Membranous Replication Factories Induced by Plus-Strand RNA Viruses


Abstract
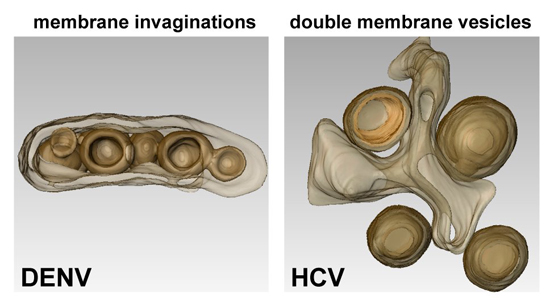
:
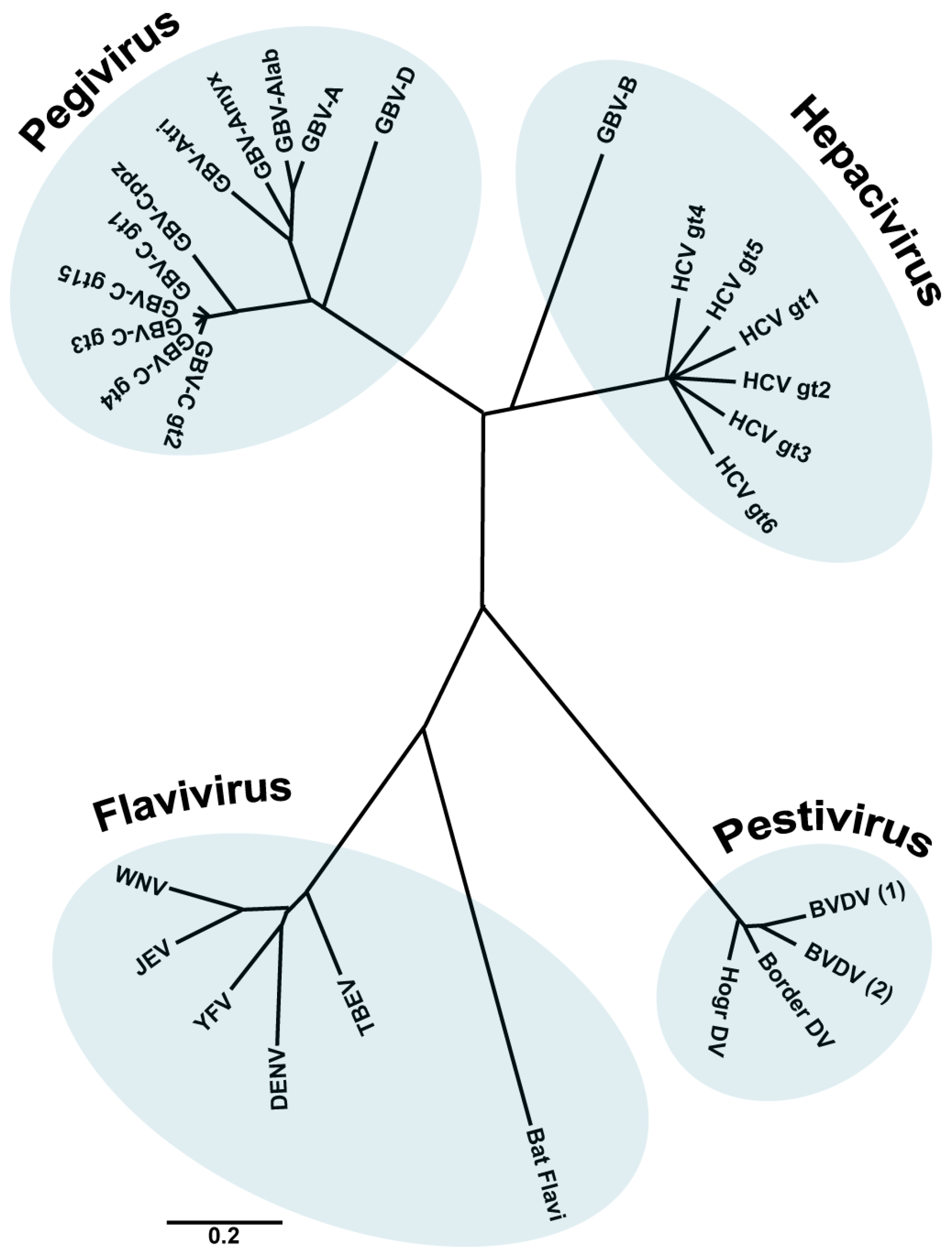
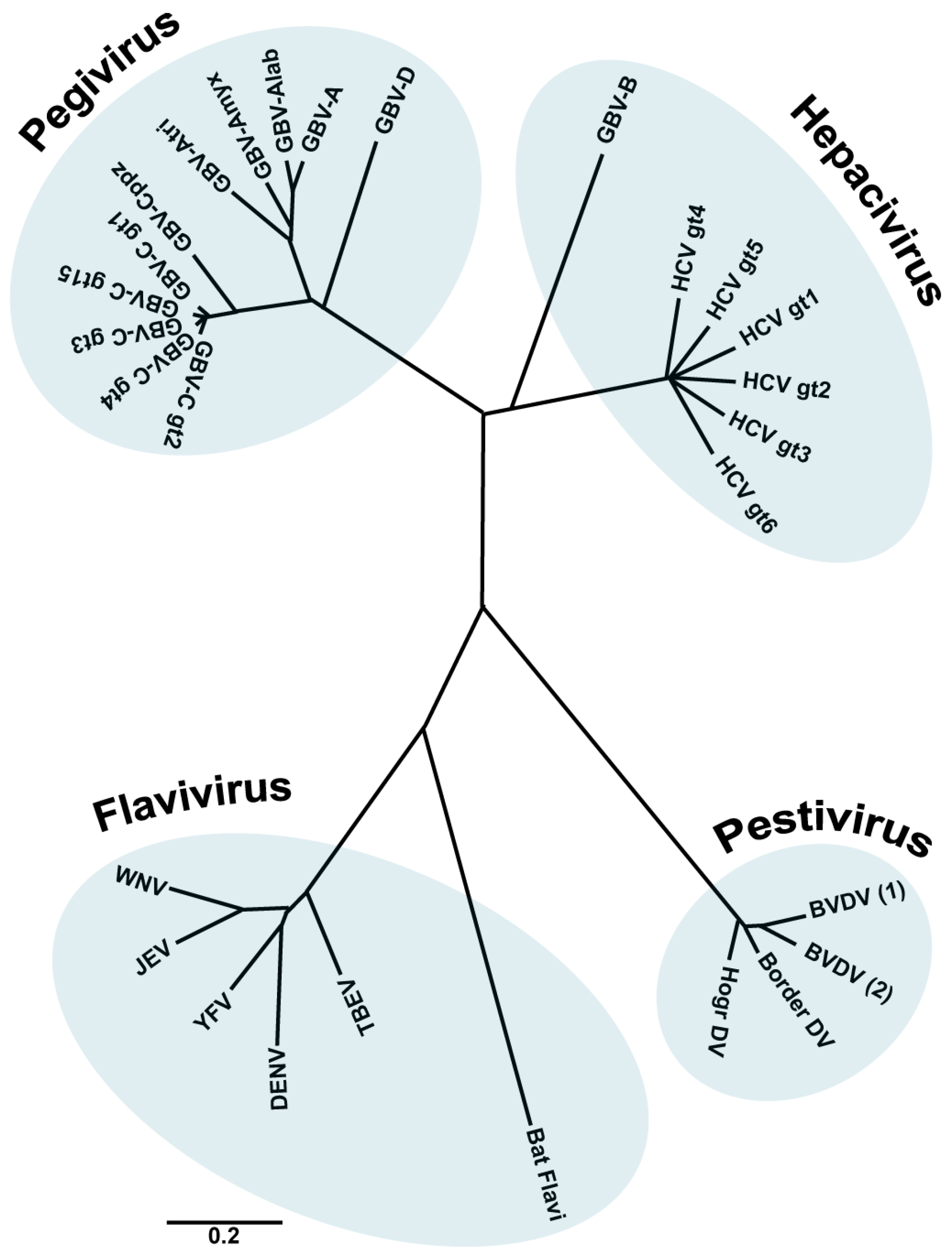
1. The Family Flaviviridae
2. Architecture and Properties of the Replication Factories of Members of the Flaviviridae Family
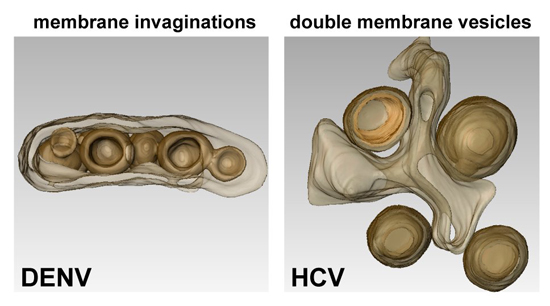
2.1. Flaviviruses

2.2. Hepaciviruses
2.3. Pestiviruses
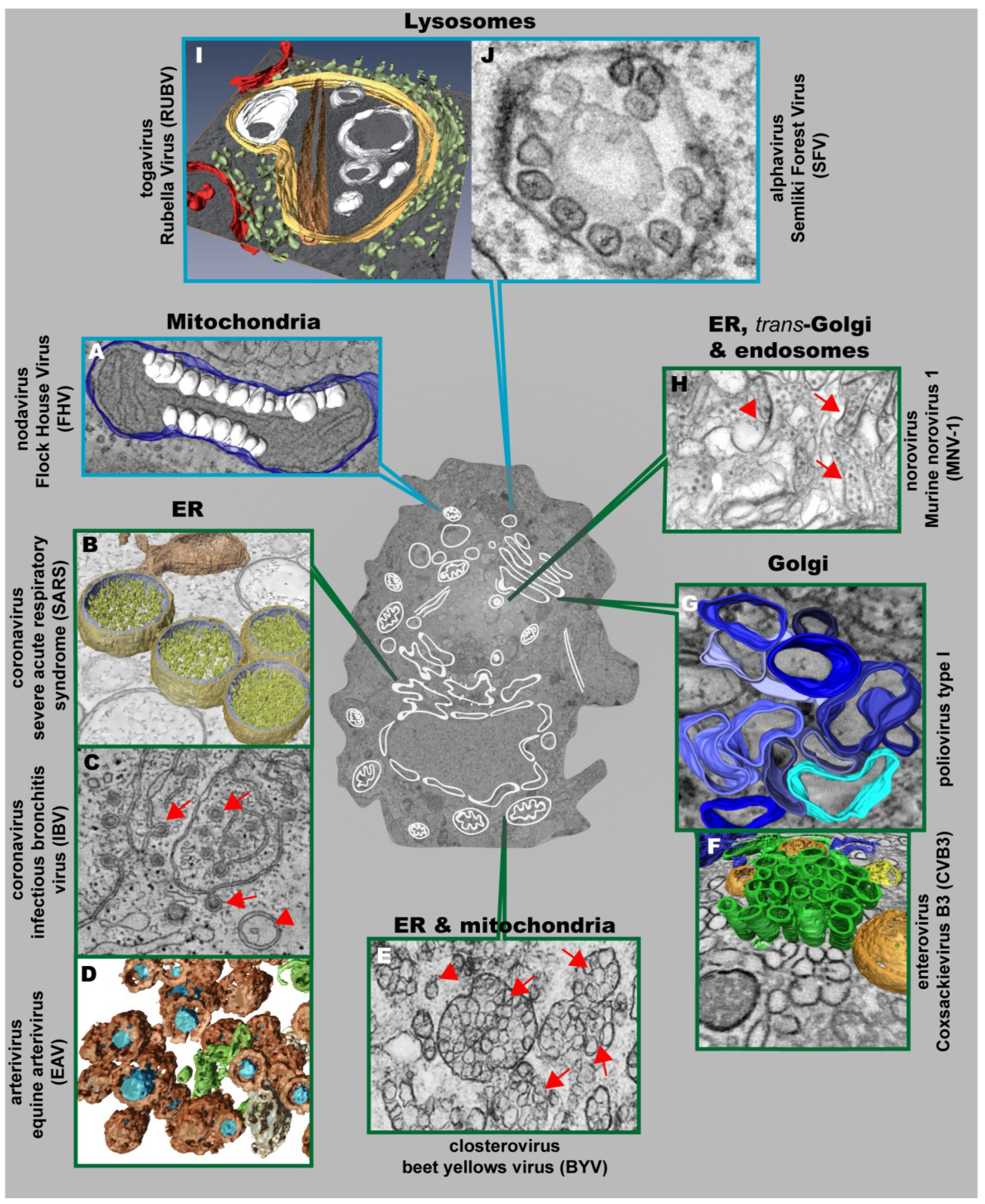
3. Architecture and Properties of the Replication Factories of Other (+) Strand RNA Viruses
3.1. Nodaviruses
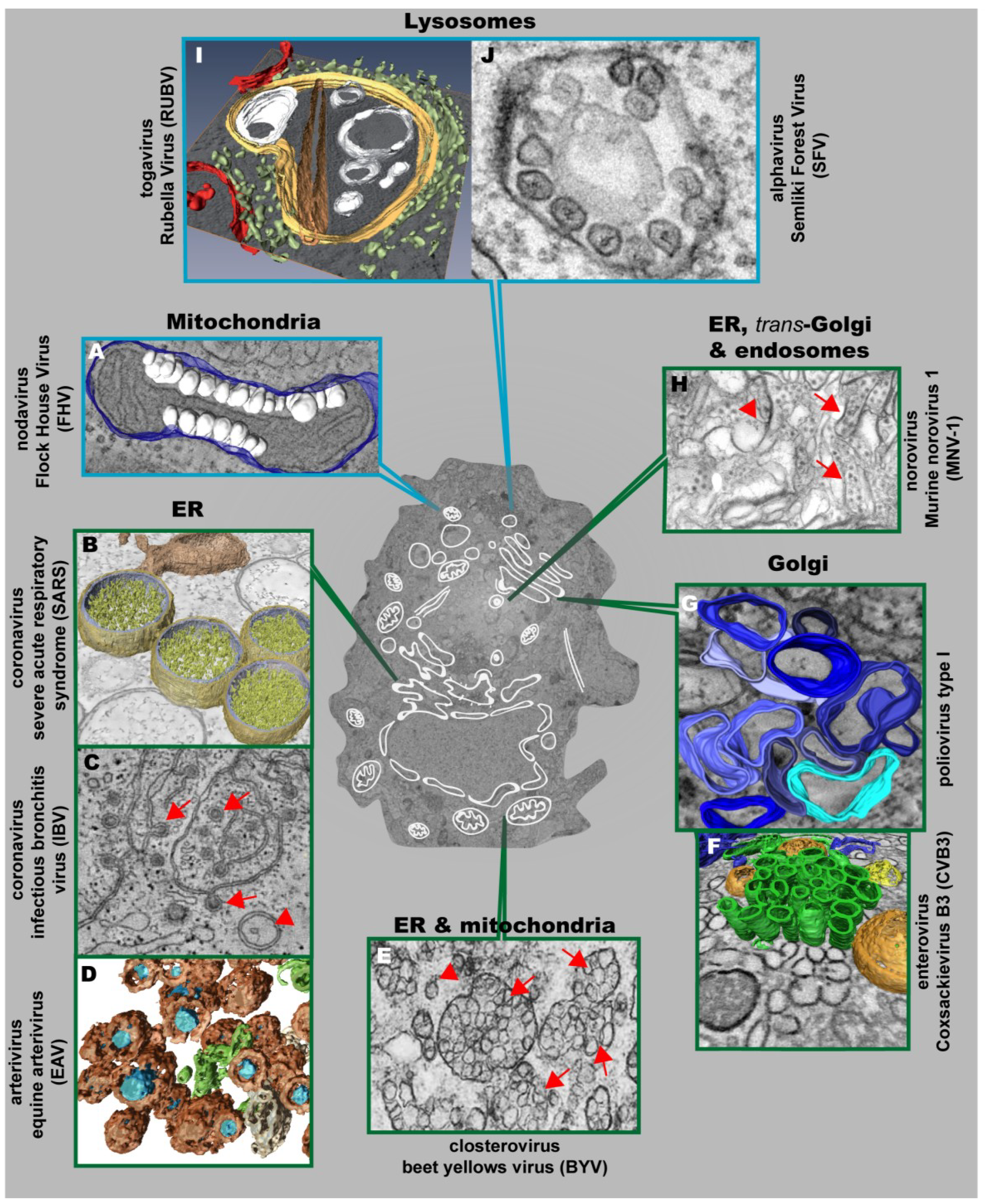
3.4. Togaviruses
3.5. Caliciviruses
3.6. Plant Viruses
4. Similarities and Differences between the Replication Factories of Flaviviruses and other (+) Strand RNA Viruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Morphotype | Virus Group and Representative Member | Name of Membrane Alteration | Size/Diameter | Membrane Source | |
|---|---|---|---|---|---|
| Single-membrane vesicles | Nodaviruses | FHV | Spherules | ~50 nm | Mitochondria |
| Bromoviruses | BMV | ~60 nm | ER (adjacent to the nucleus) | ||
| Togaviruses | RUBV | Cytoplasmic vacuoles (CPVs) | 600–2000 nm | Lysosomes | |
| SFV | CPVs with spherules | CPVs: 600–2000 nm Spherules: 50 nm | |||
| Flaviviruses | DENV | Invaginated vesicles | 80–120 nm | ER | |
| WNVKUN | 50–100 nm | ||||
| TBEV | 60–100 nm | ||||
| MVEV, SLEV and JEV | 75–145 nm | ||||
| YFV | unknown | ||||
| Double-membrane vesicles | Hepaciviruses | HCV | ~150 nm | ER | |
| Coronaviruses | SARS-CoV | 200–300 nm | |||
| MHV | 200–350 nm | ||||
| MERS-CoV | 150–320 nm | ||||
| IBV | ~200 nm | ||||
| Arteriviruses | EAV | ~95 nm | |||
| Picornaviruses | Poliovirus | 100–300 nm | Golgi | ||
| CV3B | ~160 nm | ||||
| FMDV | ~100 nm | unknown (probably ER) | |||
| Calicivirus | MNV-1 | ~200 nm | ER/Golgi | ||
| Closterovirus | BYV | ~100 nm | ER/Mitochondria | ||
5. Role of Viral Proteins in the Formation of the Replication Organelles
5.1. Single-Membrane Vesicle Inducers
5.2. Double-Membrane Vesicle Inducers
6. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References and Notes
- Stapleton, J.T.; Foung, S.; Muerhoff, A.S.; Bukh, J.; Simmonds, P. The GB viruses: A review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J. Gen. Virol. 2011, 92, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. The Origin of Hepatitis C Virus. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy, 82nd ed.; Bartenschlager, R., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, Germany, 2013; Volume 369. [Google Scholar]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N. Engl. J. Med. 1975, 292, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Thiel, H.-J.; Rice, C.M. Flaviviridae: The Viruses and Their Replication. In Fields Virology, 5th ed.; Fields, B.N., Knipe, D.M., Howley, P., Eds.; Raven Press: New York, NY, USA, 2001; pp. 1001–1152. [Google Scholar]
- Mackenzie, J.S.; Gubler, D.J.; Petersen, L.R. Emerging flaviviruses: The spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 2004, 10, S98–S109. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Collett, M.S.; Gould, E.A.; Heinz, F.X.; Meyers, G.; Monath, A.; Pletnev, A.; Rice, C.M.; Stiasny, K.; et al. Family Flaviviridae. In Virus taxonomy: Classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Academic Press: San Diego, CA, USA, 2012; pp. 1003–1020. [Google Scholar]
- Miller, R.H.; Purcell, R.H. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses as well as members of two plant virus supergroups. Proc. Natl. Acad. Sci. USA 1990, 87, 2057–2061. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Stollar, V.; Schlesinger, R.W. Studies on the nature of dengue viruses. V. Structure and development of dengue virus in Vero cells. Virology 1971, 46, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Stohlman, S.A.; Wisseman, C.L., Jr.; Eylar, O.R.; Silverman, D.J. Dengue virus-induced modifications of host cell membranes. J. Virol. 1975, 16, 1017–1026. [Google Scholar] [PubMed]
