Incorporation of Hepatitis C Virus E1 and E2 Glycoproteins: The Keystones on a Peculiar Virion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
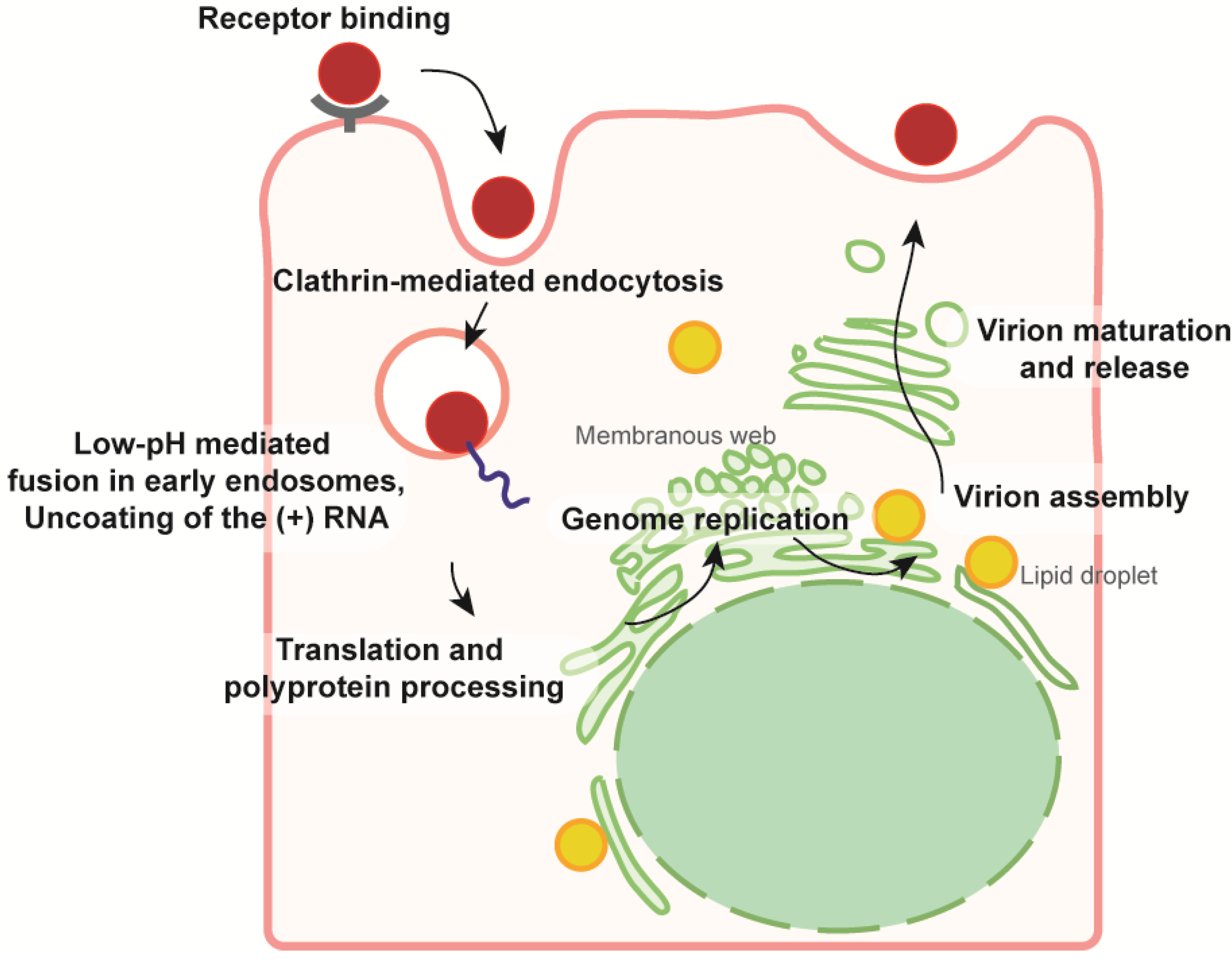
:1. Introduction
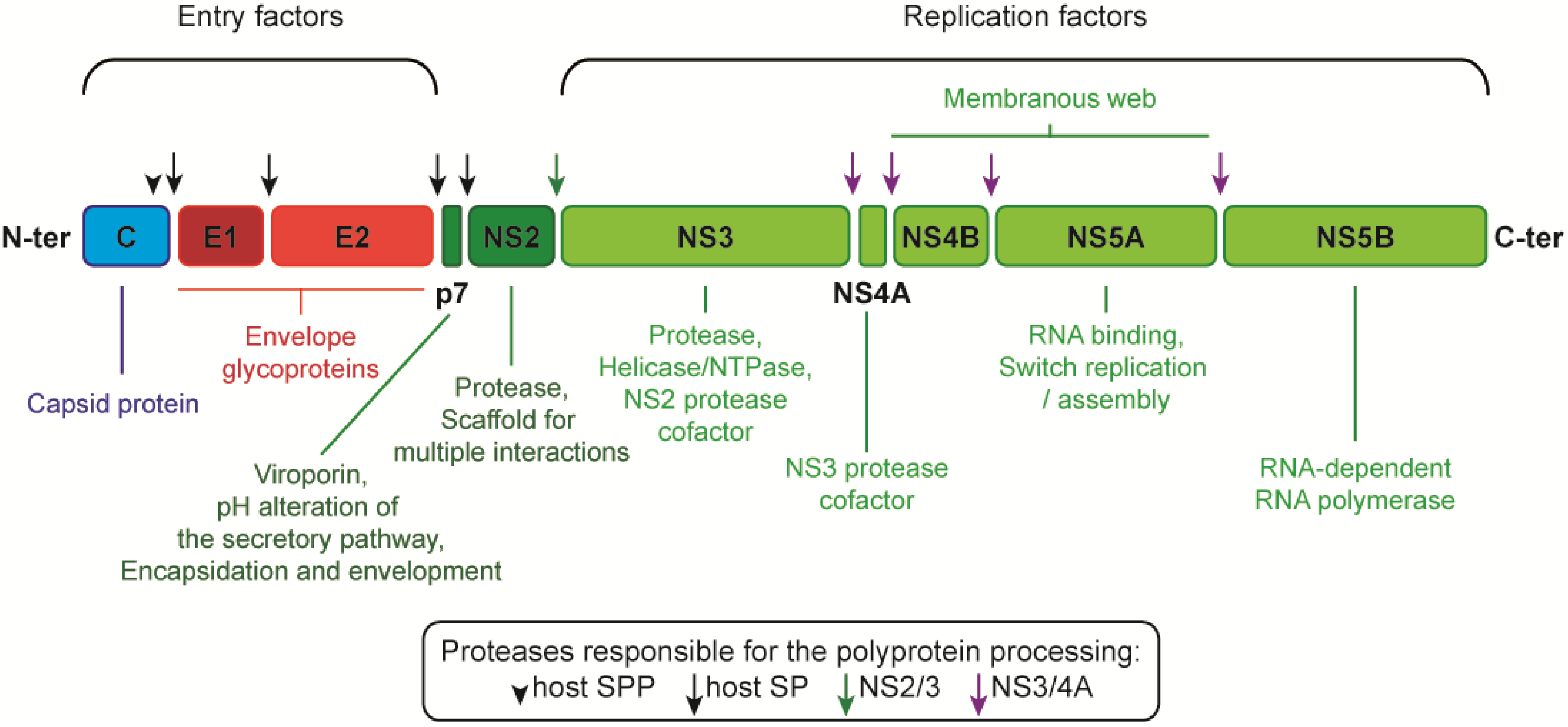
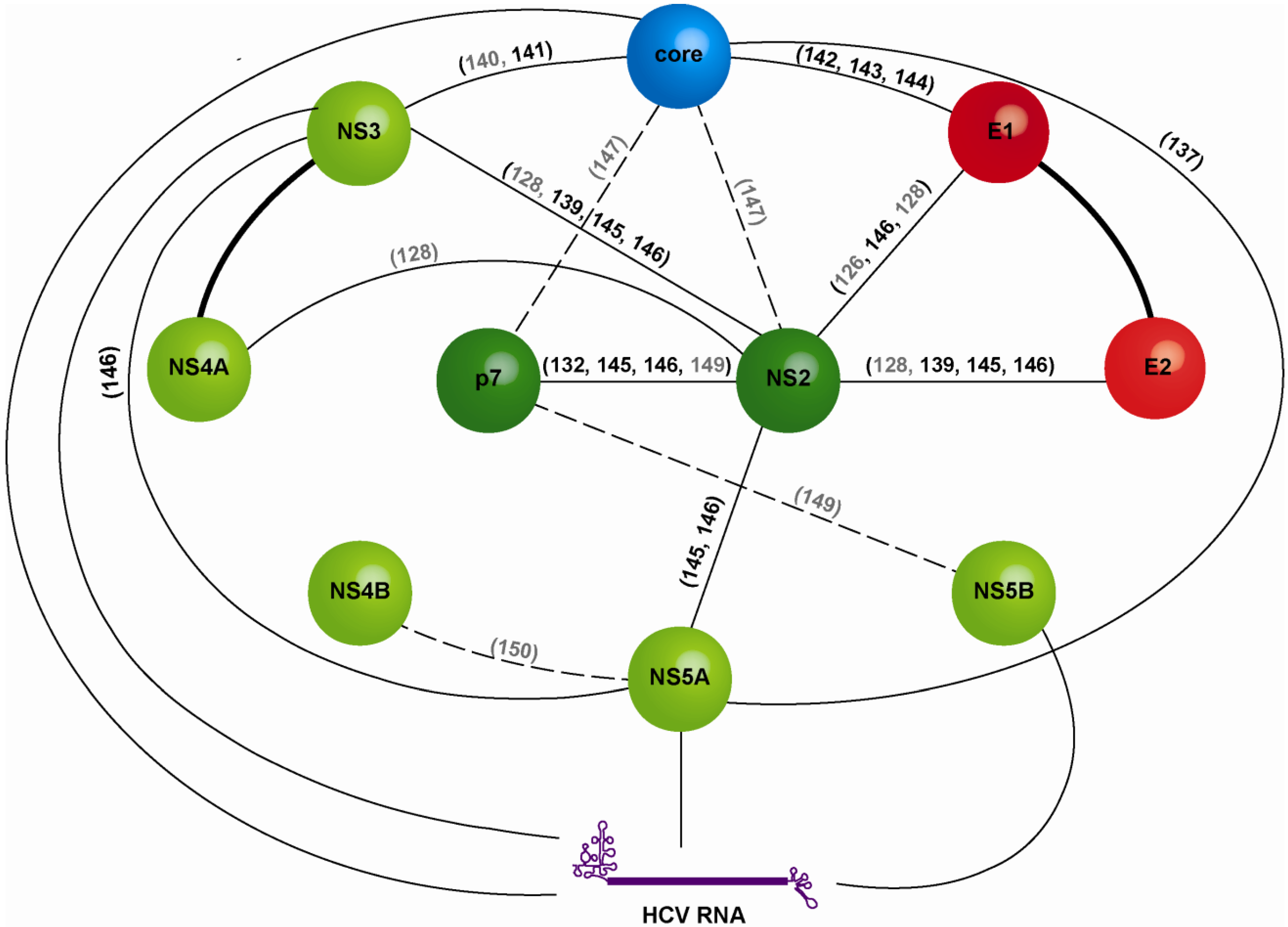
2. From the Glycoprotein Assembly-Line to a Functional Glycoprotein Heterodimer
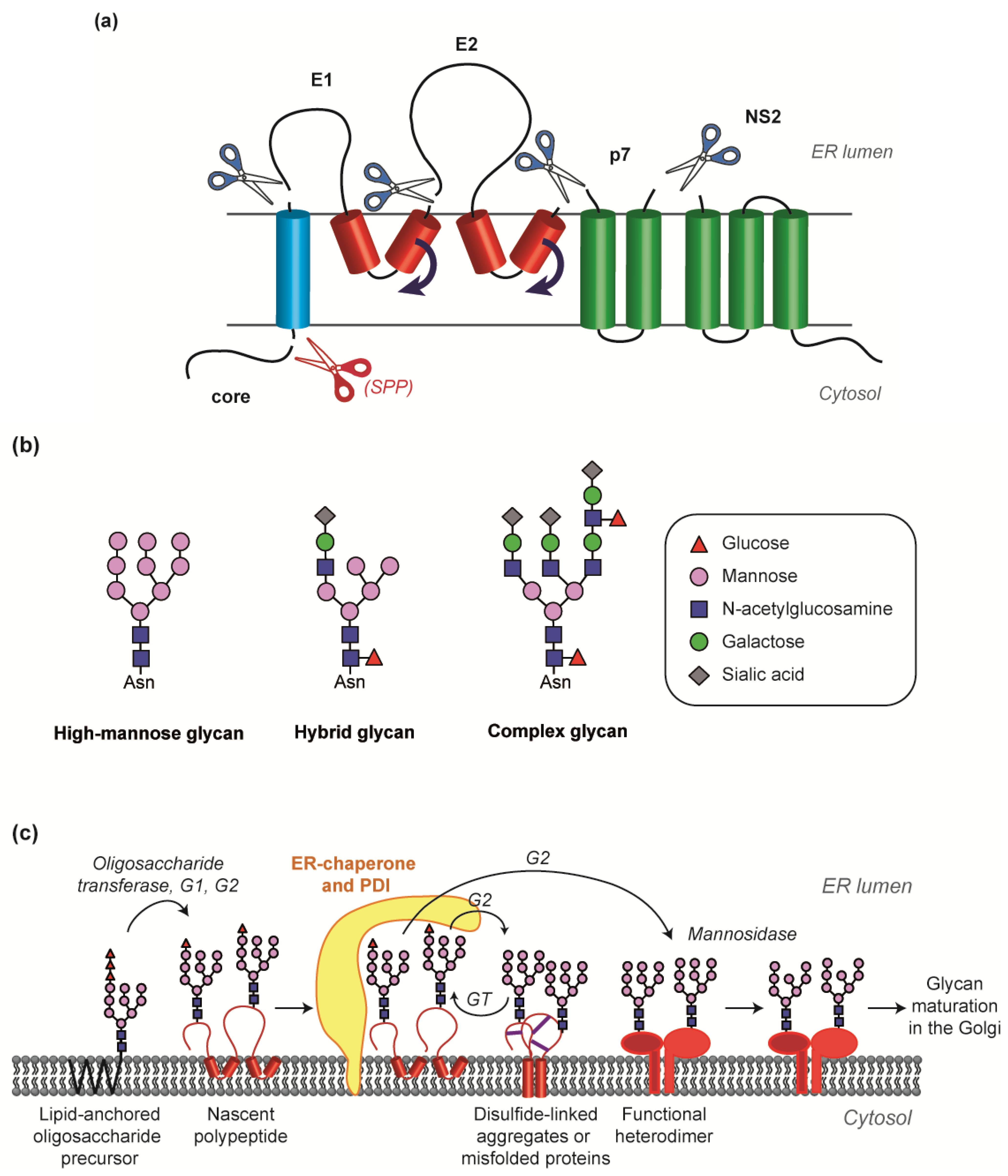
2.1. Translation and Cleavage of E1E2 from the Polyprotein Precursor

2.2. The E1E2 Tandem: Determinants for Heterodimerization
2.3. Chaperone-Mediated Folding, Disulfide Bond Formation and Glycan Maturation
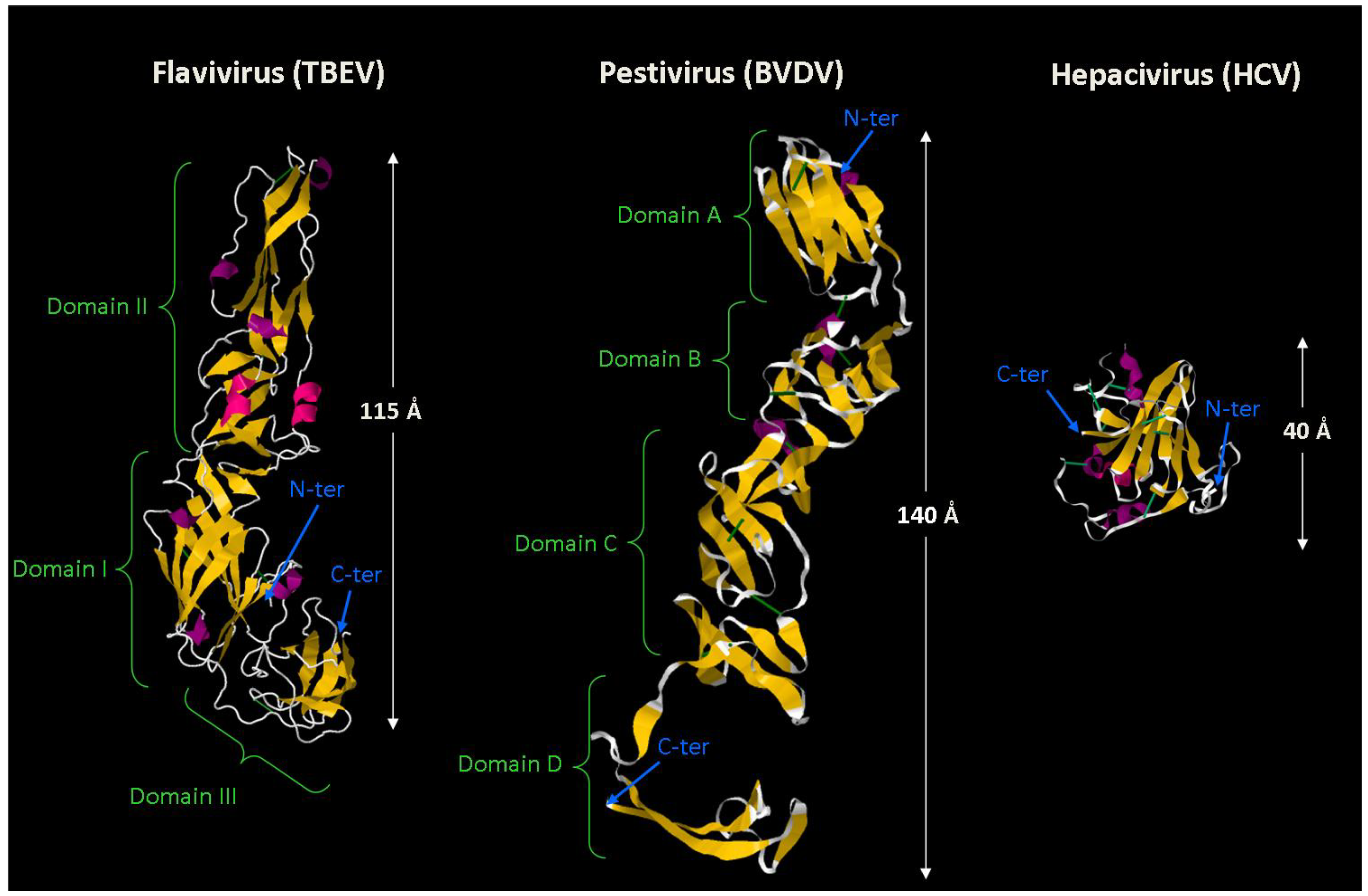
2.4. Glimpses into E1 and E2 Structures and Shift in Fusion Paradigms
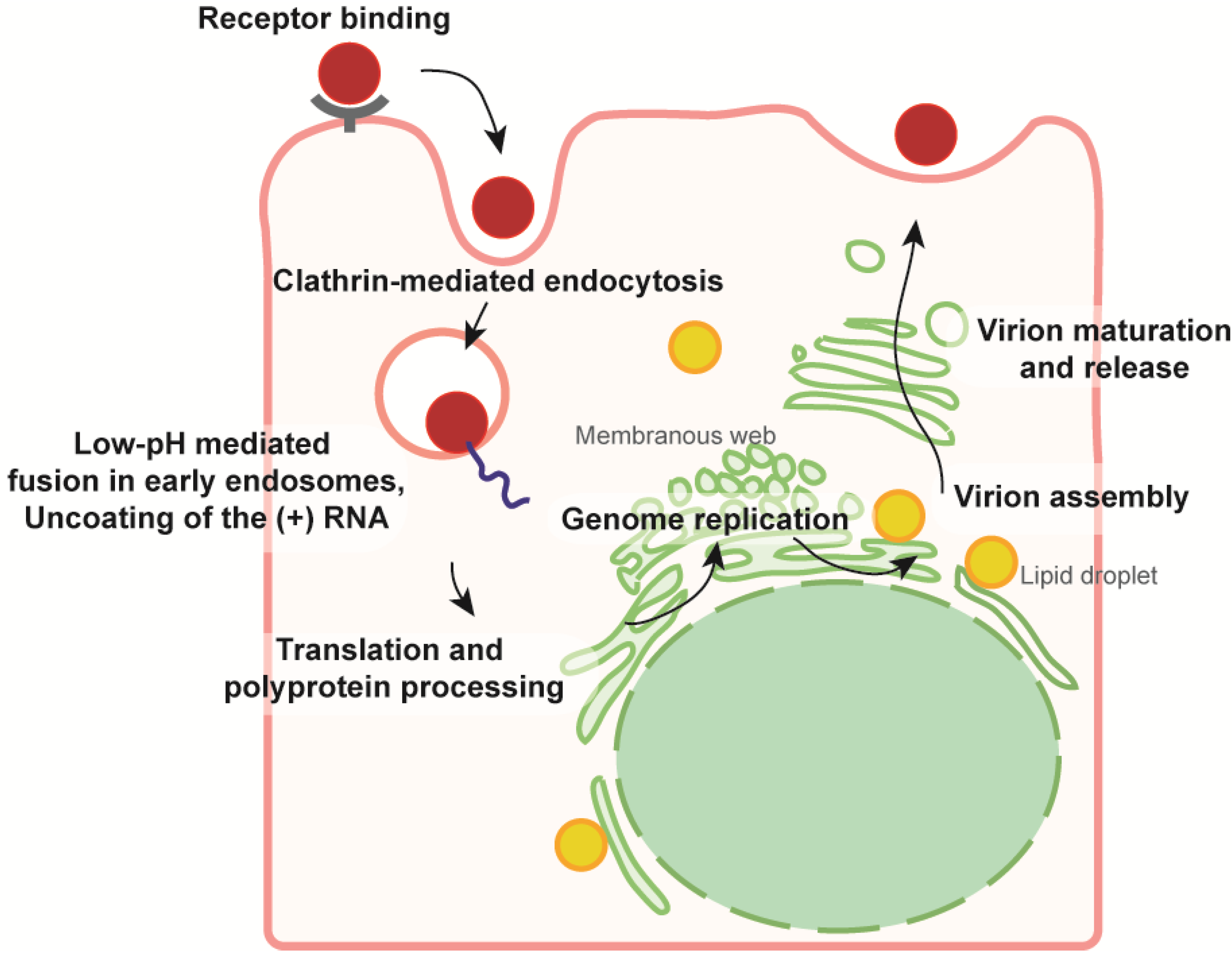
2.5. From the ER Membrane to the Surface of Secreted Viral Particles: Viral Glycoproteins on the Way Out
2.6. Systems to Study HCV Envelope Glycoproteins and Their Incorporation in the Virion
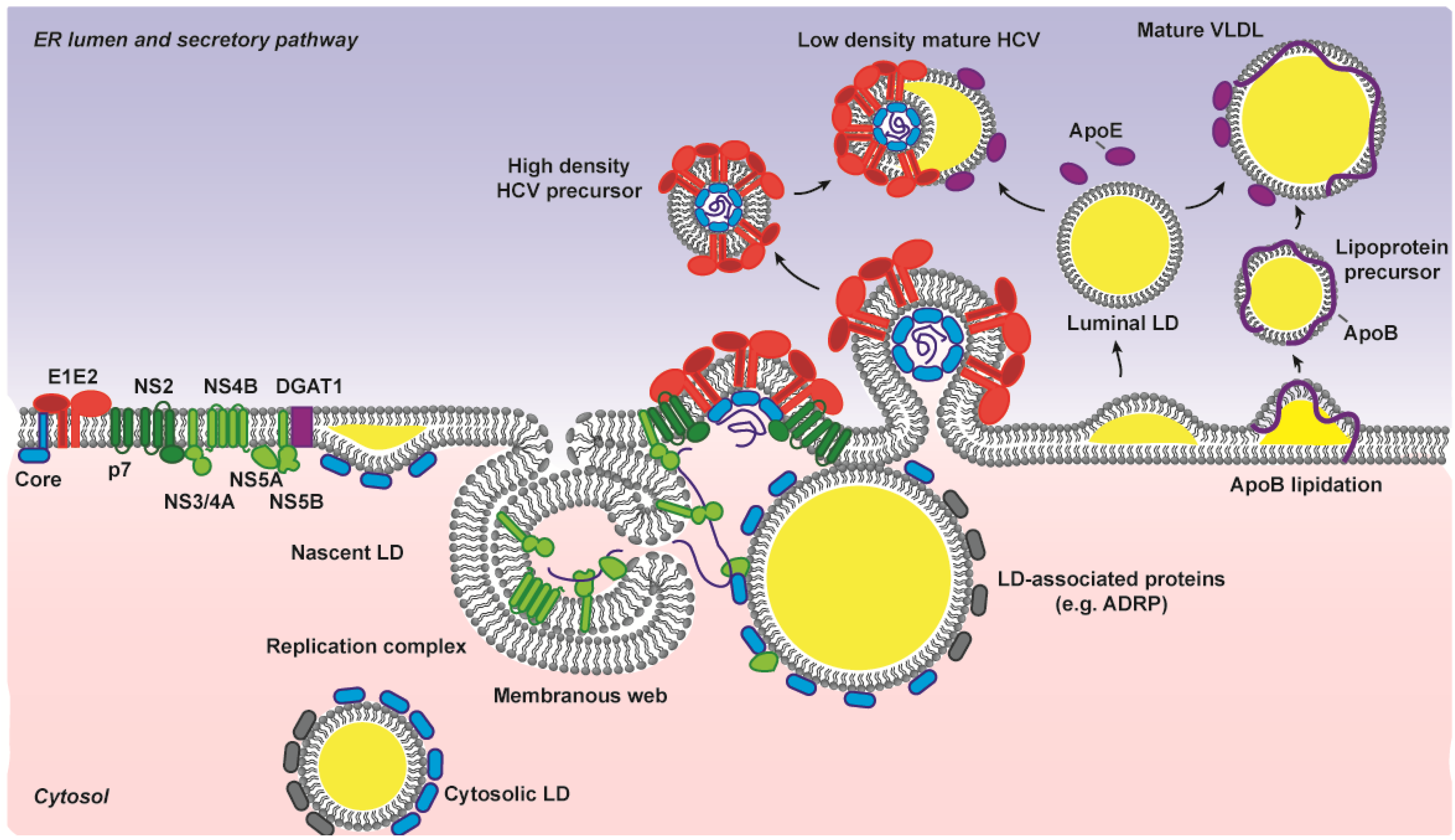
