Mechanism of West Nile Virus Neuroinvasion: A Critical Appraisal

Abstract
:1. Introduction
2. Hematogenous Route of Neuroinvasion
2.1. Blood-Brain Barrier
2.2. Transendothelial Viral Entry

2.3. Molecules for Blood-Brain Barrier Integrity

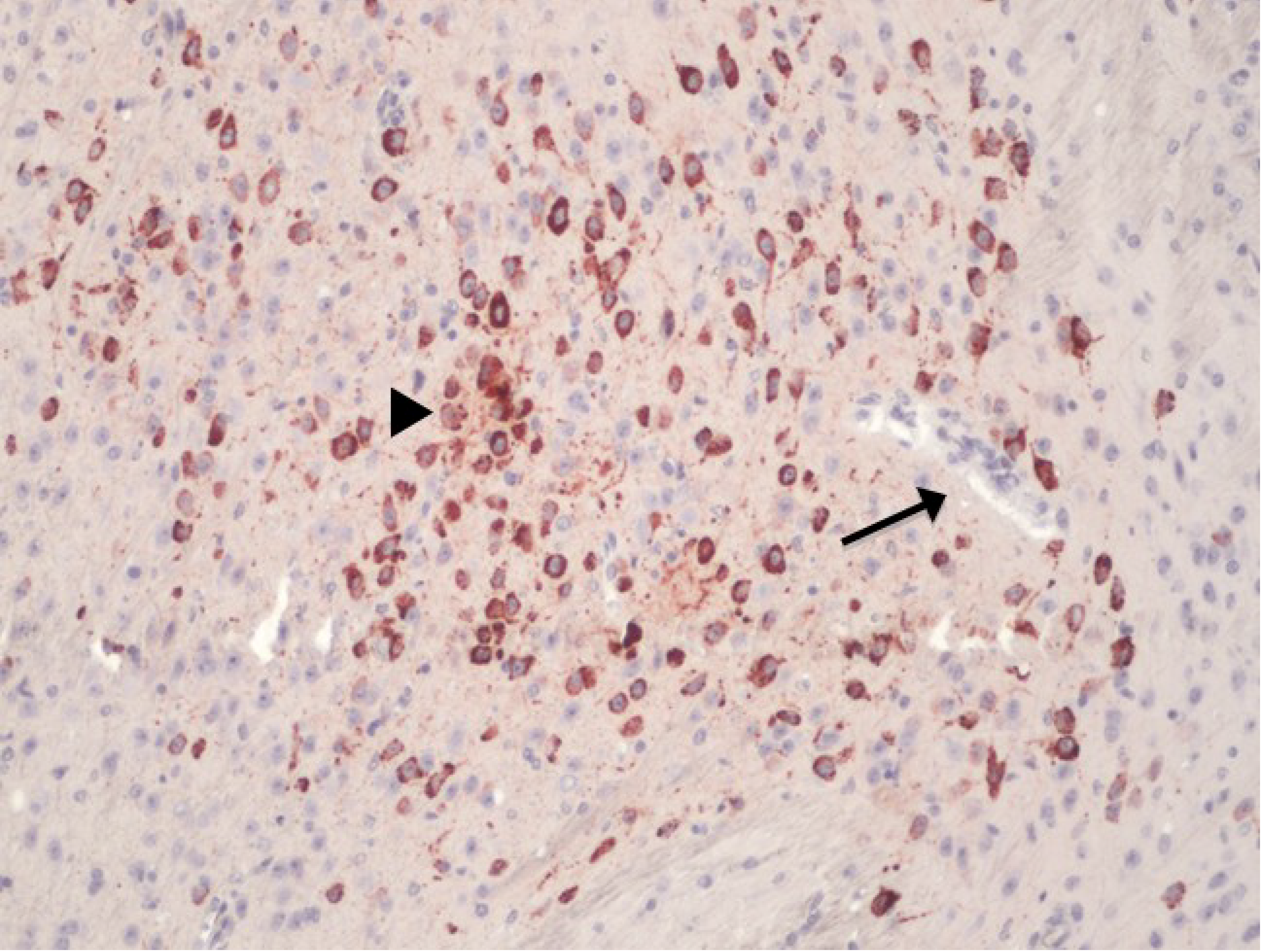{kind=link}
{kind=link}
| Junction Proteins | mRNA | Protein Level |
|---|---|---|
| Claudin-1 | ↑ [32,44];  [42] [42] | ↑ [32];  [41,42,43,44] [41,42,43,44] |
| Claudin-3 | ↑ [44] | No information |
| Claudin-4 | ↑ [44] | No information |
| Claudin-5 | NC [32] | No information |
| ZO-1 | Mild ↑ [32,44]; [42] | [41,42]; NC [43,44] |
| Occludin | Mild ↑ [32]; [42,44] | [42]; NC [44] |
| JAM-A (JAM-1) | ↑ [44] [42] | [42,44] |
| Beta-catenin | No information | [42] |
| Vascular Endothelial Cadherin | No information | [42] |
and ) indicate findings supported by in vivo evidence [42].| Cell adhesion molecules | mRNA | Protein levels |
|---|---|---|
| ICAM-1 | [48] | [45,48] |
| ICAM-3 | NC [32] | No information |
| VCAM-1 | ↑ [32]; [48] | ↑ [32,45]; [48] |
| E-Selectin | [32,48] | [45,48] |
| P-Selectin | No information | NC [45] |
| PECAM | NC [32] | No information |
and ) indicate findings supported by in vivo evidence [48].2.4. Paracellular Viral Entry
2.4.1. Via Passive Diffusion
2.4.2. Via a “Trojan Horse” Mode of Entry
3. Transneural Route of Neuroinvasion
3.1. From Peripheral Nervous System (Somatic Nerves) to Central Nervous System
3.2. From Olfactory Nerve to Olfactory Bulb
4. Multiple Routes of Neuroinvasion
5. New Avenues for Research
| Barriers | Blood-brain (neurovascular unit) | Blood-CSF (ventricular system) | Blood-CSF (arachnoid/meningeal) | Inner CSF-brain | Outer CSF-brain |
|---|---|---|---|---|---|
| Anatomical location | Cerebral blood vessels | Choroid plexus and CVO blood vessels | Dural +/− pial blood vessels | Ependyma of the ventricular system | Pia mater |
| Cellular components |
|
|
|
|
|
| Restriction point | Endothelial cells (tight junction) | Epithelial and tanycyte-like cells (tight junction) | Arachnoid epithelial cells and pial blood vessel endothelial cells (tight junction) | Neuro-ependymal cells (strap junction, only in immature brains) | Glial foot processes (various junctions, only in immature brains) |
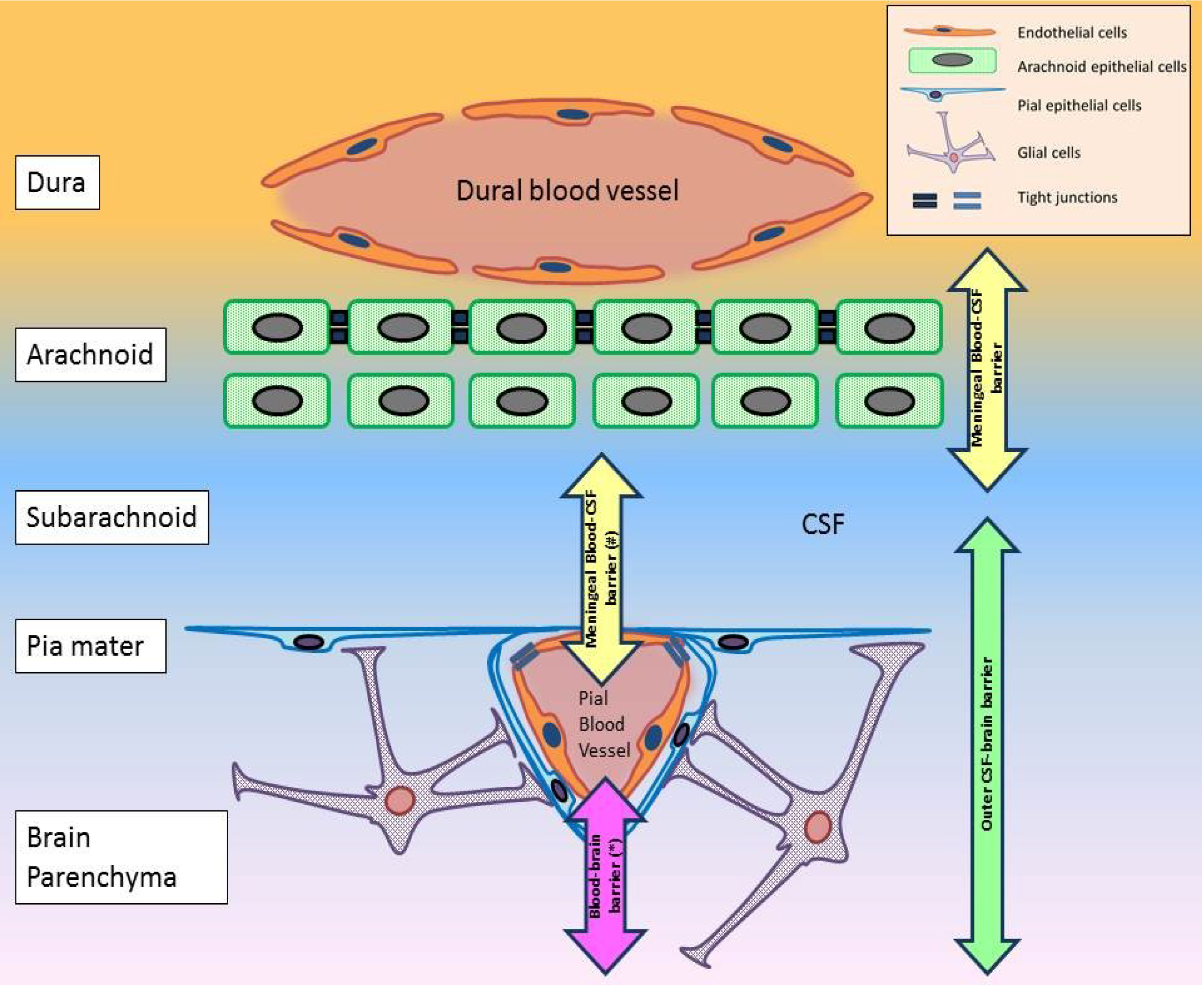
- A hypothetical blood-CSF interface in the pial blood vessels (Figure 2).
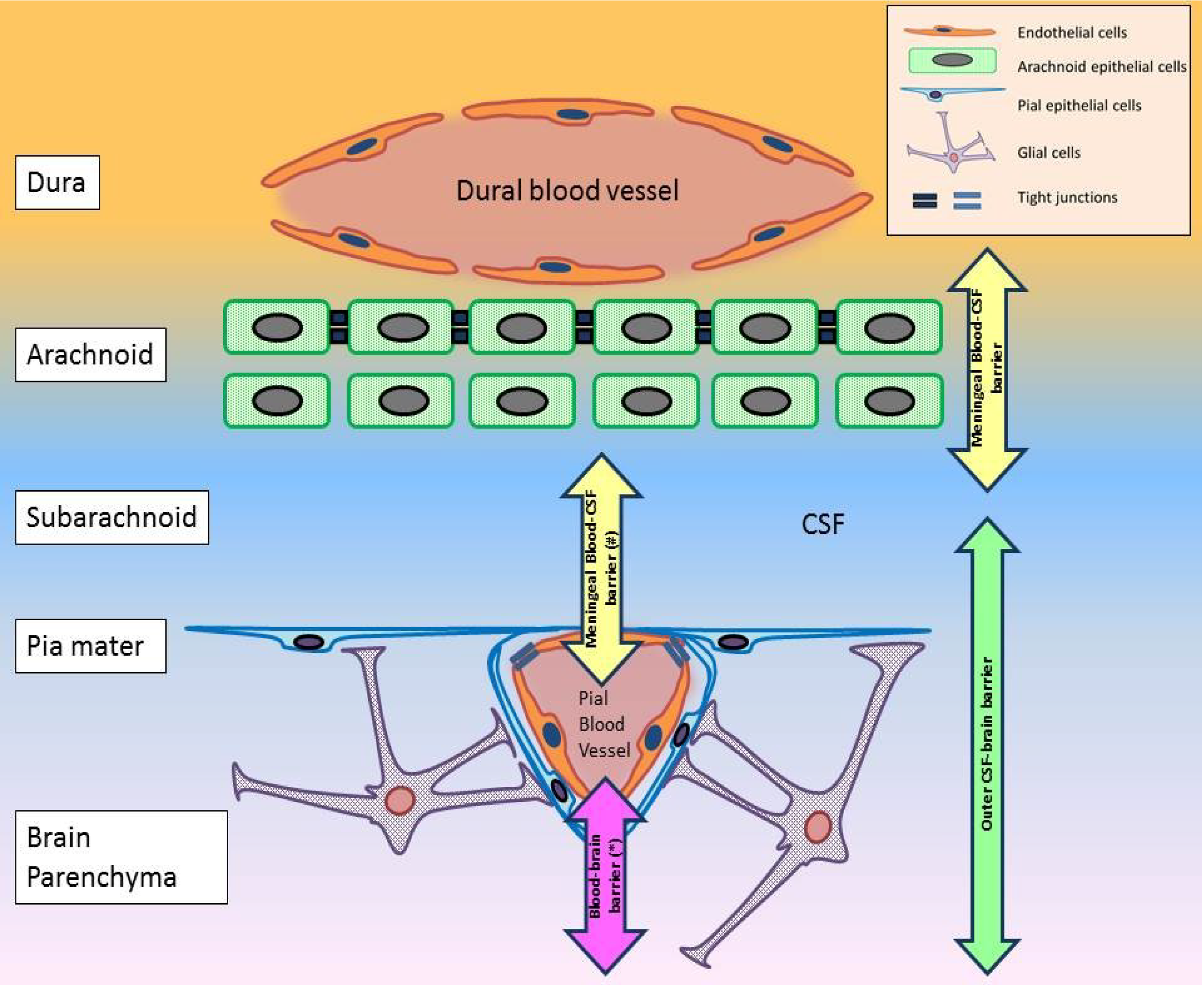
6. Molecular Determinants for WNV Neuroinvasion
| Region | Position (amino acid/nucleotide) | Mutation studied | Effect on in vivo virulence | WNV strain | Citations |
|---|---|---|---|---|---|
| prM | 141 # | Isoleucine → Threonine | Attenuation (birds) | NY99 and TM171-03-pp1 (Mexico) | [100] |
| E | Whole prM-E | prM-E of ETH76a inserted into NY99 backbone | Attenuation (mice) | ETH76a and NY99 | [97] |
| 154–156 (Glycosylation motif) | Asparagine → Serine (154) $ | Attenuation (mice) | NY99 | [101] | |
| Serine → Proline (156) # | Attenuation (birds) | NY99 and TM171-03-pp1 (Mexico) | [100] | ||
| NS1 | 250 | Proline → Leucine | Attenuation (mice) | Kunjin | [102] |
| 130-2, 175 and 207 (Glycosylation sites) $ | Asparagine → Serine/Glutamine/Alanine (130-2); Asparagine → Alanine (175 and 207) | Attenuation (mice) | NY99 (382-99) | [101,103] | |