- Hase, T.; Summers, P.L.; Eckels, K.H.; Baze, W.B. An electron and immunoelectron microscopic study of dengue-2 virus infection of cultured mosquito cells: Maturation events. Arch. Virol. 1987, 92, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.C.; Chen, C.C.; Hou, R.F. Ultrastructural studies on the reproductive system of male Aedes aegypti (Diptera: Culicidae) infected with dengue 2 virus. J. Med. Entomol. 1998, 35, 71–76. [Google Scholar] [PubMed]
- Grief, C.; Galler, R.; Cortes, L.M.; Barth, O.M. Intracellular localisation of dengue-2 RNA in mosquito cell culture using electron microscopic in situ hybridisation. Arch. Virol. 1997, 142, 2347–2357. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Improved membrane preservation of flavivirus-infected cells with cryosectioning. J. Virol. Methods 1996, 56, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell. Host. Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef]
- Junjhon, J.; Pennington, J.G.; Edwards, T.J.; Perera, R.; Lanman, J.; Kuhn, R.J. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J. Virol. 2014, 88, 4687–4697. [Google Scholar] [CrossRef] [PubMed]
- Shahar, A.; Lustig, S.; Akov, Y.; David, Y.; Schneider, P.; Friedmann, A.; Levin, R. West Nile virions aligned along myelin lamellae in organotypic spinal cord cultures. J. Neurosci. Res. 1990, 26, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Westaway, E.G.; Mackenzie, J.M.; Kenney, M.T.; Jones, M.K.; Khromykh, A.A. Ultrastructure of Kunjin virus-infected cells: Colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 1997, 71, 6650–6661. [Google Scholar] [PubMed]
- Mackenzie, J.M.; Westaway, E.G. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 2001, 75, 10787–10799. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996, 220, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Westaway, E.G. Markers for trans-Golgi membranes and the intermediate compartment localize to induced membranes with distinct replication functions in flavivirus-infected cells. J. Virol. 1999, 73, 9555–9567. [Google Scholar] [PubMed]
- Mackenzie, J.M.; Kenney, M.T.; Westaway, E.G. West Nile virus strain Kunjin NS5 polymerase is a phosphoprotein localized at the cytoplasmic site of viral RNA synthesis. J. Gen. Virol. 2007, 88, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Khromykh, A.A.; Jones, M.K.; Westaway, E.G. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 1998, 245, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Popov, V.L.; Niedrig, M.; Weber, F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J. Virol. 2010, 84, 8470–8483. [Google Scholar] [CrossRef]
- Pichlmair, A.; Reis e Sousa, C. Innate recognition of viruses. Immunity 2007, 27, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Matsumura, T.; Masuda, K.; Kanemura, K.; Fukunaga, T. Maturation site of dengue type 2 virus in cultured mosquito C6/36 cells and Vero cells. Kobe J. Med. Sci. 1998, 44, 65–79. [Google Scholar] [PubMed]
- Senigl, F.; Grubhoffer, L.; Kopecky, J. Differences in maturation of tick-borne encephalitis virus in mammalian and tick cell line. Intervirology 2006, 49, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS One 2012, 7, e47912. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohn, C.; Blair, C.D. Persistent infection of cultured mammalian cells by Japanese encephalitis virus. J. Virol. 1977, 24, 580–589. [Google Scholar] [PubMed]
- Brinton, M.A.; Fernandez, A.V. A replication-efficient mutant of West Nile virus is insensitive to DI particle interference. Virology 1983, 129, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Poidinger, M.; Coelen, R.J.; Mackenzie, J.S. Persistent infection of Vero cells by the flavivirus Murray Valley encephalitis virus. J. Gen. Virol. 1991, 72, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef] [PubMed]
