
Oncolytic Replication of E1b-Deleted Adenoviruses


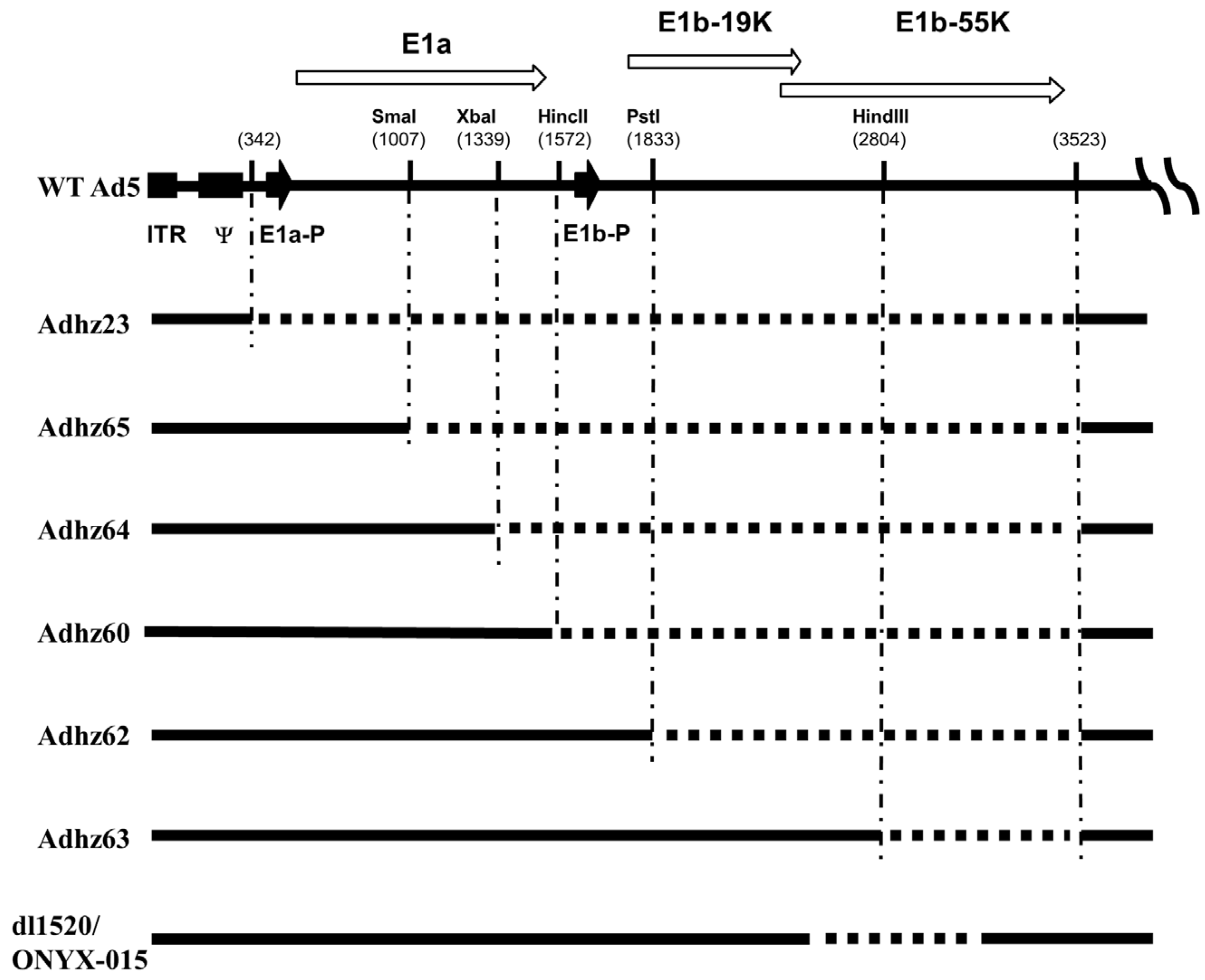
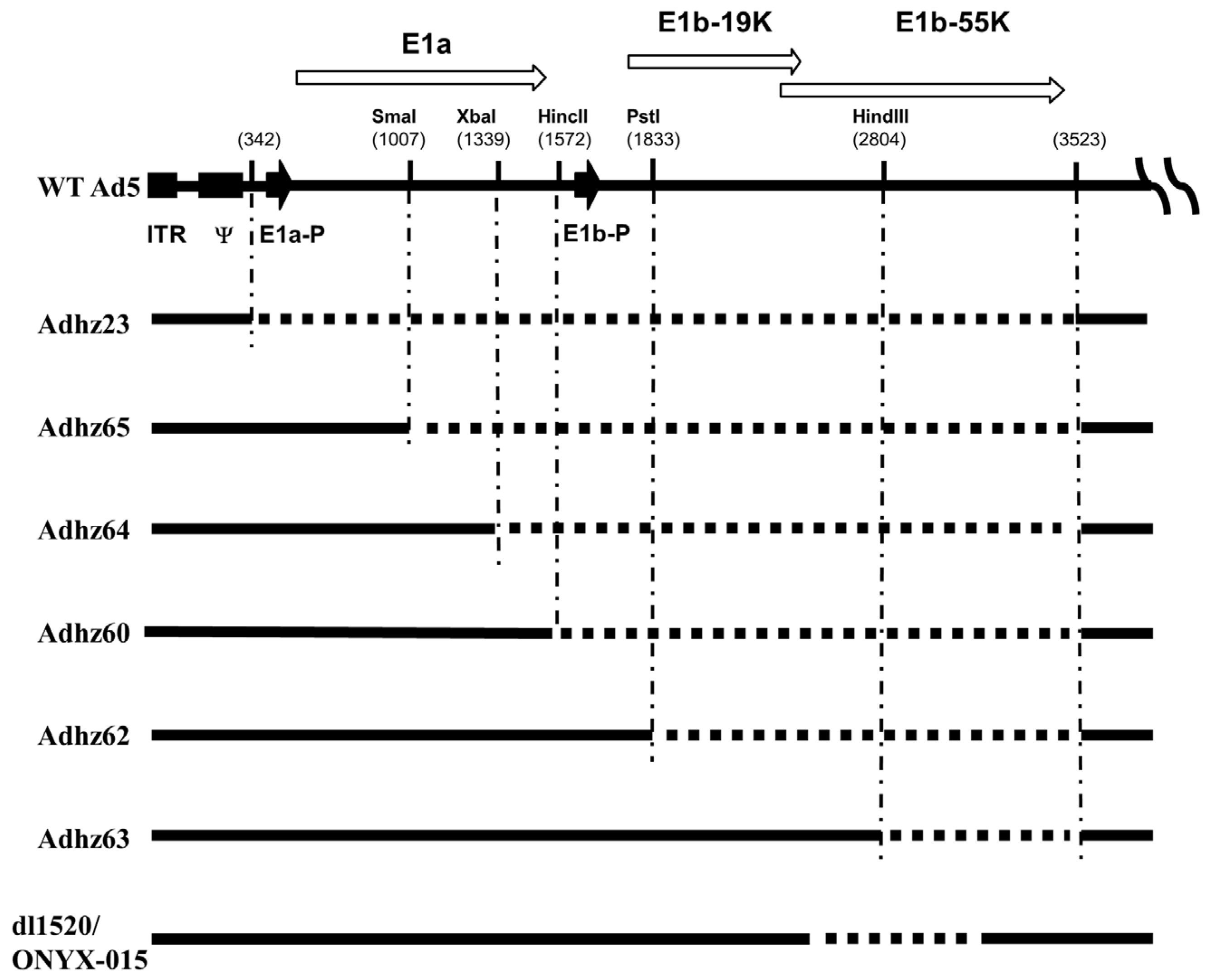{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oncolytic Adenoviruses
3. Selective Replication of E1b55K-Deleted Ad in Cancer Cells
4. Cancer Selectivity of E1b55K-Deleted Ads Based on p53 Deficiency
5. Cancer Selectivity of E1b55K-Deleted Ads Based on Late Viral mRNA Export
6. Cancer Selectivity of E1b55K-Deleted Ads Based on Cyclin E or Cell Cycle Dysregulation
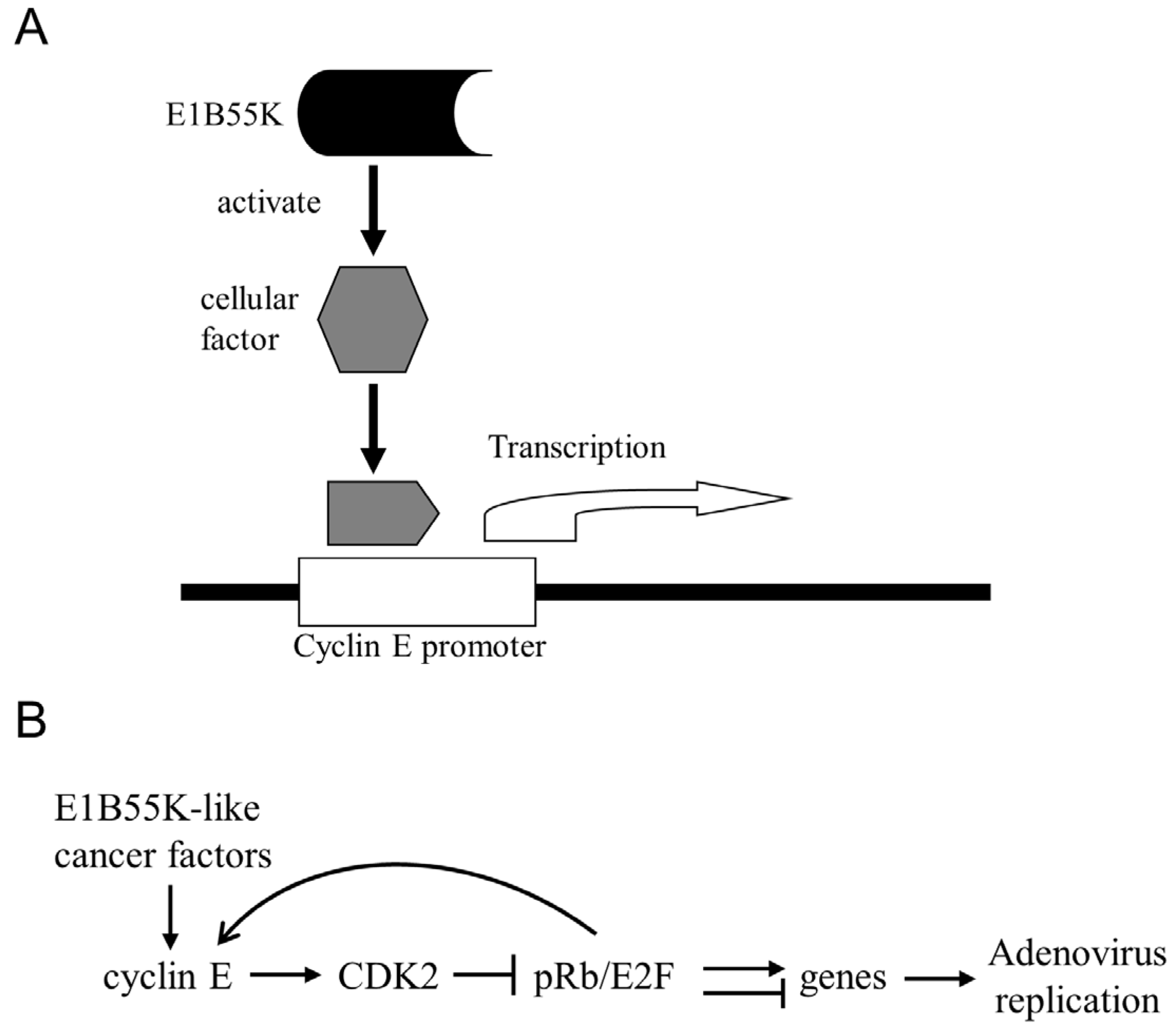
7. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA. Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.; Martuza, R.L.; Zwiebel, J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat. Med. 2001, 7, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hallden, G.; Hill, R.; Anand, A.; Liu, T.C.; Francis, J.; Brooks, G.; Lemoine, N.; Kirn, D. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat. Biotechnol. 2003, 21, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.N.; Bauer, D.F.; Cody, J.J.; Markert, J.M. Oncolytic viral therapy of malignant glioma. Neurotherapeutics 2009, 6, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Nettelbeck, D.M. Cellular genetic tools to control oncolytic adenoviruses for virotherapy of cancer. J. Mol. Med. 2008, 86, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Berk, A.J. Adenoviridae: The viruses and their replication. In Fields Virology, 5th ed.; Howley, D.M.K.P., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006; pp. 2355–2394. [Google Scholar]
- Mulvihill, S.; Warren, R.; Venook, A.; Adler, A.; Randlev, B.; Heise, C.; Kirn, D. Safety and feasibility of injection with an E1B-55 KDa gene-deleted, replication-selective adenovirus (ONYX-015) into primary carcinomas of the pancreas: A phase I trial. Gene Ther. 2001, 8, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Seth, P. Adenoviral vectors. Adv. Exp. Med. Biol. 2000, 465, 13–22. [Google Scholar] [PubMed]
- Barker, D.D.; Berk, A.J. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology 1987, 156, 107–121. [Google Scholar] [CrossRef]
- Stillman, B. Functions of the adenovirus E1B tumour antigens. Cancer Surv. 1986, 5, 389–404. [Google Scholar] [PubMed]
- Pilder, S.; Moore, M.; Logan, J.; Shenk, T. The adenovirus E1B-55K transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol. Cell. Biol. 1986, 6, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Debbas, M.; White, E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993, 7, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Debbas, M.; Sabbatini, P.; Hockenbery, D.; Korsmeyer, S.; White, E. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-KDa and Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 7742–7746. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Sabbatini, P.; Debbas, M.; Wold, W.S.; Kusher, D.I.; Gooding, L.R. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor alpha. Mol. Cell. Biol. 1992, 12, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Cuconati, A.; Degenhardt, K.; Sundararajan, R.; Anschel, A.; White, E. Bak and Bax function to limit adenovirus replication through apoptosis induction. J. Virol. 2002, 76, 4547–4558. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.M.; Tseng, M.T.; Zheng, X.; Dong, Y.; Jamshidi-Parsian, A.; Thompson, T.C.; Brenner, M.K.; McMasters, K.M.; Zhou, H.S. E1A-induced apoptosis does not prevent replication of adenoviruses with deletion of e1b in majority of infected cancer cells. Cancer Gene Ther. 2004, 11, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.M.; Zheng, X.; Waigel, S.; Zacharias, W.; McMasters, K.M.; Zhou, H.S. Gene expression profiles of normal human lung cells affected by adenoviral e1b. Virology 2006, 350, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Wiethoff, C.M.; Nemerow, G.R. Adenovirus membrane penetration: Tickling the tail of a sleeping dragon. Virology 2015, 479–480, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Willetts, M.; Webster, P.; Helenius, A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 1993, 75, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Davidson, B.L.; Breakefield, X.O. Viral vectors for gene delivery to the nervous system. Nat. Rev. Neurosci. 2003, 4, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Seth, P. Mechanism of adenovirus-mediated endosome lysis: Role of the intact adenovirus capsid structure. Biochem. Biophys. Res. Commun. 1994, 205, 1318–1324. [Google Scholar] [CrossRef] [PubMed]
- Lupold, S.E.; Rodriguez, R. Adenoviral gene therapy, radiation, and prostate cancer. Rev. Urol. 2005, 7, 193–202. [Google Scholar] [PubMed]
- Huebner, R.J.; Rowe, W.P.; Schatten, W.E.; Smith, R.R.; Thomas, L.B. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer 1956, 9, 1211–1218. [Google Scholar] [PubMed]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D. Clinical research results with dl1520 (ONYX-015), a replication-selective adenovirus for the treatment of cancer: What have we learned? Gene Ther. 2001, 8, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zheng, S.; Li, X.F.; Huang, J.J.; Zheng, X.; Li, Z. Intra-tumor injection of H101, a recombinant adenovirus, in combination with chemotherapy in patients with advanced cancers: A pilot phase II clinical trial. World J. Gastroenterol. 2004, 10, 3634–3638. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Liu, H.L.; Zhang, X.R.; Xu, J.P.; Hu, W.K.; Liang, M.; Chen, S.Y.; Hu, F.; Chu, D.T. A phase I trial of intratumoral administration of recombinant oncolytic adenovirus overexpressing HSP70 in advanced solid tumor patients. Gene Ther. 2009, 16, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Crompton, A.M.; Kirn, D.H. From ONYX-015 to armed vaccinia viruses: The education and evolution of oncolytic virus development. Curr. Cancer Drug Targets 2007, 7, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Frew, S.E.; Sammut, S.M.; Shore, A.F.; Ramjist, J.K.; Al-Bader, S.; Rezaie, R.; Daar, A.S.; Singer, P.A. Chinese health biotech and the billion-patient market. Nat. Biotechnol. 2008, 26, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.T.; Tanabe, K.K. Viral oncolysis. Oncologist 2002, 7, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.; Atencio, I.; Huang, W.M.; Horn, M.; Ramachandra, M. Overexpression of adenovirus E3–11.6K protein induces cell killing by both caspase-dependent and caspase-independent mechanisms. Virology 2004, 326, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, T. Induction of apoptosis by adenovirus E4orf4 protein. Apoptosis 2000, 5, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.H.; Lian, S.; Zhao, R.; Rao, X.M.; McMasters, K.M.; Zhou, H.S. Combination of autophagy inducer rapamycin and oncolytic adenovirus improves antitumor effect in cancer cells. Virol. J. 2013, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Gomez-Gutierrez, J.G.; Garcia-Garcia, A.; Rao, X.M.; Chen, L.; McMasters, K.M.; Zhou, H.S. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 2011, 416, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Hallden, G.; Hill, R.; Wang, Y.; Anand, A.; Liu, T.C.; Lemoine, N.R.; Francis, J.; Hawkins, L.; Kirn, D. Novel immunocompetent murine tumor models for the assessment of replication-competent oncolytic adenovirus efficacy. Mol. Ther. 2003, 8, 412–424. [Google Scholar] [CrossRef]
- Ungerechts, G.; Springfeld, C.; Frenzke, M.E.; Lampe, J.; Parker, W.B.; Sorscher, E.J.; Cattaneo, R. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol. Ther. 2007, 15, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Heise, C.; Sampson-Johannes, A.; Williams, A.; McCormick, F.; Von Hoff, D.D.; Kirn, D.H. Onyx-015, an e1b gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat. Med. 1997, 3, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Ben-Israel, H.; Kleinberger, T. Adenovirus and cell cycle control. Front. Biosci. 2002, 7, d1369–d1395. [Google Scholar] [CrossRef] [PubMed]