| NS1' and NS2A | Frameshifting motif (NS2A) | Silent mutation | Partial attenuation (mice) | Kunjin | [104] |
| 30 | Alanine → Proline | Attenuation (mice) | Kunjin | [104,105] | |
| NS3 | 249 (Helicase domain) | Threonine → Proline | Enhanced virulence (birds) | KN-3829 (Kenyan) and NY99 (382-99) | [106] |
| NS4A (and 2K) | 9 (2K protein) | Valine → Methionine | Enhanced virulence(mice and birds) | North American (Texas 2003 and NY 2002 and 1999) | [107,108,109] |
| NS4B | 38, 166 and 480 | Proline → Glycine (38); Threonine → Isoleucine (116); Asparagine → Histidine (480) | Attenuation (mice) | NY99 | [110,111] |
| 249 | Glutamate → Glycine at 249 | Attenuation (mice and bird) | North American (Texas 2003 and NY 2002 and 1999) | [109,112,113] | |
| 5'UTR | 50–52 @ | NY99’s 5'UTR substitutes KUN’s | Enhanced virulence (mice) | Kunjin and NY99 | [90] |
| Region | Position (amino acid/nucleotide) | Mutation studied | Effect of mutation | In vivo/vitro | WNV strain | Citations |
|---|---|---|---|---|---|---|
| C | Unknown | N/A | Immune evasion | In vitro | NY99 | [114] |
| Unknown | N/A | Neuroinflammation | In vivo (rats) | NY99 | [115] | |
| Unknown | N/A | Tight junction protein degradation | In vitro | NY99 (385-99) | [43] | |
| prM/M | ectoM domain | N/A | Interacts with Tctex-1 | In vitro | IS-98-ST1 and other flaviviruses | [69] |
| E | 156–160 (αA’ structure of domain I) | N/A | Affinity to DC-SIGN/DC-SIGNR | In vitro | NY99 and Egypt | [33] |
| RGD motif (domain III) | N/A | Interacts with cellular integrin | In vitro | Sarafend and DEN-2 (New Guinea) | [116] | |
| NS2A | 30 | Alanine → Proline | Reduction in immune evasion via IFN-β and unknown antiviral pathway | In vitro and in vivo (mice) | Kunjin | [105] |
| NS3 | 365 (NTPase domain) | Serine → Glycine | Immune evasion via resistance to OAS1b | In vitro | NY99 (382-99) | [107] |
| NS4B | 22 and 24 | N/A | Immune evasion by inhibiting IFN cascade | In vitro | Subgenomic WNV replicons (without structural genes) derived from KUN | [117] |
| 38 | Proline → Glycine | Attenuation in neuroinvasiveness (mice) due to enhanced IFN and T cell response | In vivo | NY99 | [110,111] | |
| NS5 | 653 | Serine → Phenylalanine | Immune evasion by inhibiting JAK-STAT pathway | In vitro | Kunjin | [118] |
7. Animal Models for WNV Neuroinvasion
| Feature | Mouse | Hamster | Horse | Human |
|---|---|---|---|---|
| Peak Viremia | Moderate (~104 PFU/mL) [19,42,57,70] | Moderate (105 TCID50/mL) [119] | Low (101−3 PFU/mL) [120] | Very Low (only detectable by real-time RT-PCR) [121,122] |
| Peripheral tissue tropism |
|
|
|
|
| Distribution of CNS Lesions | Widespread:
| Widespread:
| Commonly:
| Commonly:
|
| WNV infection in the brain | High level (~105−7 PFU/g) [16,19,57] | High level (IHC) [119] | Low level to absent (IHC) [13,37,132] | Low level unless immuno-compromized (IHC) [76] |
| Mortality rate |
|
|
|
8. Conclusions
| Aspects of WNV neuroinvasion | Current limitations and unanswered questions |
|---|---|
| Transcytosis of virions across endothelium | Lack of in vivo evidence for endothelial infection by WNV |
| BBB permeability | Unknown whether increased permeability in the BBB precedes viral neuroinvasion or vice versa |
| Paracellular neuroinvasion by diffusion across endothelial junctions | Lack of evidence |