- Hoenninger, V.M.; Rouha, H.; Orlinger, K.K.; Miorin, L.; Marcello, A.; Kofler, R.M.; Mandl, C.W. Analysis of the effects of alterations in the tick-borne encephalitis virus 3'-noncoding region on translation and RNA replication using reporter replicons. Virology 2008, 377, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Khromykh, A.A.; Westaway, E.G. Subgenomic replicons of the flavivirus Kunjin: Construction and applications. J. Virol. 1997, 71, 1497–1505. [Google Scholar] [PubMed]
- Jones, C.T.; Patkar, C.G.; Kuhn, R.J. Construction and applications of yellow fever virus replicons. Virology 2005, 331, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.; Robertson, T.; Kendrick, T.; Abdo, M.; Papadimitriou, J.; McMinn, P. Morphological features of Murray Valley encephalitis virus infection in the central nervous system of Swiss mice. Int. J. Exp. Pathol. 2000, 81, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Hase, T.; Summers, P.L.; Ray, P. Entry and replication of Japanese encephalitis virus in cultured neurogenic cells. J. Virol. Methods 1990, 30, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Hase, T.; Summers, P.L.; Dubois, D.R. Ultrastructural changes of mouse brain neurons infected with Japanese encephalitis virus. Int. J. Exp. Pathol. 1990, 71, 493–505. [Google Scholar] [PubMed]
- Murphy, F.A.; Harrison, A.K.; Gary, G.W., Jr.; Whitfield, S.G.; Forrester, F.T. St. Louis encephalitis virus infection in mice. Electron microscopic studies of central nervous system. Lab. Investig. 1968, 19, 652–662. [Google Scholar] [PubMed]
- Whitfield, S.G.; Murphy, F.A.; Sudia, W.D. St. Louis encephalitis virus: An ultrastructural study of infection in a mosquito vector. Virology 1973, 56, 70–87. [Google Scholar] [CrossRef] [PubMed]
- McGavran, M.H.; White, J.D. Electron microscopic and immunofluorescent observations on monkey liver and tissue culture cells infected with the Asibi strain of yellow fever virus liver and tissue culture cells infected with the asibi strain of yellow fever virus. Am. J. Pathol. 1964, 45, 501–517. [Google Scholar] [PubMed]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, P.; Beaumont, E.; Uzbekov, R.; Brand, D.; Gaillard, J.; Blanchard, E.; Roingeard, P. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell. Mol. Life Sci. 2013, 70, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.L.; et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef] [PubMed]
- Chasey, D.; Roeder, P.L. Virus-like particles in bovine turbinate cells infected with bovine virus diarrhoea/mucosal disease virus. Arch. Virol. 1981, 67, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.W.; Nettleton, P.F. The ultrastructure of cell cultures infected with border disease and bovine virus diarrhoea viruses. J. Gen. Virol. 1987, 68, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Kubovicova, E.; Makarevich, A.V.; Pivko, J.; Chrenek, P.; Grafenau, P.; Riha, L.; Sirotkin, A.V.; Louda, F. Alteration in ultrastructural morphology of bovine embryos following subzonal microinjection of bovine viral diarrhea virus (BVDV). Zygote 2008, 16, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Dubovi, E.J.; Cohen-Gould, L.; Donis, R.; Szeto, H.H. Cytoplasmic vacuolization responses to cytopathic bovine viral diarrhoea virus. Virus Res. 2008, 132, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Weiskircher, E.; Aligo, J.; Ning, G.; Konan, K.V. Bovine viral diarrhea virus NS4B protein is an integral membrane protein associated with Golgi markers and rearranged host membranes. Virol. J. 2009, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, S.; Mast, J.; Thiel, H.J.; Konig, M. Morphogenesis of pestiviruses: New insights from ultrastructural studies of strain Giraffe-1. J. Virol. 2014, 88, 2717–2724. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Keppler, O.T.; Habermann, A.; Allespach, I.; Krijnse-Locker, J.; Krausslich, H.G. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 2007, 3, e36. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, L.; Berghofer, B.; Bamberg, S.; Becker, S. Multivesicular bodies as a platform for formation of the Marburg virus envelope. J. Virol. 2004, 78, 12277–12287. [Google Scholar] [CrossRef] [PubMed]