- Amundson, S.A.; Myers, T.G.; Fornace, A.J., Jr. Roles for p53 in growth arrest and apoptosis: Putting on the brakes after genotoxic stress. Oncogene 1998, 17, 3287–3299. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, A.; Nelson, C.; Skulimowski, A.; McGovern, J.; Pigott, D.; Jenkins, J. Transactivation of the p53 oncogene by E1A gene products. Virology 1990, 177, 595–605. [Google Scholar] [CrossRef]
- Lowe, S.W.; Ruley, H.E. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 1993, 7, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Morita, K.; Tsunoda, H.; Imajoh-Ohmi, S.; Tanaka, H.; Yasuda, H.; Oda, K. Stabilization of p53 by adenovirus E1A occurs through its amino-terminal region by modification of the ubiquitin-proteasome pathway. J. Biol. Chem. 1998, 273, 20036–20045. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Kao, C.C.; Yew, P.R.; Berk, A.J. Domains required for in vitro association between the cellular p53 and the adenovirus 2 E1B 55K proteins. Virology 1990, 179, 806–814. [Google Scholar] [CrossRef]
- Martin, M.E.; Berk, A.J. Adenovirus E1B 55K represses p53 activation in vitro. J. Virol. 1998, 72, 3146–3154. [Google Scholar] [PubMed]
- Nevels, M.; Rubenwolf, S.; Spruss, T.; Wolf, H.; Dobner, T. The adenovirus E4orf6 protein can promote E1A/E1B-induced focus formation by interfering with p53 tumor suppressor function. Proc. Natl. Acad. Sci. USA 1997, 94, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Querido, E.; Blanchette, P.; Yan, Q.; Kamura, T.; Morrison, M.; Boivin, D.; Kaelin, W.G.; Conaway, R.C.; Conaway, J.W.; Branton, P.E. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a cullin-containing complex. Genes Dev. 2001, 15, 3104–3117. [Google Scholar] [CrossRef] [PubMed]
- Wienzek, S.; Roth, J.; Dobbelstein, M. E1B 55-kilodalton oncoproteins of adenovirus types 5 and 12 inactivate and relocalize p53, but not p51 or p73, and cooperate with E4orf6 proteins to destabilize p53. J. Virol. 2000, 74, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Pennella, M.A.; Liu, Y.; Woo, J.L.; Kim, C.A.; Berk, A.J. Adenovirus E1B 55-kilodalton protein is a p53-sumo1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. J. Virol. 2010, 84, 12210–12225. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Pellegata, N.S.; Ranzani, G.N. The significance of p53 mutations in human cancers. Eur. J. Histochem. 1996, 40, 273–282. [Google Scholar] [PubMed]
- Dix, B.R.; Edwards, S.J.; Braithwaite, A.W. Does the antitumor adenovirus ONYX-015/dl1520 selectively target cells defective in the p53 pathway? J. Virol. 2001, 75, 5443–5447. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.D.; Ornelles, D.A. P53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J. Virol. 1998, 72, 9479–9490. [Google Scholar] [PubMed]