| Paracellular neuroinvasion by “Trojan Horse” method | Unknown function and trafficking behaviour of WNV infected leukocytes |
| Transneural neuroinvasion from peripheral somatic nerves | Use of artificial route of inoculation; Unknown whether virus can successfully reach the brain by this route; |
| Transneural neuroinvasion from olfactory nerves | Use of artificial intranasal inoculation |
| Blood-CSF barrier (choroid plexus and CVOs) | Unknown role in WNV neuroinvasion |
| CSF-brain barrier | Unknown role in WNV neuroinvasion |
| Arachnoid (meningeal) barrier | Unknown role in WNV neuroinvasion |
| BSCB | Unknown role in WNV neuroinvasion |
| Molecular determinants for the mechanism of WNV neuroinvasion | In vivo validation of suggested mechanisms is deficient. |
| Rodent models | Viremia and character of CNS infection are not representative of target hosts (human and horse). Alternative pathogenesis model should be explored. |
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Clark, D.C.; Brault, A.C.; Hunsperger, E. The contribution of rodent models to the pathological assessment of flaviviral infections of the central nervous system. Arch. Virol. 2012, 157, 1423–1440. [Google Scholar] [CrossRef]
- Steiner, I.; Kennedy, P.G. West nile virus introduction into the new world. Neurology 2013, 81, 1441–1442. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. West nile virus and other arboviral diseases—United States, 2012. In MMWR; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2013; Volume 62; pp. 513–528. [Google Scholar]
- Debiasi, R.L. West nile virus neuroinvasive disease. Curr. Infect. Dis. Rep. 2011, 13, 350–359. [Google Scholar] [CrossRef]
- Hayes, E.B.; Gubler, D.J. West nile virus: Epidemiology and clinical features of an emerging epidemic in the united states. Annu. Rev. Med. 2006, 57, 181–194. [Google Scholar] [CrossRef]
- Laperriere, V.; Brugger, K.; Rubel, F. Simulation of the seasonal cycles of bird, equine and human west nile virus cases. Prev. Vet. Med. 2011, 98, 99–110. [Google Scholar] [CrossRef]
- Tyler, K.L. Emerging viral infections of the central nervous system: Part 1. Arch. Neurol. 2009, 66, 939–948. [Google Scholar]
- Castillo-Olivares, J.; Wood, J. West nile virus infection of horses. Vet. Res. 2004, 35, 467–483. [Google Scholar] [CrossRef]
- Brinton, M.A. The molecular biology of west nile virus: A new invader of the western hemisphere. Annu. Rev. Microbiol. 2002, 56, 371–402. [Google Scholar] [CrossRef]
- Martin-Acebes, M.A.; Saiz, J.C. West nile virus: A re-emerging pathogen revisited. World J. Virol. 2012, 1, 51–70. [Google Scholar] [CrossRef]
- Crowder, D.W.; Dykstra, E.A.; Brauner, J.M.; Duffy, A.; Reed, C.; Martin, E.; Peterson, W.; Carriere, Y.; Dutilleul, P.; Owen, J.P. West nile virus prevalence across landscapes is mediated by local effects of agriculture on vector and host communities. PLoS One 2013, 8, e55006. [Google Scholar]
- Sampson, B.A.; Ambrosi, C.; Charlot, A.; Reiber, K.; Veress, J.F.; Armbrustmacher, V. The pathology of human west nile virus infection. Hum. Pathol. 2000, 31, 527–531. [Google Scholar]
- Angenvoort, J.; Brault, A.C.; Bowen, R.A.; Groschup, M.H. West nile viral infection of equids. Vet. Microbiol. 2013.