3. HCV Envelopment and Glycoprotein Acquisition
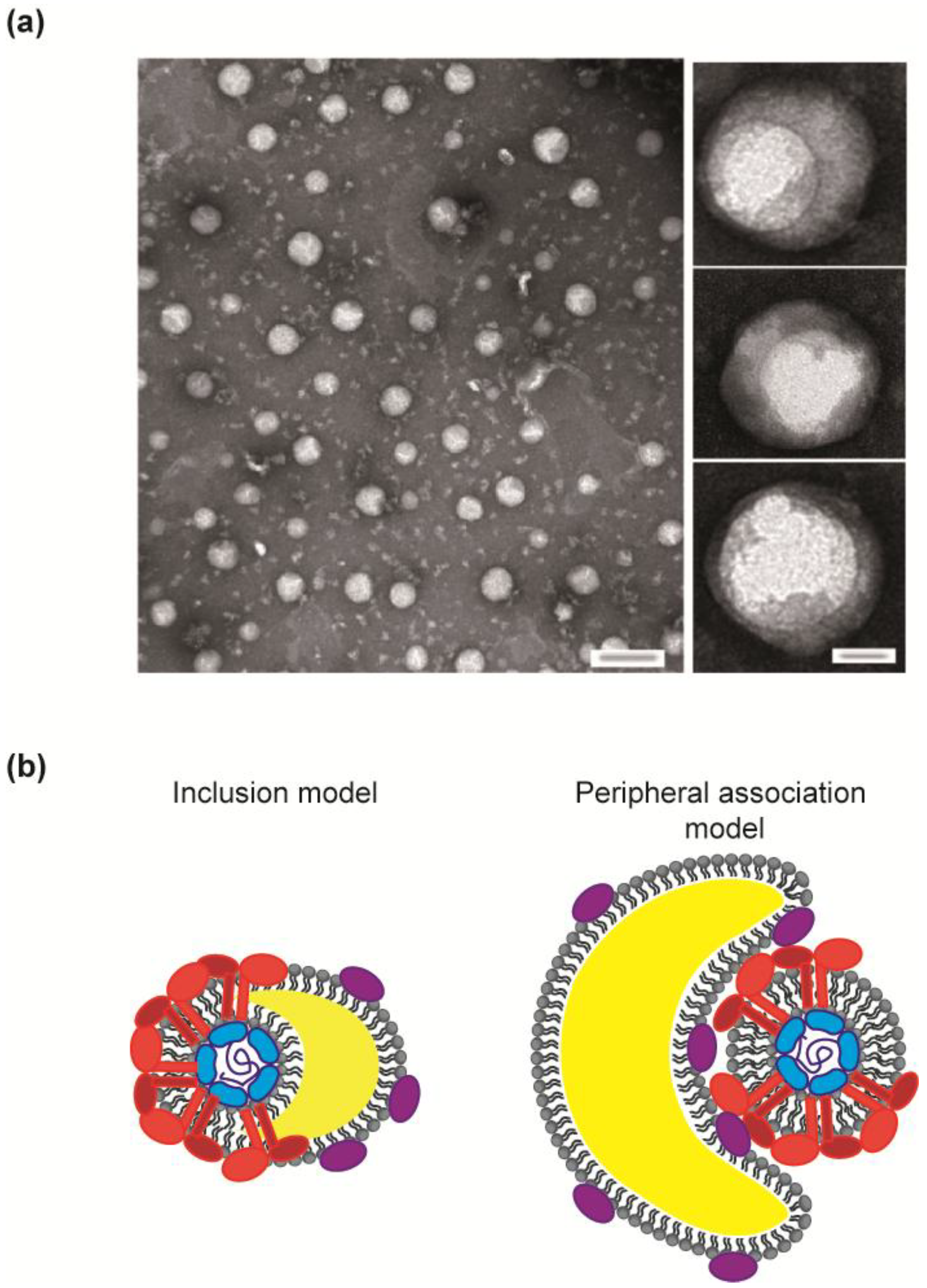
3.1. HCV Virions, a Heterogeneous Population
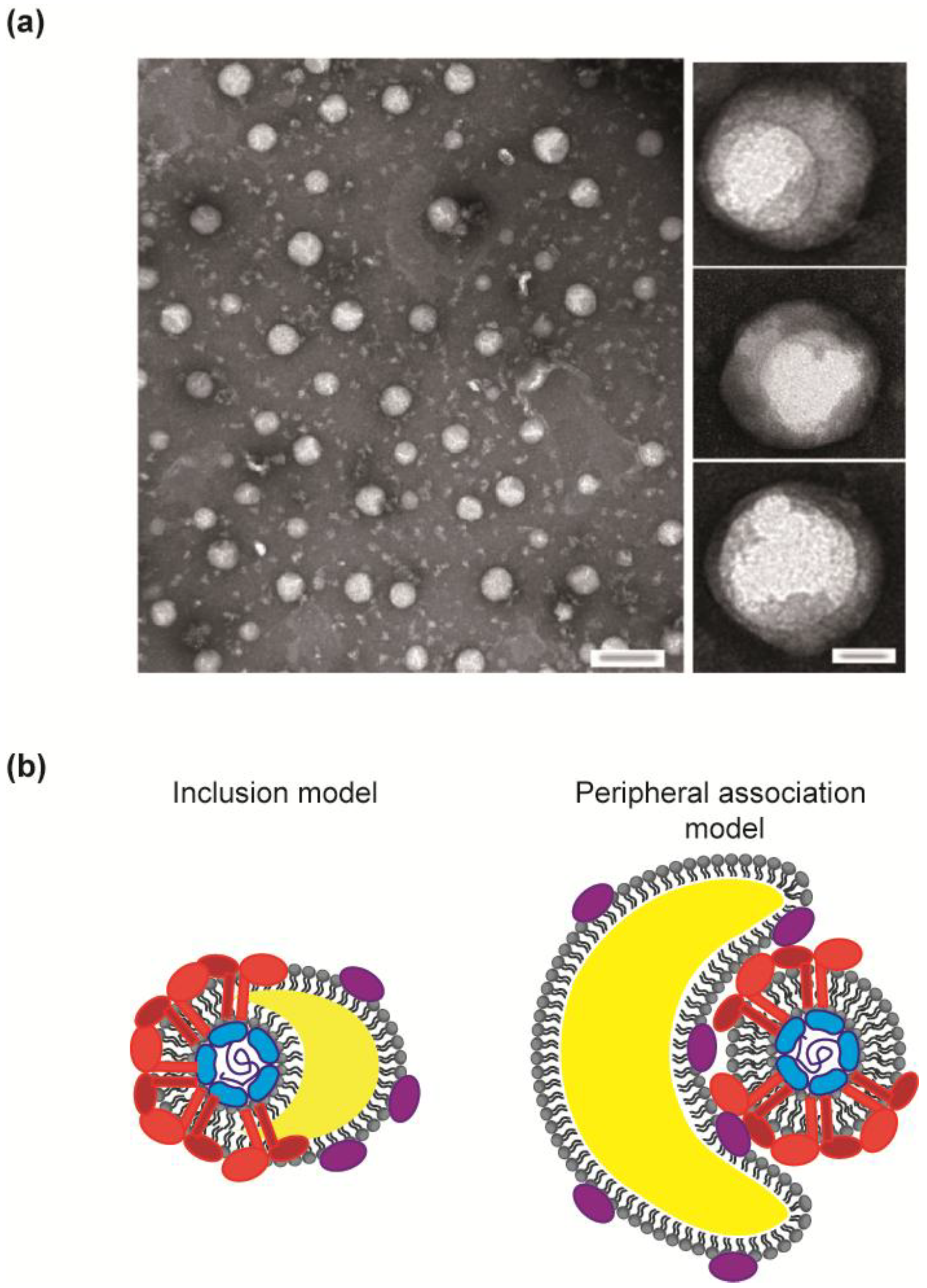
3.2. Non-Structural Proteins Bring Together the Bricks for Assembly
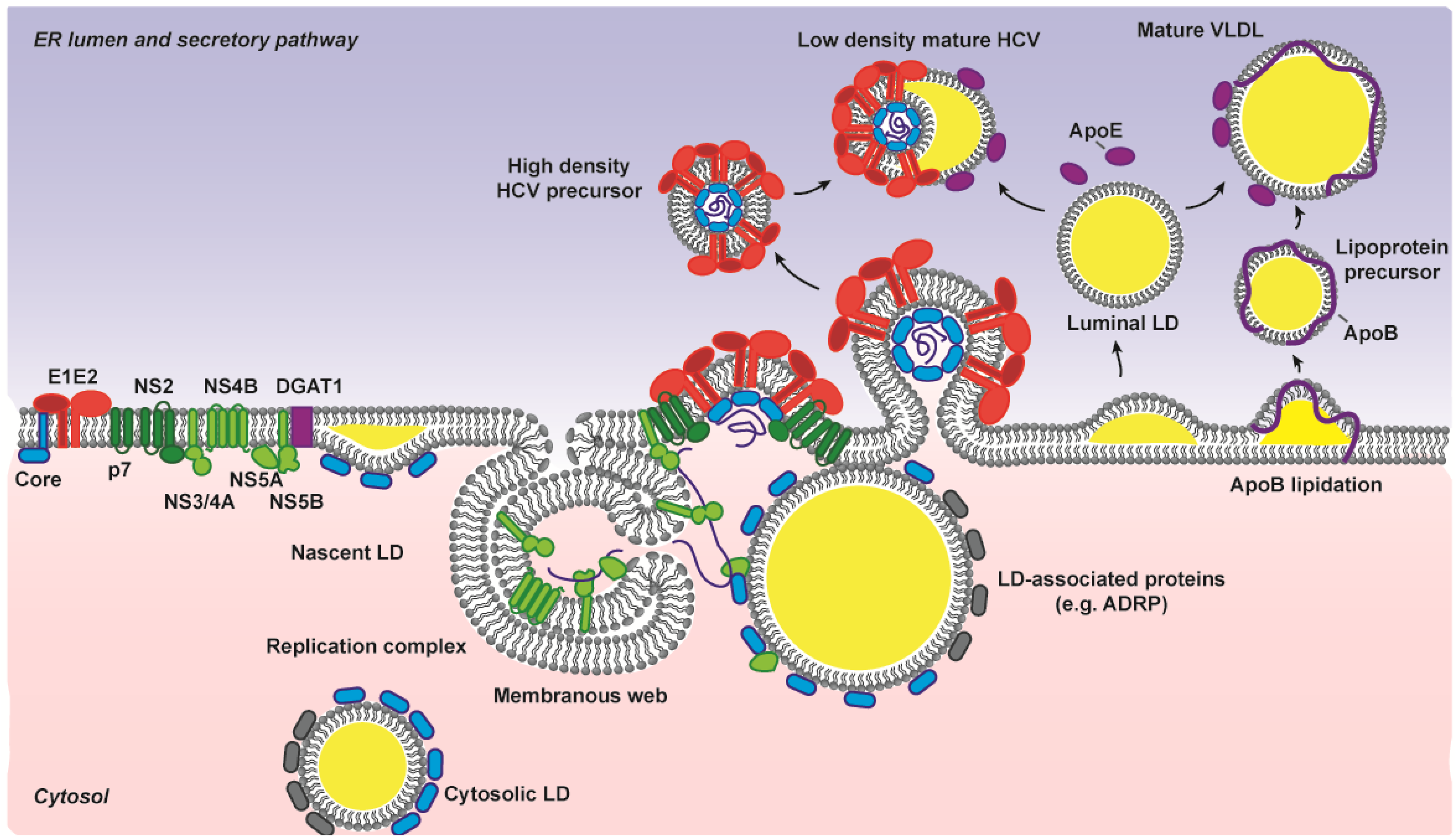
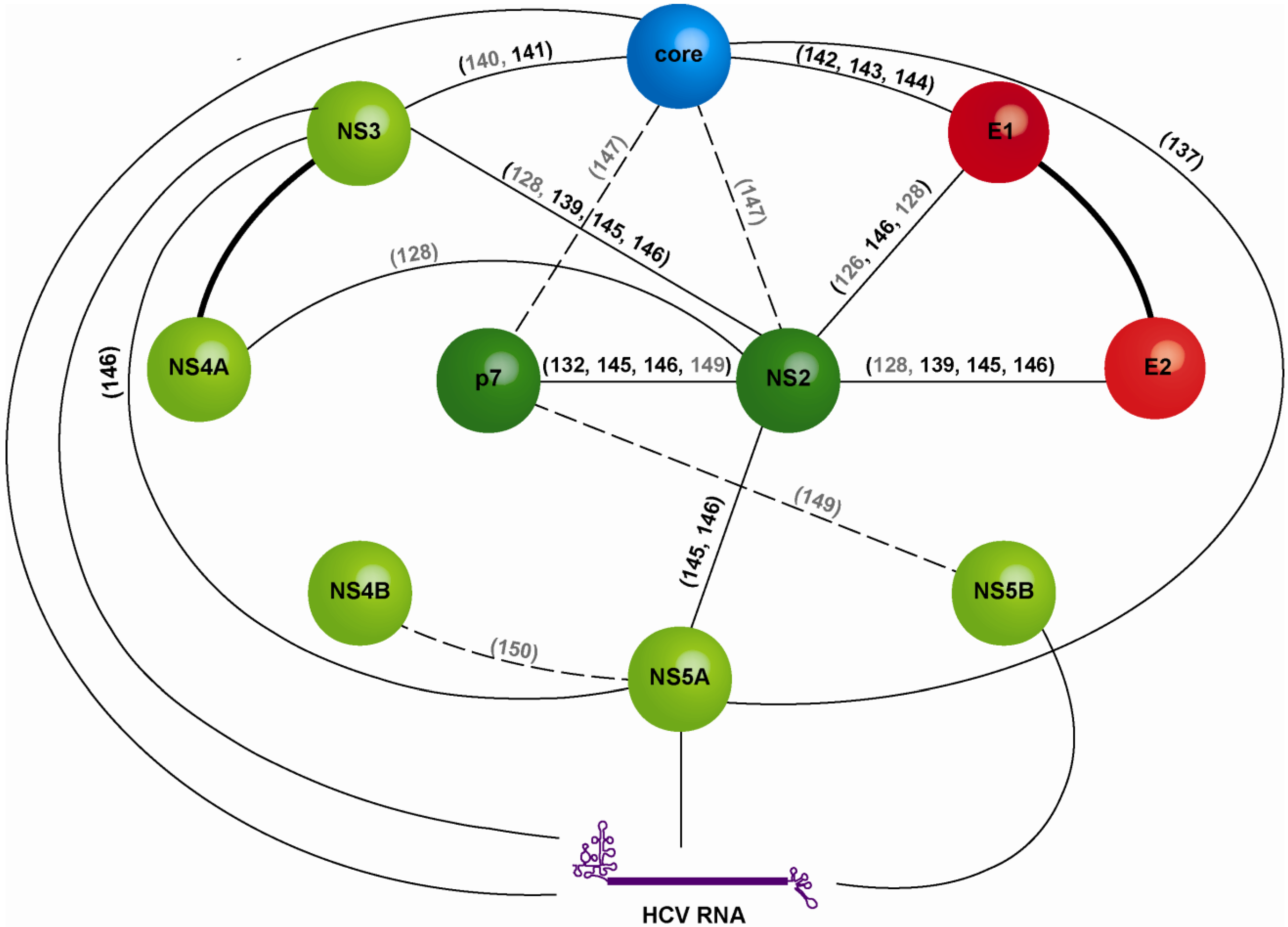
3.3. E1E2 Determinants for HCV Assembly
3.4. The Driving Force for Virion Budding

3.5. Participation of the Host Lipoprotein Machinery in HCV Assembly and Entry Process
4. Exposure of the E1E2 Functional Complex at the Virion Surface
4.1. Travelling Incognito: The Virion with Lipoprotein and Glycan Masks on
4.2. E1E2 Oligomerization at the Virion Surface
4.3. From Assembly to Entry: The Issue of Packaging an Entry-Competent Glycoprotein Complex

5. Conclusions and Future Challenges
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011, 17, 107–115. [Google Scholar] [CrossRef]
- Hoofnagle, J.H. Course and outcome of hepatitis C. Hepatology 2002, 36, S21–S29. [Google Scholar] [CrossRef]