- Geoerger, B.; Grill, J.; Opolon, P.; Morizet, J.; Aubert, G.; Terrier-Lacombe, M.J.; Bressac De-Paillerets, B.; Barrois, M.; Feunteun, J.; Kirn, D.H.; et al. Oncolytic activity of the E1B-55 KDa-deleted adenovirus ONYX-015 is independent of cellular p53 status in human malignant glioma xenografts. Cancer Res. 2002, 62, 764–772. [Google Scholar] [PubMed]
- Rothmann, T.; Hengstermann, A.; Whitaker, N.J.; Scheffner, M.; Zur Hausen, H. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J. Virol. 1998, 72, 9470–9478. [Google Scholar] [PubMed]
- Zheng, X.; Rao, X.M.; Gomez-Gutierrez, J.G.; Hao, H.; McMasters, K.M.; Zhou, H.S. Adenovirus E1B55K region is required to enhance cyclin E expression for efficient viral DNA replication. J. Virol. 2008, 82, 3415–3427. [Google Scholar] [CrossRef] [PubMed]
- Ries, S.J.; Brandts, C.H.; Chung, A.S.; Biederer, C.H.; Hann, B.C.; Lipner, E.M.; McCormick, F.; Korn, W.M. Loss of p14Arf in tumor cells facilitates replication of the adenovirus mutant dl1520 (ONYX-015). Nat. Med. 2000, 6, 1128–1133. [Google Scholar] [PubMed]
- McCormick, F. ONYX-015 selectivity and the p14arf pathway. Oncogene 2000, 19, 6670–6672. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.J.; Dix, B.R.; Myers, C.J.; Dobson-Le, D.; Huschtscha, L.; Hibma, M.; Royds, J.; Braithwaite, A.W. Evidence that replication of the antitumor adenovirus ONYX-015 is not controlled by the p53 and p14(Arf) tumor suppressor genes. J. Virol. 2002, 76, 12483–12490. [Google Scholar] [CrossRef] [PubMed]
- Dix, B.R.; O’Carroll, S.J.; Myers, C.J.; Edwards, S.J.; Braithwaite, A.W. Efficient induction of cell death by adenoviruses requires binding of E1B55K and p53. Cancer Res. 2000, 60, 2666–2672. [Google Scholar] [PubMed]
- Royds, J.A.; Hibma, M.; Dix, B.R.; Hananeia, L.; Russell, I.A.; Wiles, A.; Wynford-Thomas, D.; Braithwaite, A.W. P53 promotes adenoviral replication and increases late viral gene expression. Oncogene 2006, 25, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Hobom, U.; Dobbelstein, M. E1B-55-kilodalton protein is not required to block p53-induced transcription during adenovirus infection. J. Virol. 2004, 78, 7685–7697. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, C.C.; Johnson, L.; Bagus, B.; Choi, S.; Nicholas, C.; Shen, A.; Boyle, L.; Pandey, K.; Soria, C.; Kunich, J.; et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 2004, 6, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Flint, S.J.; Gonzalez, R.A. Regulation of mRNA production by the adenoviral E1B 55-KDa and E4 orf6 proteins. Curr. Top. Microbiol. Immunol. 2003, 272, 287–330. [Google Scholar] [PubMed]
- Leppard, K.N.; Shenk, T. The adenovirus E1B 55 Kd protein influences mRNA transport via an intranuclear effect on RNA metabolism. EMBO J. 1989, 8, 2329–2336. [Google Scholar] [PubMed]