- Bowen, R.A.; Nemeth, N.M. Experimental infections with west nile virus. Curr. Opin. Infect. Dis. 2007, 20, 293–297. [Google Scholar] [CrossRef]
- Brown, A.N.; Kent, K.A.; Bennett, C.J.; Bernard, K.A. Tissue tropism and neuroinvasion of west nile virus do not differ for two mouse strains with different survival rates. Virology 2007, 368, 422–430. [Google Scholar] [CrossRef]
- Garcia-Tapia, D.; Hassett, D.E.; Mitchell, W.J., Jr.; Johnson, G.C.; Kleiboeker, S.B. West nile virus encephalitis: Sequential histopathological and immunological events in a murine model of infection. J. Neurovirol. 2007, 13, 130–138. [Google Scholar] [CrossRef]
- Lim, P.Y.; Louie, K.L.; Styer, L.M.; Shi, P.Y.; Bernard, K.A. Viral pathogenesis in mice is similar for west nile virus derived from mosquito and mammalian cells. Virology 2010, 400, 93–103. [Google Scholar] [CrossRef]
- Weiner, L.P.; Cole, G.A.; Nathanson, N. Experimental encephalitis following peripheral inoculation of west nile virus in mice of different ages. J. Hyg. (Lond.) 1970, 68, 435–446. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B cells and antibody play critical roles in the immediate defense of disseminated infection by west nile encephalitis virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar]
- Ben-Nathan, D.; Lustig, S.; Feuerstein, G. The influence of cold or isolation stress on neuroinvasiveness and virulence of an attenuated variant of west nile virus. Arch. Virol. 1989, 109, 1–10. [Google Scholar]
- Ben-Nathan, D.; Lustig, S.; Kobiler, D. Cold stress-induced neuroinvasiveness of attenuated arboviruses is not solely mediated by corticosterone. Arch. Virol. 1996, 141, 1221–1229. [Google Scholar] [CrossRef]
- Diamond, M.S.; Sitati, E.M.; Friend, L.D.; Higgs, S.; Shrestha, B.; Engle, M. A critical role for induced igm in the protection against west nile virus infection. J. Exp. Med. 2003, 198, 1853–1862. [Google Scholar]
- Wang, T.; Scully, E.; Yin, Z.; Kim, J.H.; Wang, S.; Yan, J.; Mamula, M.; Anderson, J.F.; Craft, J.; Fikrig, E. Ifn-gamma-producing gamma delta t cells help control murine west nile virus infection. J. Immunol. 2003, 171, 2524–2531. [Google Scholar] [CrossRef]
- Shrestha, B.; Diamond, M.S. Role of cd8(+) t cells in control of west nile virus infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Alpha/beta interferon protects against lethal west nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar]
- Mehlhop, E.; Diamond, M.S. Protective immune responses against west nile virus are primed by distinct complement activation pathways. J. Exp. Med. 2006, 203, 1371–1381. [Google Scholar] [CrossRef]
- Samuel, M.A.; Whitby, K.; Keller, B.C.; Marri, A.; Barchet, W.; Williams, B.R.; Silverman, R.H.; Gale, M., Jr.; Diamond, M.S. Pkr and rnase l contribute to protection against lethal west nile virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 2006, 80, 7009–7019. [Google Scholar] [CrossRef]
- Shrestha, B.; Wang, T.; Samuel, M.A.; Whitby, K.; Craft, J.; Fikrig, E.; Diamond, M.S. Gamma interferon plays a crucial early antiviral role in protection against west nile virus infection. J. Virol. 2006, 80, 5338–5348. [Google Scholar]
- Wang, T.; Gao, Y.; Scully, E.; Davis, C.T.; Anderson, J.F.; Welte, T.; Ledizet, M.; Koski, R.; Madri, J.A.; Barrett, A.; et al. Gamma delta t cells facilitate adaptive immunity against west nile virus infection in mice. J. Immunol. 2006, 177, 1825–1832. [Google Scholar] [CrossRef]
- Correale, J.; Villa, A. Cellular elements of the blood-brain barrier. Neurochem. Res. 2009, 34, 2067–2077. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology 2009, 385, 425–433. [Google Scholar] [CrossRef]
- Hasebe, R.; Suzuki, T.; Makino, Y.; Igarashi, M.; Yamanouchi, S.; Maeda, A.; Horiuchi, M.; Sawa, H.; Kimura, T. Transcellular transport of west nile virus-like particles across human endothelial cells depends on residues 156 and 159 of envelope protein. BMC Microbiol. 2010, 10, 165–165. [Google Scholar] [CrossRef]
- Liou, M.L.; Hsu, C.Y. Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res. 1998, 293, 389–394. [Google Scholar] [CrossRef]
- Dropulic, B.; Masters, C.L. Entry of neurotropic arboviruses into the central nervous system: An in vitro study using mouse brain endothelium. J. Infect. Dis. 1990, 161, 685–691. [Google Scholar] [CrossRef]
- German, A.C.; Myint, K.S.; Mai, N.T.; Pomeroy, I.; Phu, N.H.; Tzartos, J.; Winter, P.; Collett, J.; Farrar, J.; Barrett, A.; et al. A preliminary neuropathological study of japanese encephalitis in humans and a mouse model. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 1135–1145. [Google Scholar]
- Cantile, C.; Del Piero, F.; di Guardo, G.; Arispici, M. Pathologic and immunohistochemical findings in naturally occuring west nile virus infection in horses. Vet. Pathol. 2001, 38, 414–421. [Google Scholar] [CrossRef]
- Kong, K.F.; Delroux, K.; Wang, X.; Qian, F.; Arjona, A.; Malawista, S.E.; Fikrig, E.; Montgomery, R.R. Dysregulation of tlr3 impairs the innate immune response to west nile virus in the elderly. J. Virol. 2008, 82, 7613–7623. [Google Scholar] [CrossRef]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates west nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef]
- Arjona, A.; Foellmer, H.G.; Town, T.; Leng, L.; McDonald, C.; Wang, T.; Wong, S.J.; Montgomery, R.R.; Fikrig, E.; Bucala, R. Abrogation of macrophage migration inhibitory factor decreases west nile virus lethality by limiting viral neuroinvasion. J. Clin. Invest. 2007, 117, 3059–3066. [Google Scholar] [CrossRef]
- Verma, S.; Kumar, M.; Gurjav, U.; Lum, S.; Nerurkar, V.R. Reversal of west nile virus-induced blood–brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 2010, 397, 130–138. [Google Scholar] [CrossRef]
- Roe, K.; Kumar, M.; Lum, S.; Orillo, B.; Nerurkar, V.R.; Verma, S. West nile virus-induced disruption of the blood-brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J. Gen. Virol. 2012, 93, 1193–1203. [Google Scholar]
- Medigeshi, G.R.; Hirsch, A.J.; Brien, J.D.; Uhrlaub, J.L.; Mason, P.W.; Wiley, C.; Nikolich-Zugich, J.; Nelson, J.A. West nile virus capsid degradation of claudin proteins disrupts epithelial barrier function. J. Virol. 2009, 83, 6125–6134. [Google Scholar] [CrossRef]