- Liang, T.J.; Ghany, M.G. Current and future therapies for hepatitis C virus infection. New Engl. J. Med. 2013, 368, 1907–1917. [Google Scholar] [CrossRef]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef]
- Lindenbach, B.D. Virion assembly and release. Curr. Top. Microbiol. Immunol. 2013, 369, 199–218. [Google Scholar]
- Murray, C.L.; Jones, C.T.; Rice, C.M. Architects of assembly: Roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat. Rev. Microbiol. 2008, 6, 699–708. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef]
- Lohmann, V. Hepatitis C virus RNA replication. Curr. Top. Microbiol. Immunol. 2013, 369, 167–198. [Google Scholar]
- Voisset, C.; Dubuisson, J. Functional hepatitis C virus envelope glycoproteins. Biol. Cell 2004, 96, 413–420. [Google Scholar] [CrossRef]
- Vieyres, G.; Thomas, X.; Descamps, V.; Duverlie, G.; Patel, A.H.; Dubuisson, J. Characterization of the envelope glycoproteins associated with infectious hepatitis c virus. J. Virol. 2010, 84, 10159–10168. [Google Scholar] [CrossRef]
- Meertens, L.; Bertaux, C.; Dragic, T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 2006, 80, 11571–11578. [Google Scholar] [CrossRef]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis c virus for low-ph-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef]
- Gastaminza, P.; Dryden, K.A.; Boyd, B.; Wood, M.R.; Law, M.; Yeager, M.; Chisari, F.V. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J. Virol. 2010, 84, 10999–11009. [Google Scholar] [CrossRef]
- Merz, A.; Long, G.; Hiet, M.S.; Brugger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar] [CrossRef]
- Steinmann, E.; Penin, F.; Kallis, S.; Patel, A.H.; Bartenschlager, R.; Pietschmann, T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007, 3, e103. [Google Scholar] [CrossRef]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef]