- Goodrum, F.D.; Ornelles, D.A. Roles for the E4 orf6, orf3, and E1B 55-kilodalton proteins in cell cycle-independent adenovirus replication. J. Virol. 1999, 73, 7474–7488. [Google Scholar] [PubMed]
- Woo, J.L.; Berk, A.J. Adenovirus ubiquitin-protein ligase stimulates viral late mRNA nuclear export. J. Virol. 2007, 81, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Kindsmuller, K.; Groitl, P.; Hartl, B.; Blanchette, P.; Hauber, J.; Dobner, T. Intranuclear targeting and nuclear export of the adenovirus E1B-55K protein are regulated by sumo1 conjugation. Proc. Natl. Acad. Sci. USA 2007, 104, 6684–6689. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Kindsmuller, K.; Wimmer, P.; Groitl, P.; Gonzalez, R.A.; Dobner, T. The E3 ubiquitin ligase activity associated with the adenoviral E1B-55K-E4orf6 complex does not require CRM1-dependent export. J. Virol. 2011, 85, 7081–7094. [Google Scholar] [CrossRef]
- Blanchette, P.; Kindsmuller, K.; Groitl, P.; Dallaire, F.; Speiseder, T.; Branton, P.E.; Dobner, T. Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires e4orf6 ubiquitin ligase activity. J. Virol. 2008, 82, 2642–2651. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, C.C.; Soria, C.; Bagus, B.; McCormick, F. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell 2005, 8, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, R.; Xi, Q.; Schneider, R.J. Adenovirus-specific translation by displacement of kinase Mnk1 from cap-initiation complex eIF4F. EMBO J. 2000, 19, 3465–3474. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, R.; Xi, Q.; Schneider, R.J. Structural basis for competitive inhibition of eIF4g-Mnk1 interaction by the adenovirus 100-kilodalton protein. J. Virol. 2004, 78, 7707–7716. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.W.; Telling, G.C.; Myat, M.M.; Williams, J.F.; Flint, S.J. The adenovirus L4 100-kilodalton protein is necessary for efficient translation of viral late mRNA species. J. Virol. 1990, 64, 2732–2742. [Google Scholar] [PubMed]
- Yueh, A.; Schneider, R.J. Translation by ribosome shunting on adenovirus and hsp70 mRNAs facilitated by complementarity to 18S rRNA. Genes Dev. 2000, 14, 414–421. [Google Scholar] [PubMed]
- Gonzalez, R.; Huang, W.; Finnen, R.; Bragg, C.; Flint, S.J. Adenovirus E1B 55-kilodalton protein is required for both regulation of mRNA export and efficient entry into the late phase of infection in normal human fibroblasts. J. Virol. 2006, 80, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.D.; Ornelles, D.A. The early region 1B 55-kilodalton oncoprotein of adenovirus relieves growth restrictions imposed on viral replication by the cell cycle. J. Virol. 1997, 71, 548–561. [Google Scholar] [PubMed]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef] [PubMed]