- Mittler, E.; Kolesnikova, L.; Strecker, T.; Garten, W.; Becker, S. Role of the transmembrane domain of marburg virus surface protein GP in assembly of the viral envelope. J. Virol. 2007, 81, 3942–3948. [Google Scholar] [CrossRef] [PubMed]
- Kopek, B.G.; Perkins, G.; Miller, D.J.; Ellisman, M.H.; Ahlquist, P. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 2007, 5, e220. [Google Scholar] [CrossRef] [PubMed]
- Lanman, J.; Crum, J.; Deerinck, T.J.; Gaietta, G.M.; Schneemann, A.; Sosinsky, G.E.; Ellisman, M.H.; Johnson, J.E. Visualizing flock house virus infection in Drosophila cells with correlated fluorescence and electron microscopy. J. Struct. Biol. 2008, 161, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef] [PubMed]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, A.H.; Raj, V.S.; Oudshoorn, D.; Bestebroer, T.M.; van, N.S.; Limpens, R.W.; Posthuma, C.C.; van der Meer, Y.; Barcena, M.; Haagmans, B.L.; et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J. Gen. Virol. 2013, 94, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Hawes, P.C.; Cottam, E.M.; Mantell, J.; Verkade, P.; Monaghan, P.; Wileman, T.; Britton, P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. MBio 2013, 4, e00801–e00813. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.W.; van der Meer, Y.; Roos, N.; Snijder, E.J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 1999, 73, 2016–2026. [Google Scholar] [PubMed]
- Knoops, K.; Barcena, M.; Limpens, R.W.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Ultrastructural characterization of arterivirus replication structures: Reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 2012, 86, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Kallman, F.; Williams, R.C.; Dulbecco, R.; Vogt, M. Fine structure of changes produced in cultured cells sampled at specified intervals during a single growth cycle of polio virus. J. Biophys. Biochem. Cytol. 1958, 4, 301–308. [Google Scholar] [CrossRef]
- Bienz, K.; Egger, D.; Pasamontes, L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 1987, 160, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Bienz, K. Intracellular location and translocation of silent and active poliovirus replication complexes. J. Gen. Virol. 2005, 86, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Rust, R.C.; Landmann, L.; Gosert, R.; Tang, B.L.; Hong, W.; Hauri, H.P.; Egger, D.; Bienz, K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 2001, 75, 9808–9818. [Google Scholar] [CrossRef] [PubMed]
- Bienz, K.; Egger, D.; Rasser, Y.; Bossart, W. Intracellular distribution of poliovirus proteins and the induction of virus-specific cytoplasmic structures. Virology 1983, 131, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, A.; Giddings, T.H., Jr.; Ladinsky, M.S.; Kirkegaard, K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 1996, 70, 6576–6588. [Google Scholar] [PubMed]
- Bienz, K.; Egger, D.; Rasser, Y.; Bossart, W. Kinetics and location of poliovirus macromolecular synthesis in correlation to virus-induced cytopathology. Virology 1980, 100, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Limpens, R.W.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.; Barcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. MBio 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, P.; Cook, H.; Jackson, T.; Ryan, M.; Wileman, T. The ultrastructure of the developing replication site in foot-and-mouth disease virus-infected BHK-38 cells. J. Gen. Virol. 2004, 85, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Frey, T.K. Molecular biology of rubella virus. Adv. Virus Res. 1994, 44, 69–160. [Google Scholar] [PubMed]
- Fontana, J.; Tzeng, W.P.; Calderita, G.; Fraile-Ramos, A.; Frey, T.K.; Risco, C. Novel replication complex architecture in rubella replicon-transfected cells. Cell. Microbiol. 2007, 9, 875–890. [Google Scholar] [CrossRef]