- Shanmugam, S.; Yi, M. Efficiency of E2-p7 Processing Modulates Production of Infectious Hepatitis C Virus. J. Virol. 2013, 87, 11255–11266. [Google Scholar] [CrossRef]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. Curr. Top. Microbiol. Immunol. 2013, 369, 113–142. [Google Scholar]
- Cocquerel, L.; Op de Beeck, A.; Lambot, M.; Roussel, J.; Delgrange, D.; Pillez, A.; Wychowski, C.; Penin, F.; Dubuisson, J. Topological changes in the transmembrane domains of hepatitis C virus envelope glycoproteins. EMBO J. 2002, 21, 2893–2902. [Google Scholar] [CrossRef]
- Ostapchuk, P.; Hearing, P.; Ganem, D. A dramatic shift in the transmembrane topology of a viral envelope glycoprotein accompanies hepatitis B viral morphogenesis. EMBO J. 1994, 13, 1048–1057. [Google Scholar]
- Dubuisson, J.; Falson, P.; Bartosch, B.; Alsaleh, K.; Tews, B.; Ciczora, Y.; Montigny, C.; Verney, G.; Pecheur, E.-I.; Le Maire, M.; et al. E1 envelope glycoprotein associated with HCV particle exists as a transmembrane domain-dependent trimer. In Proceedings of the 19th International Symposium on Hepatitis C Virus and Related Viruses, Venice, Italy, 5–9 October 2012.