- Xu, Z.; Waeckerlin, R.; Urbanowski, M.D.; van Marle, G.; Hobman, T.C. West nile virus infection causes endocytosis of a specific subset of tight junction membrane proteins. PLoS One 2012, 7, e37886. [Google Scholar]
- Shen, J.; Tto, S.S.; Schrieber, L.; King, N.J.C. Early e-selectin, vcam-1, icam-1, and late major histocompatibility complex antigen induction on human endothelial cells by flavivirus and comodulation of adhesion molecule expression by immune cytokines. J. Virol. 1997, 71, 9323–9332. [Google Scholar]
- Dai, J.; Wang, P.; Bai, F.; Town, T.; Fikrig, E. Icam-1 participates in the entry of west nile virus into the central nervous system. J. Virol. 2008, 82, 4164–4168. [Google Scholar] [CrossRef]
- Sigmundsdottir, H.; Butcher, E.C. Environmental cues, dendritic cells and the programming of tissue-selective lymphocyte trafficking. Nat. Immunol. 2008, 9, 981–987. [Google Scholar] [CrossRef]
- Kumar, M.; Roe, K.; Nerurkar, P.V.; Orillo, B.; Thompson, K.S.; Verma, S.; Nerurkar, V.R. Reduced immune cell infiltration and increased pro-inflammatory mediators in the brain of type 2 diabetic mouse model infected with west nile virus. J. Neuroinflammation 2014, 11, 80. [Google Scholar] [CrossRef]
- Getts, D.R.; Terry, R.L.; Getts, M.T.; Muller, M.; Rana, S.; Deffrasnes, C.; Ashhurst, T.M.; Radford, J.; Hofer, M.; Thomas, S.; et al. Targeted blockade in lethal west nile virus encephalitis indicates a crucial role for very late antigen (vla)-4-dependent recruitment of nitric oxide-producing macrophages. J. Neuroinflammation 2012, 9, 1742–2094. [Google Scholar]
- Samuel, M.A.; Diamond, M.S. Pathogenesis of west nile virus infection: A balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 2006, 80, 9349–9360. [Google Scholar] [CrossRef]
- Sips, G.J.; Wilschut, J.; Smit, J.M. Neuroinvasive flavivirus infections. Rev. Med. Virol. 2012, 22, 69–87. [Google Scholar] [CrossRef]
- Kramer-Hammerle, S.; Rothenaigner, I.; Wolff, H.; Bell, J.E.; Brack-Werner, R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005, 111, 194–213. [Google Scholar] [CrossRef]
- Kumar, M.; Verma, S.; Nerurkar, V.R. Pro-inflammatory cytokines derived from west nile virus (wnv)-infected sk-n-sh cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflammation 2010, 7, 73–73. [Google Scholar] [CrossRef]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Lim, S.M.; Koraka, P.; Osterhaus, A.D.; Martina, B.E. West nile virus: Immunity and pathogenesis. Viruses 2011, 3, 811–828. [Google Scholar] [CrossRef]
- Garcia-Tapia, D.; Loiacono, C.M.; Klelboeker, S.B. Replication of west nile virus in equine peripheral blood mononuclear cells. Vet. Immunol. Immunopathol. 2006, 110, 229–244. [Google Scholar] [CrossRef]
- Hunsperger, E.A.; Roehrig, J.T. Temporal analyses of the neuropathogenesis of a west nile virus infection in mice. J. Neurovirol. 2006, 12, 129–139. [Google Scholar] [CrossRef]
- Rios, M.; Zhang, M.J.; Grinev, A.; Srinivasan, K.; Daniel, S.; Wood, O.; Hewlett, I.K.; Dayton, A.I. Monocytes-macrophages are a potential target in human infection with west nile virus through blood transfusion. Transfusion (Paris) 2006, 46, 659–667. [Google Scholar] [CrossRef]
- Wang, S.; Welte, T.; McGargill, M.; Town, T.; Thompson, J.; Anderson, J.F.; Flavell, R.A.; Fikrig, E.; Hedrick, S.M.; Wang, T. Drak2 contributes to west nile virus entry into the brain and lethal encephalitis. J. Immunol. 2008, 181, 2084–2091. [Google Scholar]
- King, N.J.; Getts, D.R.; Getts, M.T.; Rana, S.; Shrestha, B.; Kesson, A.M. Immunopathology of flavivirus infections. Immunol. Cell Biol. 2007, 85, 33–42. [Google Scholar] [CrossRef]
- Bielefeldt-Ohmann, H.; Smirnova, N.P.; Tolnay, A.E.; Webb, B.T.; Antoniazzi, A.Q.; van Campen, H.; Hansen, T.R. Neuro-invasion by a 'trojan horse' strategy and vasculopathy during intrauterine flavivirus infection. Int. J. Exp. Pathol. 2012, 93, 24–33. [Google Scholar]
- Li, J.K.; Liang, J.J.; Liao, C.L.; Lin, Y.L. Autophagy is involved in the early step of japanese encephalitis virus infection. Microbes Infect. 2012, 14, 159–168. [Google Scholar] [CrossRef]
- Hong, R.; Bai, W.Y.; Zhai, J.W.; Liu, W.; Li, X.Y.; Zhang, J.M.; Cui, X.X.; Zhao, X.; Ye, X.L.; Deng, Q.; et al. Novel recombinant hepatitis b virus vectors efficiently deliver protein and rna encoding genes into primary hepatocytes. J. Virol. 2013, 87, 6615–6624. [Google Scholar] [CrossRef]
- Samuel, M.A.; Wang, H.; Siddharthan, V.; Morrey, J.D.; Diamond, M.S. Axonal transport mediates west nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. USA 2007, 104, 17140–17145. [Google Scholar]
- Wang, H.; Siddharthan, V.; Hall, J.O.; Morrey, J.D. West nile virus preferentially transports along motor neuron axons after sciatic nerve injection of hamsters. J. Neurovirol. 2009, 15, 293–299. [Google Scholar] [CrossRef]
- Lorenz, M.D.; Coates, J.R.; Kent, M. Handbook of Veterinary Neurology, 5th ed.; Elsevier/Saunders: St. Louis, MI, USA, 2011; p. 28. [Google Scholar]
- Antoine, J.C.; Honnorat, J.; Camdessanche, J.P.; Magistris, M.; Absi, L.; Mosnier, J.F.; Petiot, P.; Kopp, N.; Michel, D. Paraneoplastic anti-cv2 antibodies react with peripheral nerve and are associated with a mixed axonal and demyelinating peripheral neuropathy. Ann. Neurol. 2001, 49, 214–221. [Google Scholar] [CrossRef]
- Wang, H.; Siddharthan, V.; Hall, J.O.; Morrey, J.D. Autonomic nervous dysfunction in hamsters infected with west nile virus. PLoS One 2011, 6, e19575. [Google Scholar]
- Brault, J.B.; Kudelko, M.; Vidalain, P.O.; Tangy, F.; Despres, P.; Pardigon, N. The interaction of flavivirus m protein with light chain tctex-1 of human dynein plays a role in late stages of virus replication. Virology 2011, 417, 369–378. [Google Scholar] [CrossRef]
- Hunsperger, E.A.; Roehrig, J.T. Nocodazole delays viral entry into the brain following footpad inoculation with west nile virus in mice. J. Neurovirol. 2009, 15, 211–218. [Google Scholar] [CrossRef]
- Monath, T.P.; Cropp, C.B.; Harrison, A.K. Mode of entry of a neurotropic arbovirus into the central nervous system. Reinvestigation of an old controversy. Lab. Invest. 1983, 48, 399–410. [Google Scholar]