- Le Cam, L.; Polanowska, J.; Fabbrizio, E.; Olivier, M.; Philips, A.; Ng Eaton, E.; Classon, M.; Geng, Y.; Sardet, C. Timing of cyclin E gene expression depends on the regulated association of a bipartite repressor element with a novel E2F complex. EMBO J. 1999, 18, 1878–1890. [Google Scholar] [CrossRef] [PubMed]
- Moroy, T.; Geisen, C. Cyclin e. Int. J. Biochem. Cell Biol. 2004, 36, 1424–1439. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Lee, Y.M.; Welcker, M.; Swanger, J.; Zagozdzon, A.; Winer, J.D.; Roberts, J.M.; Kaldis, P.; Clurman, B.E.; Sicinski, P. Kinase-independent function of cyclin E. Mol. Cell 2007, 25, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Geisen, C.; Moroy, T. The oncogenic activity of cyclin E is not confined to Cdk2 activation alone but relies on several other, distinct functions of the protein. J. Biol. Chem. 2002, 277, 39909–39918. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.C.; Clurman, B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Loeb, K.R.; Kostner, H.; Firpo, E.; Norwood, T.; Tsuchiya, K.D.; Clurman, B.E.; Roberts, J.M. A mouse model for cyclin E-dependent genetic instability and tumorigenesis. Cancer Cell 2005, 8, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Donnellan, R.; Chetty, R. Cyclin E in human cancers. FASEB J. 1999, 13, 773–780. [Google Scholar] [PubMed]
- Ma, Y.; Fiering, S.; Black, C.; Liu, X.; Yuan, Z.; Memoli, V.A.; Robbins, D.J.; Bentley, H.A.; Tsongalis, G.J.; Demidenko, E.; et al. Transgenic cyclin E triggers dysplasia and multiple pulmonary adenocarcinomas. Proc. Natl. Acad. Sci. USA 2007, 104, 4089–4094. [Google Scholar] [CrossRef] [PubMed]
- Marone, M.; Scambia, G.; Giannitelli, C.; Ferrandina, G.; Masciullo, V.; Bellacosa, A.; Benedetti-Panici, P.; Mancuso, S. Analysis of cyclin E and CDK2 in ovarian cancer: Gene amplification and RNA overexpression. Int. J. Cancer 1998, 75, 34–39. [Google Scholar] [CrossRef]
- Sawasaki, T.; Shigemasa, K.; Shiroyama, Y.; Kusuda, T.; Fujii, T.; Parmley, T.H.; O’Brien, T.J.; Ohama, K. Cyclin E mRNA overexpression in epithelial ovarian cancers: Inverse correlation with p53 protein accumulation. J. Soc. Gynecol. Investig. 2001, 8, 179–185. [Google Scholar] [CrossRef]
- Strohmaier, H.; Spruck, C.H.; Kaiser, P.; Won, K.A.; Sangfelt, O.; Reed, S.I. Human F-box protein hCdc4 targets cyclin e for proteolysis and is mutated in a breast cancer cell line. Nature 2001, 413, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Akli, S.; Zheng, P.J.; Multani, A.S.; Wingate, H.F.; Pathak, S.; Zhang, N.; Tucker, S.L.; Chang, S.; Keyomarsi, K. Tumor-specific low molecular weight forms of cyclin E induce genomic instability and resistance to p21, p27, and antiestrogens in breast cancer. Cancer Res. 2004, 64, 3198–3208. [Google Scholar] [CrossRef] [PubMed]