- Dubuisson, J.; Hsu, H.H.; Cheung, R.C.; Greenberg, H.B.; Russell, D.G.; Rice, C.M. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 1994, 68, 6147–6160. [Google Scholar]
- Duvet, S.; Cocquerel, L.; Pillez, A.; Cacan, R.; Verbert, A.; Moradpour, D.; Wychowski, C.; Dubuisson, J. Hepatitis C virus glycoprotein complex localization in the endoplasmic reticulum involves a determinant for retention and not retrieval. J. Biol. Chem. 1998, 273, 32088–32095. [Google Scholar] [CrossRef]
- Rouille, Y.; Helle, F.; Delgrange, D.; Roingeard, P.; Voisset, C.; Blanchard, E.; Belouzard, S.; McKeating, J.; Patel, A.H.; Maertens, G.; Wakita, T.; Wychowski, C.; Dubuisson, J. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. 2006, 80, 2832–2841. [Google Scholar] [CrossRef]
- Albecka, A.; Montserret, R.; Krey, T.; Tarr, A.W.; Diesis, E.; Ball, J.K.; Descamps, V.; Duverlie, G.; Rey, F.; Penin, F.; Dubuisson, J. Identification of new functional regions in hepatitis C virus envelope glycoprotein E2. J. Virol. 2011, 85, 1777–1792. [Google Scholar] [CrossRef]
- Maurin, G.; Fresquet, J.; Granio, O.; Wychowski, C.; Cosset, F.L.; Lavillette, D. Identification of interactions in the E1E2 heterodimer of hepatitis C virus important for cell entry. J. Biol. Chem. 2011, 286, 23865–23876. [Google Scholar]
- Douam, F.; Thi, V.L.; Maurin, G.; Fresquet, J.; Mompelat, D.; Zeisel, M.B.; Baumert, T.F.; Cosset, F.L.; Lavillette, D. A critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 2014, 59, 776–788. [Google Scholar] [CrossRef]
- Carlsen, T.H.; Scheel, T.K.; Ramirez, S.; Foung, S.K.; Bukh, J. Characterization of hepatitis C virus recombinants with chimeric E1/E2 envelope proteins and identification of single amino acids in the E2 stem region important for entry. J. Virol. 2013, 87, 1385–1399. [Google Scholar] [CrossRef]
- Ralston, R.; Thudium, K.; Berger, K.; Kuo, C.; Gervase, B.; Hall, J.; Selby, M.; Kuo, G.; Houghton, M.; Choo, Q.L. Characterization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia viruses. J. Virol. 1993, 67, 6753–6761. [Google Scholar]
- Cocquerel, L.; Wychowski, C.; Minner, F.; Penin, F.; Dubuisson, J. Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J. Virol. 2000, 74, 3623–3633. [Google Scholar] [CrossRef]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, E.; Pietschmann, T. Institute of Experimental Virology, Twincore, Center for Experimental and Clinical Infection Research, a Joint Venture between the Medical School Hannover and the Helmholtz Center for Infection Research, 30625 Hannover, Germany. Unpublished work. 2014. [Google Scholar]
- Deleersnyder, V.; Pillez, A.; Wychowski, C.; Blight, K.; Xu, J.; Hahn, Y.S.; Rice, C.M.; Dubuisson, J. Formation of native hepatitis C virus glycoprotein complexes. J. Virol. 1997, 71, 697–704. [Google Scholar]
- Op De Beeck, A.; Voisset, C.; Bartosch, B.; Ciczora, Y.; Cocquerel, L.; Keck, Z.; Foung, S.; Cosset, F.L.; Dubuisson, J. Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 2004, 78, 2994–3002. [Google Scholar] [CrossRef]
- Flint, M.; Logvinoff, C.; Rice, C.M.; McKeating, J.A. Characterization of infectious retroviral pseudotype particles bearing hepatitis C virus glycoproteins. J. Virol. 2004, 78, 6875–6882. [Google Scholar] [CrossRef]
- Cocquerel, L.; Duvet, S.; Meunier, J.C.; Pillez, A.; Cacan, R.; Wychowski, C.; Dubuisson, J. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol. 1999, 73, 2641–2649. [Google Scholar]
- Cocquerel, L.; Meunier, J.C.; Pillez, A.; Wychowski, C.; Dubuisson, J. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 1998, 72, 2183–2191. [Google Scholar]
- Michalak, J.P.; Wychowski, C.; Choukhi, A.; Meunier, J.C.; Ung, S.; Rice, C.M.; Dubuisson, J. Characterization of truncated forms of hepatitis C virus glycoproteins. J. Gen. Virol. 1997, 78, 2299–2306. [Google Scholar]
- Patel, J.; Patel, A.H.; McLauchlan, J. The transmembrane domain of the hepatitis C virus E2 glycoprotein is required for correct folding of the E1 glycoprotein and native complex formation. Virology 2001, 279, 58–68. [Google Scholar] [CrossRef]
- Ciczora, Y.; Callens, N.; Montpellier, C.; Bartosch, B.; Cosset, F.L.; Op de Beeck, A.; Dubuisson, J. Contribution of the charged residues of hepatitis C virus glycoprotein E2 transmembrane domain to the functions of the E1E2 heterodimer. J. Gen. Virol. 2005, 86, 2793–2798. [Google Scholar] [CrossRef]
- Ciczora, Y.; Callens, N.; Penin, F.; Pecheur, E.I.; Dubuisson, J. Transmembrane domains of hepatitis C virus envelope glycoproteins: Residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J. Virol. 2007, 81, 2372–2381. [Google Scholar]
- Op De Beeck, A.; Montserret, R.; Duvet, S.; Cocquerel, L.; Cacan, R.; Barberot, B.; Le Maire, M.; Penin, F.; Dubuisson, J. The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J. Biol. Chem. 2000, 275, 31428–31437. [Google Scholar] [CrossRef]
- Molenkamp, R.; Kooi, E.A.; Lucassen, M.A.; Greve, S.; Thijssen, J.C.; Spaan, W.J.; Bredenbeek, P.J. Yellow fever virus replicons as an expression system for hepatitis C virus structural proteins. J. Virol. 2003, 77, 1644–1648. [Google Scholar] [CrossRef]
- Drummer, H.E.; Poumbourios, P. Hepatitis C virus glycoprotein E2 contains a membrane-proximal heptad repeat sequence that is essential for E1E2 glycoprotein heterodimerization and viral entry. J. Biol. Chem. 2004, 279, 30066–30072. [Google Scholar] [CrossRef]
- McCaffrey, K.; Gouklani, H.; Boo, I.; Poumbourios, P.; Drummer, H.E. The variable regions of hepatitis C virus glycoprotein E2 have an essential structural role in glycoprotein assembly and virion infectivity. J. Gen. Virol. 2011, 92, 112–121. [Google Scholar] [CrossRef]
- Yi, M.; Nakamoto, Y.; Kaneko, S.; Yamashita, T.; Murakami, S. Delineation of regions important for heteromeric association of hepatitis C virus E1 and E2. Virology 1997, 231, 119–129. [Google Scholar] [CrossRef]
- Choukhi, A.; Pillez, A.; Drobecq, H.; Sergheraert, C.; Wychowski, C.; Dubuisson, J. Characterization of aggregates of hepatitis C virus glycoproteins. J. Gen. Virol. 1999, 80, 3099–3107. [Google Scholar]
- Goffard, A.; Callens, N.; Bartosch, B.; Wychowski, C.; Cosset, F.L.; Montpellier, C.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 2005, 79, 8400–8409. [Google Scholar] [CrossRef]
- Helle, F.; Goffard, A.; Morel, V.; Duverlie, G.; McKeating, J.; Keck, Z.Y.; Foung, S.; Penin, F.; Dubuisson, J.; Voisset, C. The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 2007, 81, 8101–8111. [Google Scholar] [CrossRef]
- Fournillier Jacob, A.; Cahour, A.; Escriou, N.; Girard, M.; Wychowski, C. Processing of the E1 glycoprotein of hepatitis C virus expressed in mammalian cells. J. Gen. Virol. 1996, 77, 1055–1064. [Google Scholar] [CrossRef]
