.; Danthi, P.; Eby, E.A.; Guglielmi, K.M.; Holm, G.H.; Johnson, E.M.; Maginnis, M.S.; Naik, S.; et al. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 2007, 1, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, R.M.; Lahti, L.; Boehme, K.W.; Ikizler, M.; Doyle, J.D.; Dermody, T.S.; Schiff, L.A. Genetic determinants of reovirus pathogenesis in a murine model of respiratory infection. J. Virol. 2013, 87, 9279–9289. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; Hengel, H.; Abel, T.W.; Dermody, T.S. Apoptosis induction influences reovirus replication and virulence in newborn mice. J. Virol. 2013, 87, 12980–12989. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Danthi, P. The μ1 72–96 loop controls conformational transitions during reovirus cell entry. J. Virol. 2013, 87, 13532–13542. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Johnston, R.N.; Shmulevitz, M. Potential for improving potency and specificity of reovirus oncolysis with next-generation reovirus variants. Viruses 2015, 7, 6251–6278. [Google Scholar] [CrossRef] [PubMed]
- MacNeill, A. On the potential of oncolytic virotherapy for the treatment of canine cancers. Oncolytic. Virother. 2015, 4, 95–107. [Google Scholar]
- van Den Wollenberg, D.J.M.; van Den Hengel, S.K.; Dautzenberg, I.J.C.; Cramer, S.J.; Kranenburg, O.; Hoeben, R.C. A strategy for genetic modification of the spike-encoding segment of human reovirus T3D for reovirus targeting. Gene Ther. 2008, 15, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- Brochu-Lafontaine, V.; Lemay, G. Addition of exogenous polypeptides on the mammalian reovirus outer capsid using reverse genetics. J. Virol. Methods 2012, 179, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, A.A.; Blattman, J.N.; Blattman, N.N.; Greenberg, P.D.; Nibert, M.L. Engineering recombinant reoviruses with tandem repeats and a tetravirus 2A-like element for exogenous polypeptide expression. Proc. Natl. Acad. Sci. USA 2013, 110, E1867–E1876. [Google Scholar] [CrossRef] [PubMed]
- Roner, M.R.; Joklik, W.K. Reovirus reverse genetics: Incorporation of the cat gene into the reovirus genome. Proc. Natl. Acad. Sci. USA 2001, 98, 8036–8041. [Google Scholar] [CrossRef] [PubMed]
- Kapikian, A.Z.; Shope, R.E. Rotaviruses, reoviruses, coltiviruses, and orbiviruses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; The University of Texas Medical Branch at Galveston: Galveston TX, Mexico, 1996. [Google Scholar]
- Montufar-Solis, D.; Klein, J.R. Experimental intestinal reovirus infection of mice: What we know, what we need to know. Immunol. Res. 2005, 33, 257–265. [Google Scholar] [CrossRef]
- Major, A.S.; Rubin, D.H.; Cuff, C.F. Mucosal immunity to reovirus infection. In Reoviruses II; Tyler, K., Oldstone, M.A., Eds.; Springer-Verlag Berlin Heidelberg: Heidelberg, Germany, 1998; Volume 233/2, pp. 163–177. [Google Scholar]
- Tyler, K.L. Pathogenesis of reovirus infections of the central nervous system. In Reoviruses II; Tyler, K., Oldstone, M.A., Eds.; Springer-Verlag Berlin Heidelberg: Heidelberg, Germany, 1998; Volume 233/2, pp. 93–124. [Google Scholar]
- Organ, E.L.; Rubin, D.H. Pathogenesis of reovirus gastrointestinal and hepatobiliary disease. In Reoviruses II; Tyler, K., Oldstone, M.A., Eds.; Springer-Verlag Berlin Heidelberg: Heidelberg, Germany, 1998; Volume 233/2, pp. 67–83. [Google Scholar]
- Smakman, N.; van Der Bilt, J.D.W.; van Den Wollenberg, D.J.M.; Hoeben, R.C.; Borel Rinkes, I.H.M.; Kranenburg, O. Immunosuppression promotes reovirus therapy of colorectal liver metastases. Cancer Gene Ther. 2006, 13, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Westberg, S.; Sadeghi, A.; Svensson, E.; Segall, T.; Dimopoulou, M.; Korsgren, O.; Hemminki, A.; Loskog, A.S.; Totterman, T.H.; von Euler, H. Treatment efficacy and immune stimulation by adcd40l gene therapy of spontaneous canine malignant melanoma. J. Immunother. 2013, 36, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Debatin, K.M.; Krammer, P.H. Death receptors in chemotherapy and cancer. Oncogene 2004, 23, 2950–2966. [Google Scholar] [CrossRef] [PubMed]
- Van Valen, F.; Fulda, S.; Truckenbrod, B.; Eckervogt, V.; Sonnemann, J.; Hillmann, A.; Rodl, R.; Hoffmann, C.; Winkelmann, W.; Schafer, L.; et al. Apoptotic responsiveness of the Ewing's sarcoma family of tumours to tumour necrosis factor-related apoptosis-inducing ligand (trail). Int. J. Cancer 2000, 88, 252–259. [Google Scholar] [CrossRef]
- Van Den Wollenberg, D.J.M. Unpublished observations. manuscript in preparation.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kemp, V.; Hoeben, R.C.; Van den Wollenberg, D.J.M. Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus. Viruses 2016, 8, 4. https://doi.org/10.3390/v8010004
Kemp V, Hoeben RC, Van den Wollenberg DJM. Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus. Viruses. 2016; 8(1):4. https://doi.org/10.3390/v8010004
Chicago/Turabian StyleKemp, Vera, Rob C. Hoeben, and Diana J. M. Van den Wollenberg. 2016. "Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus" Viruses 8, no. 1: 4. https://doi.org/10.3390/v8010004
APA StyleKemp, V., Hoeben, R. C., & Van den Wollenberg, D. J. M. (2016). Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus. Viruses, 8(1), 4. https://doi.org/10.3390/v8010004