- Keyomarsi, K.; Tucker, S.L.; Buchholz, T.A.; Callister, M.; Ding, Y.; Hortobagyi, G.N.; Bedrosian, I.; Knickerbocker, C.; Toyofuku, W.; Lowe, M.; et al. Cyclin E and survival in patients with breast cancer. N. Engl. J. Med. 2002, 347, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Wingate, H.; Puskas, A.; Duong, M.; Bui, T.; Richardson, D.; Liu, Y.; Tucker, S.L.; van Pelt, C.; Meijer, L.; Hunt, K.; et al. Low molecular weight cyclin E is specific in breast cancer and is associated with mechanisms of tumor progression. Cell Cycle 2009, 8, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Spruck, C.H.; Won, K.A.; Reed, S.I. Deregulated cyclin E induces chromosome instability. Nature 1999, 401, 297–300. [Google Scholar] [PubMed]
- Minella, A.C.; Swanger, J.; Bryant, E.; Welcker, M.; Hwang, H.; Clurman, B.E. P53 and p21 form an inducible barrier that protects cells against cyclin E-cdk2 deregulation. Curr. Biol. 2002, 12, 1817–1827. [Google Scholar] [CrossRef]
- Cheng, P.H.; Rao, X.M.; McMasters, K.M.; Zhou, H.S. Molecular basis for viral selective replication in cancer cells: Activation of CDK2 by adenovirus-induced cyclin E. PLoS ONE 2013, 8, e57340. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gutierrez, J.G.; Rao, X.M.; Zhou, H.S.; McMasters, K.M. Enhanced cancer cell killing by truncated E2F-1 used in combination with oncolytic adenovirus. Virology 2012, 433, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.H.; Rao, X.M.; Duan, X.; Li, X.F.; Egger, M.E.; McMasters, K.M.; Zhou, H.S. Virotherapy targeting cyclin E overexpression in tumors with adenovirus-enhanced cancer-selective promoter. J. Mol. Med. 2015, 93, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.H.; Rao, X.M.; Wechman, S.L.; Li, X.F.; McMasters, K.M.; Zhou, H.S. Oncolytic adenovirus targeting cyclin E overexpression repressed tumor growth in syngeneic immunocompetent mice. BMC Cancer 2015, 15, 716. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, P.-H.; Wechman, S.L.; McMasters, K.M.; Zhou, H.S. Oncolytic Replication of E1b-Deleted Adenoviruses. Viruses 2015, 7, 5767-5779. https://doi.org/10.3390/v7112905
Cheng P-H, Wechman SL, McMasters KM, Zhou HS. Oncolytic Replication of E1b-Deleted Adenoviruses. Viruses. 2015; 7(11):5767-5779. https://doi.org/10.3390/v7112905
Chicago/Turabian StyleCheng, Pei-Hsin, Stephen L. Wechman, Kelly M. McMasters, and Heshan Sam Zhou. 2015. "Oncolytic Replication of E1b-Deleted Adenoviruses" Viruses 7, no. 11: 5767-5779. https://doi.org/10.3390/v7112905