- Nir, Y.; Beemer, A.; Goldwasser, R.A. West nile virus infection in mice following exposure to a viral aerosol. Br. J. Exp. Pathol. 1965, 46, 443. [Google Scholar]
- McMinn, P.C.; Dalgarno, L.; Weir, R.C. A comparison of the spread of murray valley encephalitis viruses of high or low neuroinvasiveness in the tissues of swiss mice after peripheral inoculation. Virology 1996, 220, 414–423. [Google Scholar] [CrossRef]
- Grevers, G.; Herrmann, U. Fenestrated endothelia in vessels of the nasal mucosa. An electron-microscopic study in the rabbit. Arch. Otorhinolaryngol. 1987, 244, 55–60. [Google Scholar] [CrossRef]
- Yamada, M.; Nakamura, K.; Yoshii, M.; Kaku, Y.; Narita, M. Brain lesions induced by experimental intranasal infection of japanese encephalitis virus in piglets. J. Comp. Pathol. 2009, 141, 156–162. [Google Scholar] [CrossRef]
- Guarner, J.; Shieh, W.J.; Hunter, S.; Paddock, C.D.; Morken, T.; Campbell, G.L.; Marfin, A.A.; Zaki, S.R. Clinicopathologic study and laboratory diagnosis of 23 cases with west nile virus encephalomyelitis. Hum. Pathol. 2004, 35, 983–990. [Google Scholar] [CrossRef]
- Morrey, J.D.; Olsen, A.L.; Siddharthan, V.; Motter, N.E.; Wang, H.; Taro, B.S.; Chen, D.; Ruffner, D.; Hall, J.O. Increased blood-brain barrier permeability is not a primary determinant for lethality of west nile virus infection in rodents. J. Gen. Virol. 2008, 89, 467–473. [Google Scholar]
- Ichikawa, H.; Itoh, K. Blood-arachnoid barrier disruption in experimental rat meningitis detected using gadolinium-enhancement ratio imaging. Brain Res. 2011, 1390, 142–149. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Saunders, N.R.; Habgood, M.D.; Dziegielewska, K.M. Barrier mechanisms in the brain, ii. Immature brain. Clin. Exp. Pharmacol. Physiol. 1999, 26, 85–91. [Google Scholar] [CrossRef]
- Saunders, N.R.; Liddelow, S.A.; Dziegielewska, K.M. Barrier mechanisms in the developing brain. Front. Pharmacol. 2012, 3, 1–18. [Google Scholar]
- Saunders, N.R.; Ek, C.J.; Habgood, M.D.; Dziegielewska, K.M. Barriers in the brain: A renaissance? Trends Neurosci. 2008, 31, 279–286. [Google Scholar] [CrossRef]
- Segal, M.B. The choroid plexuses and the barriers between the blood and the cerebrospinal fluid. Cell. Mol. Neurobiol. 2000, 20, 183–196. [Google Scholar] [CrossRef]
- Langlet, F.; Mullier, A.; Bouret, S.G.; Prevot, V.; Dehouck, B. Tanycyte-like cells form a blood-cerebrospinal fluid barrier in the circumventricular organs of the mouse brain. J. Comp. Neurol. 2013, 521, 3389–3405. [Google Scholar] [CrossRef]
- Allt, G.; Lawrenson, J.G. Is the pial microvessel a good model for blood-brain barrier studies? Brain Res. Brain Res. Rev. 1997, 24, 67–76. [Google Scholar] [CrossRef]
- Reina, M.A.; De Leon Casasola Ode, L.; Villanueva, M.C.; Lopez, A.; Maches, F.; de Andres, J.A. Ultrastructural findings in human spinal pia mater in relation to subarachnoid anesthesia. Anesth. Analg. 2004, 98, 1479–1485. [Google Scholar]
- Clay, C.C.; Rodrigues, D.S.; Ho, Y.S.; Fallert, B.A.; Janatpour, K.; Reinhart, T.A.; Esser, U. Neuroinvasion of fluorescein-positive monocytes in acute simian immunodeficiency virus infection. J. Virol. 2007, 81, 12040–12048. [Google Scholar] [CrossRef]
- Chaves, A.J.; Busquets, N.; Valle, R.; Rivas, R.; Vergara-Alert, J.; Dolz, R.; Ramis, A.; Darji, A.; Majo, N. Neuropathogenesis of a highly pathogenic avian influenza virus (h7n1) in experimentally infected chickens. Vet. Res. 2011, 42, 106. [Google Scholar] [CrossRef] [Green Version]
- Frost, M.J.; Zhang, J.; Edmonds, J.H.; Prow, N.A.; Gu, X.; Davis, R.; Hornitzky, C.; Arzey, K.E.; Finlaison, D.; Hick, P.; et al. Characterization of virulent west nile virus kunjin strain, australia, 2011. Emerg. Infect. Dis. 2012, 18, 792–800. [Google Scholar]
- Audsley, M.; Edmonds, J.; Liu, W.; Mokhonov, V.; Mokhonova, E.; Melian, E.B.; Prow, N.; Hall, R.A.; Khromykh, A.A. Virulence determinants between new york 99 and kunjin strains of west nile virus. Virology 2011, 414, 63–73. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sengillo, J.D.; Bell, R.D.; Wang, J.; Zlokovic, B.V. Blood-spinal cord barrier pericyte reductions contribute to increased capillary permeability. J. Cereb. Blood Flow Metab. 2012, 32, 1841–1852. [Google Scholar] [CrossRef]
- Bartanusz, V.; Jezova, D.; Alajajian, B.; Digicaylioglu, M. The blood-spinal cord barrier: Morphology and clinical implications. Ann. Neurol. 2011, 70, 194–206. [Google Scholar] [CrossRef]
- Prow, N.A.; Irani, D.N. The opioid receptor antagonist, naloxone, protects spinal motor neurons in a murine model of alphavirus encephalomyelitis. Exp. Neurol. 2007, 205, 461–470. [Google Scholar] [CrossRef]
- Brault, A.C. Changing patterns of west nile virus transmission: Altered vector competence and host susceptibility. Vet. Res. 2009, 40, 1–19. [Google Scholar] [CrossRef]
- May, F.J.; Davis, C.T.; Tesh, R.B.; Barrett, A.D. Phylogeography of west nile virus: From the cradle of evolution in africa to eurasia, australia, and the americas. J. Virol. 2011, 85, 2964–2974. [Google Scholar]
- McMullen, A.R.; Albayrak, H.; May, F.J.; Davis, C.T.; Beasley, D.W.; Barrett, A.D. Molecular evolution of lineage 2 west nile virus. J. Gen. Virol. 2013, 94, 318–325. [Google Scholar] [CrossRef]
- Beasley, D.W.C.; Whiteman, M.C.; Zhang, S.L.; Huang, C.Y.H.; Schneider, B.S.; Smith, D.R.; Gromowski, G.D.; Higgs, S.; Kinney, R.M.; Barrett, A.D.T. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 west nile virus strains. J. Virol. 2005, 79, 8339–8347. [Google Scholar] [CrossRef]
- Appler, K.K.; Brown, A.N.; Stewart, B.S.; Behr, M.J.; Demarest, V.L.; Wong, S.J.; Bernard, K.A. Persistence of west nile virus in the central nervous system and periphery of mice. PLoS One 2010, 5, e10649. [Google Scholar]
- Morrey, J.D.; Siddharthan, V.; Wang, H.; Hall, J.O. Respiratory insufficiency correlated strongly with mortality of rodents infected with west nile virus. PLoS One 2012, 7, e38672. [Google Scholar]