- Lee, J.Y.; Marshall, J.A.; Bowden, D.S. Characterization of rubella virus replication complexes using antibodies to double-stranded RNA. Virology 1994, 200, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Magliano, D.; Marshall, J.A.; Bowden, D.S.; Vardaxis, N.; Meanger, J.; Lee, J.Y. Rubella virus replication complexes are virus-modified lysosomes. Virology 1998, 240, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Lopez-Iglesias, C.; Tzeng, W.P.; Frey, T.K.; Fernandez, J.J.; Risco, C. Three-dimensional structure of Rubella virus factories. Virology 2010, 405, 579–591. [Google Scholar] [CrossRef]
- Risco, C.; Carrascosa, J.L.; Frey, T.K. Structural maturation of rubella virus in the Golgi complex. Virology 2003, 312, 261–269. [Google Scholar] [CrossRef]
- Froshauer, S.; Kartenbeck, J.; Helenius, A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell. Biol. 1988, 107, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Kujala, P.; Ikaheimonen, A.; Ehsani, N.; Vihinen, H.; Auvinen, P.; Kaariainen, L. Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol. 2001, 75, 3873–3884. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.M.; Levin, J.G.; Grimley, P.M.; Berezesky, I.K. Membrane-associated replication complex in arbovirus infection. J. Virol. 1972, 10, 504–515. [Google Scholar]
- Grimley, P.M.; Berezesky, I.K.; Friedman, R.M. Cytoplasmic structures associated with an arbovirus infection: Loci of viral ribonucleic acid synthesis. J. Virol. 1968, 2, 1326–1338. [Google Scholar] [PubMed]
- Grimley, P.M.; Levin, J.G.; Berezesky, I.K.; Friedman, R.M. Specific membranous structures associated with the replication of group A arboviruses. J. Virol. 1972, 10, 492–503. [Google Scholar]
- Spuul, P.; Balistreri, G.; Kaariainen, L.; Ahola, T. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest Virus replication complexes from the plasma membrane to modified lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef] [PubMed]
- Green, K.Y.; Chanock, R.M.; Kapikian, A.Z. Human caliciviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2001; pp. 841–874. [Google Scholar]
- Wobus, C.E.; Karst, S.M.; Thackray, L.B.; Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Krug, A.; Mackenzie, J.M.; Green, K.Y.; Virgin, H.W. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004, 2, e432. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.L.; Sosnovtsev, S.V.; Green, K.Y.; Wobus, C.; Virgin, H.W.; Mackenzie, J.M. Mouse norovirus replication is associated with virus-induced vesicle clusters originating from membranes derived from the secretory pathway. J. Virol. 2009, 83, 9709–9719. [Google Scholar] [CrossRef] [PubMed]
- Duizer, E.; Schwab, K.J.; Neill, F.H.; Atmar, R.L.; Koopmans, M.P.; Estes, M.K. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 2004, 85, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Green, K.Y.; Mory, A.; Fogg, M.H.; Weisberg, A.; Belliot, G.; Wagner, M.; Mitra, T.; Ehrenfeld, E.; Cameron, C.E.; Sosnovtsev, S.V. Isolation of enzymatically active replication complexes from feline calicivirus-infected cells. J. Virol. 2002, 76, 8582–8595. [Google Scholar] [CrossRef] [PubMed]
- Mas, P.; Beachy, R.N. Replication of tobacco mosaic virus on endoplasmic reticulum and role of the cytoskeleton and virus movement protein in intracellular distribution of viral RNA. J. Cell. Biol. 1999, 147, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Schaad, M.C.; Jensen, P.E.; Carrington, J.C. Formation of plant RNA virus replication complexes on membranes: Role of an endoplasmic reticulum-targeted viral protein. EMBO J. 1997, 16, 4049–4059. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.A.; Sit, T.L.; Callaway, A.S.; Allen, N.S.; Lommel, S.A. Red clover necrotic mosaic virus replication proteins accumulate at the endoplasmic reticulum. Virology 2004, 320, 276–290. [Google Scholar] [CrossRef] [PubMed]