- Meunier, J.C.; Fournillier, A.; Choukhi, A.; Cahour, A.; Cocquerel, L.; Dubuisson, J.; Wychowski, C. Analysis of the glycosylation sites of hepatitis C virus (HCV) glycoprotein E1 and the influence of E1 glycans on the formation of the HCV glycoprotein complex. J. Gen. Virol. 1999, 80, 887–896. [Google Scholar]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef]
- Dubuisson, J.; Rice, C.M. Hepatitis C virus glycoprotein folding: Disulfide bond formation and association with calnexin. J. Virol. 1996, 70, 778–786. [Google Scholar]
- Choukhi, A.; Ung, S.; Wychowski, C.; Dubuisson, J. Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus glycoproteins. J. Virol. 1998, 72, 3851–3858. [Google Scholar]
- Wahid, A.; Helle, F.; Descamps, V.; Duverlie, G.; Penin, F.; Dubuisson, J. Disulfide bonds in hepatitis C virus glycoprotein E1 control the assembly and entry functions of E2 glycoprotein. J. Virol. 2013, 87, 1605–1617. [Google Scholar] [CrossRef]
- McCaffrey, K.; Boo, I.; Tewierek, K.; Edmunds, M.L.; Poumbourios, P.; Drummer, H.E. Role of conserved cysteine residues in hepatitis C virus glycoprotein e2 folding and function. J. Virol. 2012, 86, 3961–3974. [Google Scholar] [CrossRef]
- Lavie, M.; Goffard, A.; Dubuisson, J. Assembly of a functional HCV glycoprotein heterodimer. Curr. Issues Mol. Biol. 2007, 9, 71–86. [Google Scholar]
- Brazzoli, M.; Helenius, A.; Foung, S.K.; Houghton, M.; Abrignani, S.; Merola, M. Folding and dimerization of hepatitis C virus E1 and E2 glycoproteins in stably transfected CHO cells. Virology 2005, 332, 438–453. [Google Scholar] [CrossRef]
- Krey, T.; d'Alayer, J.; Kikuti, C.M.; Saulnier, A.; Damier-Piolle, L.; Petitpas, I.; Johansson, D.X.; Tawar, R.G.; Baron, B.; Robert, B.; et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 2010, 6, e1000762. [Google Scholar] [CrossRef] [Green Version]
- Yagnik, A.T.; Lahm, A.; Meola, A.; Roccasecca, R.M.; Ercole, B.B.; Nicosia, A.; Tramontano, A. A model for the hepatitis C virus envelope glycoprotein E2. Proteins 2000, 40, 355–366. [Google Scholar] [CrossRef]
- Garry, R.F.; Dash, S. Proteomics computational analyses suggest that hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class II fusion proteins. Virology 2003, 307, 255–265. [Google Scholar] [CrossRef]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef]
- Lavillette, D.; Pecheur, E.I.; Donot, P.; Fresquet, J.; Molle, J.; Corbau, R.; Dreux, M.; Penin, F.; Cosset, F.L. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 2007, 81, 8752–8765. [Google Scholar] [CrossRef]
- Flint, M.; Thomas, J.M.; Maidens, C.M.; Shotton, C.; Levy, S.; Barclay, W.S.; McKeating, J.A. Functional analysis of cell surface-expressed hepatitis C virus E2 glycoprotein. J. Virol. 1999, 73, 6782–6790. [Google Scholar]
- Drummer, H.E.; Boo, I.; Poumbourios, P. Mutagenesis of a conserved fusion peptide-like motif and membrane-proximal heptad-repeat region of hepatitis C virus glycoprotein E1. J. Gen. Virol. 2007, 88, 1144–1148. [Google Scholar] [CrossRef]
- Russell, R.S.; Kawaguchi, K.; Meunier, J.C.; Takikawa, S.; Faulk, K.; Bukh, J.; Purcell, R.H.; Emerson, S.U. Mutational analysis of the hepatitis C virus E1 glycoprotein in retroviral pseudoparticles and cell-culture-derived H77/JFH1 chimeric infectious virus particles. J. Viral Hepat. 2009, 16, 621–632. [Google Scholar] [CrossRef]
- Li, H.F.; Huang, C.H.; Ai, L.S.; Chuang, C.K.; Chen, S.S. Mutagenesis of the fusion peptide-like domain of hepatitis C virus E1 glycoprotein: Involvement in cell fusion and virus entry. J. Biomed. Sci. 2009, 16, 89. [Google Scholar] [CrossRef]
- Garry, C.E.; Garry, R.F. Proteomics computational analyses suggest that the carboxyl terminal glycoproteins of Bunyaviruses are class II viral fusion protein (beta-penetrenes). Theor. Biol. Med. Model. 2004, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Tischler, N.D.; Gonzalez, A.; Perez-Acle, T.; Rosemblatt, M.; Valenzuela, P.D. Hantavirus Gc glycoprotein: Evidence for a class II fusion protein. J. Gen. Virol. 2005, 86, 2937–2947. [Google Scholar] [CrossRef]
- Vaney, M.C.; Rey, F.A. Class II enveloped viruses. Cell Microbiol. 2011, 13, 1451–1459. [Google Scholar] [CrossRef]
- Kielian, M.; Rey, F.A. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. Microbiol. 2006, 4, 67–76. [Google Scholar] [CrossRef]
- Dessau, M.; Modis, Y. Crystal structure of glycoprotein C from Rift Valley fever virus. Proc. Natl. Acad. Sci. USA 2013, 110, 1696–1701. [Google Scholar] [CrossRef]
- El Omari, K.; Iourin, O.; Harlos, K.; Grimes, J.M.; Stuart, D.I. Structure of a pestivirus envelope glycoprotein E2 clarifies its role in cell entry. Cell Rep. 2013, 3, 30–35. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Kanai, R.; Modis, Y. Crystal structure of glycoprotein E2 from bovine viral diarrhea virus. Proc. Natl. Acad. Sci. USA 2013, 110, 6805–6810. [Google Scholar] [CrossRef]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; Wilson, I.A.; Law, M. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef]
- Helle, F.; Vieyres, G.; Elkrief, L.; Popescu, C.I.; Wychowski, C.; Descamps, V.; Castelain, S.; Roingeard, P.; Duverlie, G.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 2010, 84, 11905–11915. [Google Scholar] [CrossRef]
- Helle, F.; Duverlie, G.; Dubuisson, J. The hepatitis C virus glycan shield and evasion of the humoral immune response. Viruses 2011, 3, 1909–1932. [Google Scholar] [CrossRef]
- Owsianka, A.M.; Timms, J.M.; Tarr, A.W.; Brown, R.J.; Hickling, T.P.; Szwejk, A.; Bienkowska-Szewczyk, K.; Thomson, B.J.; Patel, A.H.; Ball, J.K. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 2006, 80, 8695–8704. [Google Scholar] [CrossRef]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef]
- Dubuisson, J.; Penin, F.; Moradpour, D. Interaction of hepatitis C virus proteins with host cell membranes and lipids. Trends Cell Biol. 2002, 12, 517–523. [Google Scholar] [CrossRef]
- Vieyres, G.; Brohm, C.; Friesland, M.; Gentzsch, J.; Wolk, B.; Roingeard, P.; Steinmann, E.; Pietschmann, T. Subcellular localization and function of an epitope-tagged p7 viroporin in hepatitis C virus-producing cells. J. Virol. 2013, 87, 1664–1678. [Google Scholar] [CrossRef]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef]
- Counihan, N.A.; Rawlinson, S.M.; Lindenbach, B.D. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 2011, 7, e1002302. [Google Scholar] [CrossRef]
- Coller, K.E.; Heaton, N.S.; Berger, K.L.; Cooper, J.D.; Saunders, J.L.; Randall, G. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 2012, 8, e1002466. [Google Scholar] [CrossRef]
- Fiches, G.N.; Eyre, N.S.; Turville, S.G.; Beard, M.R. Dynamic imaging of HCV Viral RNA traffic and localisation. In Proceeding of the 19th International Symposium on Hepatitis C Virus and Related Viruses, Venice, Italy, 5–9 October 2012.