- Langevin, S.A.; Bowen, R.A.; Ramey, W.N.; Sanders, T.A.; Maharaj, P.D.; Fang, Y.; Cornelius, J.; Barker, C.M.; Reisen, W.K.; Beasley, D.W.; et al. Envelope and pre-membrane protein structural amino acid mutations mediate diminished avian growth and virulence of a mexican west nile virus isolate. J. Gen. Virol. 2011, 92, 2810–2820. [Google Scholar] [CrossRef]
- Whiteman, M.C.; Li, L.; Wicker, J.A.; Kinney, R.M.; Huang, C.; Beasley, D.W.; Chung, K.M.; Diamond, M.S.; Solomon, T.; Barrett, A.D. Development and characterization of non-glycosylated e and ns1 mutant viruses as a potential candidate vaccine for west nile virus. Vaccine 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Hall, R.A.; Khromykh, A.A.; Mackenzie, J.M.; Scherret, J.H.; Khromykh, T.I.; Mackenzie, J.S. Loss of dimerisation of the nonstructural protein ns1 of kunjin virus delays viral replication and reduces virulence in mice, but still allows secretion of ns1. Virology 1999, 264, 66–75. [Google Scholar] [CrossRef]
- Whiteman, M.C.; Wicker, J.A.; Kinney, R.M.; Huang, C.Y.; Solomon, T.; Barrett, A.D. Multiple amino acid changes at the first glycosylation motif in ns1 protein of west nile virus are necessary for complete attenuation for mouse neuroinvasiveness. Vaccine 2011, 29, 9702–9710. [Google Scholar] [CrossRef]
- Melian, E.B.; Hinzman, E.; Nagasaki, T.; Firth, A.E.; Wills, N.M.; Nouwens, A.S.; Blitvich, B.J.; Leung, J.; Funk, A.; Atkins, J.F.; et al. Ns1' of flaviviruses in the japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J. Virol. 2010, 84, 1641–1647. [Google Scholar] [CrossRef]
- Liu, W.J.; Wang, X.J.; Clark, D.C.; Lobigs, M.; Hall, R.A.; Khromykh, A.A. A single amino acid substitution in the west nile virus nonstructural protein ns2a disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J. Virol. 2006, 80, 2396–2404. [Google Scholar]
- Brault, A.C.; Huang, C.Y.; Langevin, S.A.; Kinney, R.M.; Bowen, R.A.; Ramey, W.N.; Panella, N.A.; Holmes, E.C.; Powers, A.M.; Miller, B.R. A single positively selected west nile viral mutation confers increased virogenesis in american crows. Nat. Genet. 2007, 39, 1162–1166. [Google Scholar] [CrossRef]
- Mertens, E.; Kajaste-Rudnitski, A.; Torres, S.; Funk, A.; Frenkiel, M.-P.; Iteman, I.; Khromykh, A.A.; Desprès, P. Viral determinants in the ns3 helicase and 2k peptide that promote west nile virus resistance to antiviral action of 2',5'-oligoadenylate synthetase 1b. Virology 2010, 399, 176–185. [Google Scholar]
- Zou, G.; Puig-Basagoiti, F.; Zhang, B.; Qing, M.; Chen, L.; Pankiewicz, K.W.; Felczak, K.; Yuan, Z.; Shi, P.Y. A single-amino acid substitution in west nile virus 2k peptide between ns4a and ns4b confers resistance to lycorine, a flavivirus inhibitor. Virology 2009, 384, 242–252. [Google Scholar] [CrossRef]
- Davis, C.T.; Beasley, D.W.; Guzman, H.; Siirin, M.; Parsons, R.E.; Tesh, R.B.; Barrett, A.D. Emergence of attenuated west nile virus variants in texas, 2003. Virology 2004, 330, 342–350. [Google Scholar] [CrossRef]
- Wicker, J.A.; Whiteman, M.C.; Beasley, D.W.; Davis, C.T.; McGee, C.E.; Lee, J.C.; Higgs, S.; Kinney, R.M.; Huang, C.Y.; Barrett, A.D. Mutational analysis of the west nile virus ns4b protein. Virology 2012, 426, 22–33. [Google Scholar] [CrossRef]
- Welte, T.; Xie, G.; Wicker, J.A.; Whiteman, M.C.; Li, L.; Rachamallu, A.; Barrett, A.; Wang, T. Immune responses to an attenuated west nile virus ns4b-p38g mutant strain. Vaccine 2011, 29, 4853–4861. [Google Scholar]
- Wicker, J.A.; Whiteman, M.C.; Beasley, D.W.; Davis, C.T.; Zhang, S.; Schneider, B.S.; Higgs, S.; Kinney, R.M.; Barrett, A.D. A single amino acid substitution in the central portion of the west nile virus ns4b protein confers a highly attenuated phenotype in mice. Virology 2006, 349, 245–253. [Google Scholar] [CrossRef]
- Puig-Basagoiti, F.; Tilgner, M.; Bennett, C.J.; Zhou, Y.; Munoz-Jordan, J.L.; Garcia-Sastre, A.; Bernard, K.A.; Shi, P.Y. A mouse cell-adapted ns4b mutation attenuates west nile virus rna synthesis. Virology 2007, 361, 229–241. [Google Scholar] [CrossRef]
- Hunt, T.A.; Urbanowski, M.D.; Kakani, K.; Law, L.M.; Brinton, M.A.; Hobman, T.C. Interactions between the west nile virus capsid protein and the host cell-encoded phosphatase inhibitor, i2pp2a. Cell. Microbiol. 2007, 9, 2756–2766. [Google Scholar] [CrossRef]
- van Marle, G.; Antony, J.; Ostermann, H.; Dunham, C.; Hunt, T.; Halliday, W.; Maingat, F.; Urbanowski, M.D.; Hobman, T.; Peeling, J.; et al. West nile virus-induced neuroinflammation: Glial infection and capsid protein-mediated neurovirulence. J. Virol. 2007, 81, 10933–10949. [Google Scholar] [CrossRef]
- Chu, J.J.H.; Rajamanonmani, R.; Li, J.; Bhuvanakantham, R.; Lescar, J.; Ng, M.L. Inhibition of west nile virus entry by using a recombinant domain iii from the envelope glycoprotein. J. Gen. Virol. 2005, 86, 405–412. [Google Scholar] [CrossRef]
- Evans, J.D.; Seeger, C. Differential effects of mutations in ns4b on west nile virus replication and inhibition of interferon signaling. J. Virol. 2007, 81, 11809–11816. [Google Scholar] [CrossRef]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The ns5 protein of the virulent west nile virus ny99 strain is a potent antagonist of type i interferon-mediated jak-stat signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar]
- Xiao, S.Y.; Guzman, H.; Zhang, H.; Travassos da Rosa, A.P.; Tesh, R.B. West nile virus infection in the golden hamster (mesocricetus auratus): A model for west nile encephalitis. Emerg. Infect. Dis. 2001, 7, 714–721. [Google Scholar]
- Bunning, M.L.; Bowen, R.A.; Cropp, C.B.; Sullivan, K.G.; Davis, B.S.; Komar, N.; Godsey, M.S.; Baker, D.; Hettler, D.L.; Holmes, D.A.; et al. Experimental infection of horses with west nile virus. Emerg. Infect. Dis. 2002, 8, 380–386. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Marfin, A.A. Manifestations of west nile neuroinvasive disease. Rev. Med. Virol. 2006, 16, 209–224. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kerst, A.J.; Nasci, R.S.; Godsey, M.S.; Mitchell, C.J.; Savage, H.M.; Komar, N.; Panella, N.A.; Allen, B.C.; Volpe, K.E.; et al. Rapid detection of west nile virus from human clinical specimens, field-collected mosquitoes, and avian samples by a taqman reverse transcriptase-pcr assay. J. Clin. Microbiol. 2000, 38, 4066–4071. [Google Scholar]