- De, G.M.; Coscoy, L.; Jaspars, E.M. Localization and biochemical characterization of alfalfa mosaic virus replication complexes. Virology 1993, 194, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Hayes, R.J.; Buck, K.W. Complete replication of a eukaryotic virus RNA in vitro by a purified RNA-dependent RNA polymerase. Cell 1990, 63, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Panavas, T.; Hawkins, C.M.; Panaviene, Z.; Nagy, P.D. The role of the p33:p33/p92 interaction domain in RNA replication and intracellular localization of p33 and p92 proteins of Cucumber necrosis tombusvirus. Virology 2005, 338, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Weber-Lotfi, F.; Dietrich, A.; Russo, M.; Rubino, L. Mitochondrial targeting and membrane anchoring of a viral replicase in plant and yeast cells. J. Virol. 2002, 76, 10485–10496. [Google Scholar] [CrossRef]
- Restrepo-Hartwig, M.A.; Ahlquist, P. Brome mosaic virus helicase- and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J. Virol. 1996, 70, 8908–8916. [Google Scholar] [PubMed]
- Den Boon, J.A.; Chen, J.; Ahlquist, P. Identification of sequences in Brome mosaic virus replicase protein 1a that mediate association with endoplasmic reticulum membranes. J. Virol. 2001, 75, 12370–12381. [Google Scholar] [CrossRef]
- Schwartz, M.; Chen, J.; Janda, M.; Sullivan, M.; den, B.J.; Ahlquist, P. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell. 2002, 9, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Gushchin, V.A.; Solovyev, A.G.; Erokhina, T.N.; Morozov, S.Y.; Agranovsky, A.A. Beet yellows virus replicase and replicative compartments: Parallels with other RNA viruses. Front. Microbiol. 2013, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Cronshaw, J.; Hoefert, L.; Esau, K. Ultrastructural features of Beta leaves infected with beet yellows virus. J. Cell. Biol. 1966, 31, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Esau, K.; Cronshaw, J.; Hoefert, L.L. Relation of beet yellows virus to the phloem and to movement in the sieve tube. J. Cell. Biol. 1967, 32, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Erokhina, T.N.; Vitushkina, M.V.; Zinovkin, R.A.; Lesemann, D.E.; Jelkmann, W.; Koonin, E.V.; Agranovsky, A.A. Ultrastructural localization and epitope mapping of the methyltransferase-like and helicase-like proteins of Beet yellows virus. J. Gen. Virol. 2001, 82, 1983–1994. [Google Scholar] [PubMed]
- Zinovkin, R.A.; Erokhina, T.N.; Lesemann, D.E.; Jelkmann, W.; Agranovsky, A.A. Processing and subcellular localization of the leader papain-like proteinase of Beet yellows closterovirus. J. Gen. Virol. 2003, 84, 2265–2270. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Stewart, L.R.; Kiss, Z.; Falk, B.W. Lettuce infectious yellows virus (LIYV) RNA 1-encoded P34 is an RNA-binding protein and exhibits perinuclear localization. Virology 2010, 403, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Gonsalves, D.; Teliz, D.; Lee, K.W. Ultrastructure and mitochondrial vesiculation associated with closterovirus-like particles in leafroll-diseased grapevines. Phytopathology 1989, 79, 357–360. [Google Scholar] [CrossRef]
- Faoro, F.; Carzaniga, R. Cyotchemistry and immuno-cytochemistry of the inclusion bodies induced by grapevine leafroll-associated closteroviruses GLRaV-1 and GLRaV-3. Riv. Patol. Veg. 1995, 5, 85–94. [Google Scholar]
- Koonin, E.V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 1991, 72, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V. Evolution and taxonomy of positive-strand RNA viruses: Implications of comparative analysis of amino acid sequences. Crit Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Kuhn, R.J.; Yang, C.F.; Takegami, T.; Wimmer, E. Initiation of poliovirus plus-strand RNA synthesis in a membrane complex of infected HeLa cells. J. Virol. 1986, 60, 43–53. [Google Scholar] [PubMed]
- Xiang, W.; Cuconati, A.; Hope, D.; Kirkegaard, K.; Wimmer, E. Complete protein linkage map of poliovirus P3 proteins: Interaction of polymerase 3Dpol with VPg and with genetic variants of 3AB. J. Virol. 1998, 72, 6732–6741. [Google Scholar] [PubMed]
- Wessels, E.; Notebaart, R.A.; Duijsings, D.; Lanke, K.; Vergeer, B.; Melchers, W.J.; van Kuppeveld, F.J. Structure-function analysis of the coxsackievirus protein 3A: Identification of residues important for dimerization, viral rna replication, and transport inhibition. J. Biol. Chem. 2006, 281, 28232–28243. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Krishnakumar, S.S.; Franco, D.; Paul, A.V.; London, E.; Wimmer, E. Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry 2007, 46, 5185–5199. [Google Scholar] [CrossRef] [PubMed]
- Bienz, K.; Egger, D.; Pfister, T.; Troxler, M. Structural and functional characterization of the poliovirus replication complex. J. Virol. 1992, 66, 2740–2747. [Google Scholar] [PubMed]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef] [PubMed]
- Brabec-Zaruba, M.; Berka, U.; Blaas, D.; Fuchs, R. Induction of autophagy does not affect human rhinovirus type 2 production. J. Virol. 2007, 81, 10815–10817. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Autophagy: A novel guardian of HCV against innate immune response. Autophagy 2011, 7, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef]
- Wileman, T. Aggresomes and autophagy generate sites for virus replication. Science 2006, 312, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Westaway, E.G.; Khromykh, A.A.; Kenney, M.T.; Mackenzie, J.M.; Jones, M.K. Proteins C and NS4B of the flavivirus Kunjin translocate independently into the nucleus. Virology 1997, 234, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Roosendaal, J.; Westaway, E.G.; Khromykh, A.; Mackenzie, J.M. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 2006, 80, 4623–4632. [Google Scholar] [CrossRef]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef]
- Rawson, R.B. The SREBP pathway--insights from Insigs and insects. Nat. Rev. Mol. Cell. Biol. 2003, 4, 631–640. [Google Scholar] [CrossRef]
- Salonen, A.; Ahola, T.; Kaariainen, L. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 2005, 285, 139–173. [Google Scholar] [PubMed]
- Kopek, B.G.; Settles, E.W.; Friesen, P.D.; Ahlquist, P. Nodavirus-induced membrane rearrangement in replication complex assembly requires replicase protein a, RNA templates, and polymerase activity. J. Virol. 2010, 84, 12492–12503. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.; Kaiser, W.J.; Hollinshead, M.; Moffat, K.; Chaudhry, Y.; Wileman, T.; Sosnovtsev, S.V.; Goodfellow, I.G. Feline calicivirus p32, p39 and p30 proteins localize to the endoplasmic reticulum to initiate replication complex formation. J. Gen. Virol. 2010, 91, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Stern, O.; Hung, Y.F.; Valdau, O.; Yaffe, Y.; Harris, E.; Hoffmann, S.; Willbold, D.; Sklan, E.H. An N-terminal amphipathic helix in dengue virus nonstructural protein 4A mediates oligomerization and is essential for replication. J. Virol. 2013, 87, 4080–4085. [Google Scholar] [CrossRef]
- Ahola, T.; Kaariainen, L. Reaction in alphavirus mRNA capping: Formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. USA 1995, 92, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Laakkonen, P.; Vihinen, H.; Kaariainen, L. Critical residues of Semliki Forest virus RNA capping enzyme involved in methyltransferase and guanylyltransferase-like activities. J. Virol. 1997, 71, 392–397. [Google Scholar] [PubMed]
- Laakkonen, P.; Ahola, T.; Kaariainen, L. The effects of palmitoylation on membrane association of Semliki forest virus RNA capping enzyme. J. Biol. Chem. 1996, 271, 28567–28571. [Google Scholar] [CrossRef] [PubMed]