- Shrestha, B.; Zhang, B.; Purtha, W.E.; Klein, R.S.; Diamond, M.S. Tumor necrosis factor alpha protects against lethal west nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J. Virol. 2008, 82, 8956–8964. [Google Scholar] [CrossRef]
- Tesh, R.B.; Siirin, M.; Guzman, H.; da Rosa, A.; Wu, X.Y.; Duan, T.; Lei, H.; Nunes, M.R.; Xiao, S.Y. Persistent west nile virus infection in the golden hamster: Studies on its mechanism and possible implications for other flavivirus infections. J. Infect. Dis. 2005, 192, 287–295. [Google Scholar] [CrossRef]
- Tonry, J.H.; Xiao, S.Y.; Siirin, M.; Chen, H.; da Rosa, A.P.; Tesh, R.B. Persistent shedding of west nile virus in urine of experimentally infected hamsters. Am. J. Trop. Med. Hyg. 2005, 72, 320–324. [Google Scholar]
- Ding, X.; Wu, X.; Duan, T.; Siirin, M.; Guzman, H.; Yang, Z.; Tesh, R.B.; Xiao, S.Y. Nucleotide and amino acid changes in west nile virus strains exhibiting renal tropism in hamsters. Am. J. Trop. Med. Hyg. 2005, 73, 803–807. [Google Scholar]
- Cantile, C.; di Guardo, G.; Eleni, C.; Arispici, M. Clinical and neuropathological features of west nile virus equine encephalomyelitis in italy. Equine Vet. J. 2000, 32, 31–35. [Google Scholar] [CrossRef]
- Smith, R.D.; Konoplev, S.; DeCourten-Myers, G.; Brown, T. West nile virus encephalitis with myositis and orchitis. Hum. Pathol. 2004, 35, 254–258. [Google Scholar] [CrossRef]
- Tonry, J.H.; Brown, C.B.; Cropp, C.B.; Co, J.K.G.; Bennett, S.N.; Nerurkar, V.R.; Kuberski, T.; Gubler, D.J. West nile virus detection in urine. Emerg. Infect. Dis. 2005, 11, 1294–1296. [Google Scholar] [CrossRef]
- Omalu, B.I.; Shakir, A.A.; Wang, G.; Lipkin, W.I.; Wiley, C.A. Fatal fulminant pan-meningo-polioencephalitis due to west nile virus. Brain Pathol. 2003, 13, 465–472. [Google Scholar]
- Agamanolis, D.P.; Leslie, M.J.; Caveny, E.A.; Guarner, J.; Shieh, W.J.; Zaki, S.R. Neuropathological findings in west nile virus encephalitis: A case report. Ann. Neurol. 2003, 54, 547–551. [Google Scholar] [CrossRef]
- Bielefeldt-Ohmann, H.; Bosco-Lauth, A.; Tolnay, A.E.; Prow, N.A.; Wang, W.; Khromykh, A.A.; Hall, R.A.; Bowen, R. Experimental infection of horses with the equine-pathogenic west nile virus strain wnvnsw 2011—Comparison to natural cases. University of Queensland: St. Lucia, Qld, Australia, Unpublished work; 2013. [Google Scholar]
- O'Leary, D.R.; Marfin, A.A.; Montgomery, S.P.; Kipp, A.M.; Lehman, J.A.; Biggerstaff, B.J.; Elko, V.L.; Collins, P.D.; Jones, J.E.; Campbell, G.L. The epidemic of west nile virus in the united states, 2002. Vector Borne Zoonotic Dis. 2004, 4, 61–70. [Google Scholar] [CrossRef]
- Mostashari, F.; Bunning, M.L.; Kitsutani, P.T.; Singer, D.A.; Nash, D.; Cooper, M.J.; Katz, N.; Liljebjelke, K.A.; Biggerstaff, B.J.; Fine, A.D.; et al. Epidemic west nile encephalitis, new york, 1999: Results of a household-based seroepidemiological survey. Lancet 2001, 358, 261–264. [Google Scholar] [CrossRef]
- Kleinschmidt-DeMasters, B.K.; Marder, B.A.; Levi, M.E.; Laird, S.P.; McNutt, J.T.; Escott, E.J.; Everson, G.T.; Tyler, K.L. Naturally acquired west nile virus encephalomyelitis in transplant recipients—clinical, laboratory, diagnostic, and neuropathological features. Arch. Neurol. 2004, 61, 1210–1220. [Google Scholar] [CrossRef]
- Diamond, M.S.; Mehlhop, E.; Oliphant, T.; Samuel, M.A. The host immunologic response to west nile encephalitis virus. Front. Biosci. (Landmark Ed) 2009, 14, 3024–3034. [Google Scholar]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef]
- Ratterree, M.S.; Gutierrez, R.A.; Travassos da Rosa, A.P.; Dille, B.J.; Beasley, D.W.; Bohm, R.P.; Desai, S.M.; Didier, P.J.; Bikenmeyer, L.G.; Dawson, G.J.; et al. Experimental infection of rhesus macaques with west nile virus: Level and duration of viremia and kinetics of the antibody response after infection. J. Infect. Dis. 2004, 189, 669–676. [Google Scholar] [CrossRef]
- Verstrepen, B.E.; Fagrouch, Z.; van Heteren, M.; Buitendijk, H.; Haaksma, T.; Beenhakker, N.; Palu, G.; Richner, J.M.; Diamond, M.S.; Bogers, W.M.; et al. Experimental infection of rhesus macaques and common marmosets with a european strain of west nile virus. PLoS Negl. Trop. Dis. 2014, 8, e2797. [Google Scholar] [CrossRef]
- Suen, W.; Prow, N.A.; Wang, W.; Broad, N.; Hall, R.A.; Kirkland, P.; Bielefeldt-Ohmann, H. The establishment of a rabbit model to elucidate the mechanism of neuroinvasion by an emergent australian west nile virus. In Proceedings of The 7th Australasian Virology Society Meeting, Queenstown, New Zealand, 8–11 December 2013; 2013; p. 97. [Google Scholar]
- Lee, H.H.; Hong, S.K.; Yoon, S.H.; Jang, S.J.; Bahk, Y.Y.; Song, M.D.; Park, P.J.; Lee, K.H.; Kim, C.G.; Kim, B.; et al. Immunogenicity of japanese encephalitis virus envelope protein by hyphantria cunea nuclear polyhedrosis virus vector in guinea pig. Appl. Biochem. Biotechnol. 2012, 167, 259–269. [Google Scholar] [CrossRef]
- Fenton, R.J.; Clark, A.; Potter, C.W. Immunity to influenza in ferrets. Xiv: Comparative immunity following infection or immunization with live or inactivated vaccine. Br. J. Exp. Pathol. 1981, 62, 297–307. [Google Scholar]
- Clarke, P.; Leser, J.S.; Quick, E.D.; Dionne, K.R.; Beckham, J.D.; Tyler, K.L. Death receptor-mediated apoptotic signaling is activated in the brain following infection with west nile virus in the absence of a peripheral immune response. J. Virol. 2014, 88, 1080–1089. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suen, W.W.; Prow, N.A.; Hall, R.A.; Bielefeldt-Ohmann, H. Mechanism of West Nile Virus Neuroinvasion: A Critical Appraisal. Viruses 2014, 6, 2796-2825. https://doi.org/10.3390/v6072796
Suen WW, Prow NA, Hall RA, Bielefeldt-Ohmann H. Mechanism of West Nile Virus Neuroinvasion: A Critical Appraisal. Viruses. 2014; 6(7):2796-2825. https://doi.org/10.3390/v6072796
Chicago/Turabian StyleSuen, Willy W., Natalie A. Prow, Roy A. Hall, and Helle Bielefeldt-Ohmann. 2014. "Mechanism of West Nile Virus Neuroinvasion: A Critical Appraisal" Viruses 6, no. 7: 2796-2825. https://doi.org/10.3390/v6072796