- Peranen, J.; Laakkonen, P.; Hyvonen, M.; Kaariainen, L. The alphavirus replicase protein nsP1 is membrane-associated and has affinity to endocytic organelles. Virology 1995, 208, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Lampio, A.; Auvinen, P.; Kaariainen, L. Semliki Forest virus mRNA capping enzyme requires association with anionic membrane phospholipids for activity. EMBO J. 1999, 18, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Lampio, A.; Kilpelainen, I.; Pesonen, S.; Karhi, K.; Auvinen, P.; Somerharju, P.; Kaariainen, L. Membrane binding mechanism of an RNA virus-capping enzyme. J. Biol. Chem. 2000, 275, 37853–37859. [Google Scholar] [CrossRef]
- Salonen, A.; Vasiljeva, L.; Merits, A.; Magden, J.; Jokitalo, E.; Kaariainen, L. Properly folded nonstructural polyprotein directs the semliki forest virus replication complex to the endosomal compartment. J. Virol. 2003, 77, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Kallio, K.; Hellstrom, K.; Balistreri, G.; Spuul, P.; Jokitalo, E.; Ahola, T. Template RNA length determines the size of replication complex spherules for Semliki Forest virus. J. Virol. 2013, 87, 9125–9134. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.V.; Molla, A.; Wimmer, E. Studies of a putative amphipathic helix in the N-terminus of poliovirus protein 2C. Virology 1994, 199, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Echeverri, A.C.; Dasgupta, A. Amino terminal regions of poliovirus 2C protein mediate membrane binding. Virology 1995, 208, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Van Kuppeveld, F.J.; Melchers, W.J.; Kirkegaard, K.; Doedens, J.R. Structure-function analysis of coxsackie B3 virus protein 2B. Virology 1997, 227, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Moffat, K.; Knox, C.; Howell, G.; Clark, S.J.; Yang, H.; Belsham, G.J.; Ryan, M.; Wileman, T. Inhibition of the secretory pathway by foot-and-mouth disease virus 2BC protein is reproduced by coexpression of 2B with 2C, and the site of inhibition is determined by the subcellular location of 2C. J. Virol. 2007, 81, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, J.; Penin, F.; Moradpour, D. Hepatitis C virus nonstructural protein 4B: A journey into unexplored territory. Rev. Med. Virol. 2010, 20, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, J.; Roingeard, P.; Penin, F.; Moradpour, D. Amphipathic alpha-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J. Virol. 2010, 84, 12529–12537. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Romero-Brey, I.; Gouttenoire, J.; Stoitsova, S.; Krijnse-Locker, J.; Moradpour, D.; Bartenschlager, R. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 2011, 85, 6963–6976. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; van, T.H.; Roos, N.; Pedersen, K.W. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J. Gen. Virol. 2001, 82, 985–994. [Google Scholar] [PubMed]
- Posthuma, C.C.; Pedersen, K.W.; Lu, Z.; Joosten, R.G.; Roos, N.; Zevenhoven-Dobbe, J.C.; Snijder, E.J. Formation of the arterivirus replication/transcription complex: A key role for nonstructural protein 3 in the remodeling of intracellular membranes. J. Virol. 2008, 82, 4480–4491. [Google Scholar] [CrossRef] [PubMed]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. MBio 2013, 4. [Google Scholar] [CrossRef]
- Gadlage, M.J.; Sparks, J.S.; Beachboard, D.C.; Cox, R.G.; Doyle, J.D.; Stobart, C.C.; Denison, M.R. Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function. J. Virol. 2010, 84, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Romero-Brey, I.; Bartenschlager, R. Membranous Replication Factories Induced by Plus-Strand RNA Viruses. Viruses 2014, 6, 2826-2857. https://doi.org/10.3390/v6072826
Romero-Brey I, Bartenschlager R. Membranous Replication Factories Induced by Plus-Strand RNA Viruses. Viruses. 2014; 6(7):2826-2857. https://doi.org/10.3390/v6072826
Chicago/Turabian StyleRomero-Brey, Inés, and Ralf Bartenschlager. 2014. "Membranous Replication Factories Induced by Plus-Strand RNA Viruses" Viruses 6, no. 7: 2826-2857. https://doi.org/10.3390/v6072826
APA StyleRomero-Brey, I., & Bartenschlager, R. (2014). Membranous Replication Factories Induced by Plus-Strand RNA Viruses. Viruses, 6(7), 2826-2857. https://doi.org/10.3390/v6072826