- Drummer, H.E.; Maerz, A.; Poumbourios, P. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett. 2003, 546, 385–390. [Google Scholar] [CrossRef]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 2003, 100, 7271–7276. [Google Scholar] [CrossRef]
- Sandrin, V.; Boulanger, P.; Penin, F.; Granier, C.; Cosset, F.L.; Bartosch, B. Assembly of functional hepatitis C virus glycoproteins on infectious pseudoparticles occurs intracellularly and requires concomitant incorporation of E1 and E2 glycoproteins. J. Gen. Virol. 2005, 86, 3189–3199. [Google Scholar]
- Muller, B.; Heilemann, M. Shedding new light on viruses: Super-resolution microscopy for studying human immunodeficiency virus. Trends Microbiol. 2013, 21, 522–533. [Google Scholar] [CrossRef]
- Vieyres, G.; Pietschmann, T. Entry and replication of recombinant hepatitis C viruses in cell culture. Methods 2013, 59, 233–248. [Google Scholar] [CrossRef]
- Steinmann, E.; Pietschmann, T. Cell culture systems for hepatitis C virus. Curr. Top. Microbiol. Immunol. 2013, 369, 17–48. [Google Scholar]
- Welsch, S.; Muller, B.; Krausslich, H.G. More than one door—Budding of enveloped viruses through cellular membranes. FEBS Lett. 2007, 581, 2089–2097. [Google Scholar] [CrossRef]
- Lavillette, D.; Tarr, A.W.; Voisset, C.; Donot, P.; Bartosch, B.; Bain, C.; Patel, A.H.; Dubuisson, J.; Ball, J.K.; Cosset, F.L. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 2005, 41, 265–274. [Google Scholar] [CrossRef]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.J.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef]
- Kato, T.; Date, T.; Murayama, A.; Morikawa, K.; Akazawa, D.; Wakita, T. Cell culture and infection system for hepatitis C virus. Nat. Protoc. 2006, 1, 2334–2339. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Meuleman, P.; Ploss, A.; Vanwolleghem, T.; Syder, A.J.; McKeating, J.A.; Lanford, R.E.; Feinstone, S.M.; Major, M.E.; Leroux-Roels, G.; Rice, C.M. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. USA 2006, 103, 3805–3809. [Google Scholar] [CrossRef]
- Podevin, P.; Carpentier, A.; Pene, V.; Aoudjehane, L.; Carriere, M.; Zaidi, S.; Hernandez, C.; Calle, V.; Meritet, J.F.; Scatton, O.; et al. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology 2010, 139, 1355–1364. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; Andre, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef]
- Pacini, L.; Graziani, R.; Bartholomew, L.; De Francesco, R.; Paonessa, G. Naturally occurring hepatitis C virus subgenomic deletion mutants replicate efficiently in Huh-7 cells and are trans-packaged in vitro to generate infectious defective particles. J. Virol. 2009, 83, 9079–9093. [Google Scholar] [CrossRef]
- Phan, T.; Kohlway, A.; Dimberu, P.; Pyle, A.M.; Lindenbach, B.D. The acidic domain of hepatitis C virus NS4A contributes to RNA replication and virus particle assembly. J. Virol. 2011, 85, 1193–1204. [Google Scholar] [CrossRef]
- Steinmann, E.; Brohm, C.; Kallis, S.; Bartenschlager, R.; Pietschmann, T. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J. Virol. 2008, 82, 7034–7046. [Google Scholar] [CrossRef]
- Suzuki, R.; Saito, K.; Kato, T.; Shirakura, M.; Akazawa, D.; Ishii, K.; Aizaki, H.; Kanegae, Y.; Matsuura, Y.; Saito, I.; Wakita, T.; Suzuki, T. Trans-complemented hepatitis C virus particles as a versatile tool for study of virus assembly and infection. Virology 2012, 432, 29–38. [Google Scholar] [CrossRef]
- Li, R.; Qin, Y.; He, Y.; Tao, W.; Zhang, N.; Tsai, C.; Zhou, P.; Zhong, J. Production of hepatitis C virus lacking the envelope-encoding genes for single-cycle infection by providing homologous envelope proteins or vesicular stomatitis virus glycoproteins in trans. J. Virol. 2011, 85, 2138–2147. [Google Scholar] [CrossRef]
- Brohm, C.; Steinmann, E.; Friesland, M.; Lorenz, I.C.; Patel, A.; Penin, F.; Bartenschlager, R.; Pietschmann, T. Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans-complementation. J. Virol. 2009, 83, 11682–11693. [Google Scholar] [CrossRef]
- Fournier, C.; Helle, F.; Descamps, V.; Morel, V.; Francois, C.; Dedeurwaerder, S.; Wychowski, C.; Duverlie, G.; Castelain, S. Natural selection of adaptive mutations in non-structural genes increases trans-encapsidation of hepatitis C virus replicons lacking envelope protein genes. J. Gen. Virol. 2013, 94, 996–1008. [Google Scholar] [CrossRef]
- Doerrbecker, J.; Friesland, M.; Riebesehl, N.; Ginkel, C.; Brown, R.; Ciesek, S.; Wedemeyer, H.; Sarrazin, C.; Pietschmann, T.; Steinmann, E. Incorporation of primary patient-derived glycoproteins into authentic infectious hepatitis c virus particles. Hepatology 2014. submitted for publication. [Google Scholar]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef]
- Choo, S.H.; So, H.S.; Cho, J.M.; Ryu, W.S. Association of hepatitis C virus particles with immunoglobulin: A mechanism for persistent infection. J. Gen. Virol. 1995, 76, 2337–2341. [Google Scholar] [CrossRef]
- Nielsen, S.U.; Bassendine, M.F.; Burt, A.D.; Martin, C.; Pumeechockchai, W.; Toms, G.L. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 2006, 80, 2418–2428. [Google Scholar] [CrossRef]
- Trestard, A.; Bacq, Y.; Buzelay, L.; Dubois, F.; Barin, F.; Goudeau, A.; Roingeard, P. Ultrastructural and physicochemical characterization of the hepatitis C virus recovered from the serum of an agammaglobulinemic patient. Arch. Virol. 1998, 143, 2241–2245. [Google Scholar] [CrossRef]
- Maillard, P.; Krawczynski, K.; Nitkiewicz, J.; Bronnert, C.; Sidorkiewicz, M.; Gounon, P.; Dubuisson, J.; Faure, G.; Crainic, R.; Budkowska, A. Nonenveloped nucleocapsids of hepatitis C virus in the serum of infected patients. J. Virol. 2001, 75, 8240–8250. [Google Scholar] [CrossRef]
- Thomssen, R.; Bonk, S.; Thiele, A. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. 1993, 182, 329–334. [Google Scholar]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef]
- Gastaminza, P.; Kapadia, S.B.; Chisari, F.V. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 2006, 80, 11074–11081. [Google Scholar] [CrossRef]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef]
- Jiang, J.; Luo, G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 2009, 83, 12680–12691. [Google Scholar] [CrossRef]
- Meunier, J.C.; Russell, R.S.; Engle, R.E.; Faulk, K.N.; Purcell, R.H.; Emerson, S.U. Apolipoprotein c1 association with hepatitis C virus. J. Virol. 2008, 82, 9647–9656. [Google Scholar] [CrossRef]
- Felmlee, D.J.; Sheridan, D.A.; Bridge, S.H.; Nielsen, S.U.; Milne, R.W.; Packard, C.J.; Caslake, M.J.; McLauchlan, J.; Toms, G.L.; Neely, R.D.; et al. Intravascular transfer contributes to postprandial increase in numbers of very-low-density hepatitis C virus particles. Gastroenterology 2010, 139, 1774–1783.e6. [Google Scholar] [CrossRef]
- Diaz, O.; Delers, F.; Maynard, M.; Demignot, S.; Zoulim, F.; Chambaz, J.; Trepo, C.; Lotteau, V.; Andre, P. Preferential association of Hepatitis C virus with apolipoprotein B48-containing lipoproteins. J. Gen. Virol. 2006, 87, 2983–2991. [Google Scholar] [CrossRef]
- Da Costa, D.; Turek, M.; Felmlee, D.J.; Girardi, E.; Pfeffer, S.; Long, G.; Bartenschlager, R.; Zeisel, M.B.; Baumert, T.F. Reconstitution of the entire hepatitis C virus life cycle in nonhepatic cells. J. Virol. 2012, 86, 11919–11925. [Google Scholar] [CrossRef]
- Hueging, K.; Doepke, M.; Vieyres, G.; Bankwitz, D.; Frentzen, A.; Doerrbecker, J.; Gumz, F.; Haid, S.; Wolk, B.; Kaderali, L.; Pietschmann, T. Apolipoprotein E co-determines tissue-tropism of hepatitis C virus and it is crucial for viral cell-to-cell transmission by contributing to a post-envelopment step of assembly. J. Virol. 2014, 88, 1433–1446. [Google Scholar] [CrossRef]
- Sakai, A.; Claire, M.S.; Faulk, K.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H.; Bukh, J. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc. Natl. Acad. Sci. USA 2003, 100, 11646–11651. [Google Scholar] [CrossRef]
- Dentzer, T.G.; Lorenz, I.C.; Evans, M.J.; Rice, C.M. Determinants of the hepatitis C virus nonstructural protein 2 protease domain required for production of infectious virus. J. Virol. 2009, 83, 12702–12713. [Google Scholar] [CrossRef]
- Jirasko, V.; Montserret, R.; Appel, N.; Janvier, A.; Eustachi, L.; Brohm, C.; Steinmann, E.; Pietschmann, T.; Penin, F.; Bartenschlager, R. Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J. Biol. Chem. 2008, 283, 28546–28562. [Google Scholar] [CrossRef]
- Phan, T.; Beran, R.K.; Peters, C.; Lorenz, I.C.; Lindenbach, B.D. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 2009, 83, 8379–8395. [Google Scholar] [CrossRef]
- 129. Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V., Jr.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; Walther, P.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef]
- Boulant, S.; Targett-Adams, P.; McLauchlan, J. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 2007, 88, 2204–2213. [Google Scholar] [CrossRef]
- Popescu, C.I.; Callens, N.; Trinel, D.; Roingeard, P.; Moradpour, D.; Descamps, V.; Duverlie, G.; Penin, F.; Heliot, L.; Rouille, Y.; Dubuisson, J. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 2011, 7, e1001278. [Google Scholar] [CrossRef]
- Boulant, S.; Douglas, M.W.; Moody, L.; Budkowska, A.; Targett-Adams, P.; McLauchlan, J. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 2008, 9, 1268–1282. [Google Scholar] [CrossRef]
- Boson, B.; Granio, O.; Bartenschlager, R.; Cosset, F.L. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog. 2011, 7, e1002144. [Google Scholar] [CrossRef]
- Neveu, G.; Barouch-Bentov, R.; Ziv-Av, A.; Gerber, D.; Jacob, Y.; Einav, S. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLoS Pathog. 2012, 8, e1002845. [Google Scholar] [CrossRef]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; Suzuki, T. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef]
- Stapleford, K.A.; Lindenbach, B.D. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 2011, 85, 1706–1717. [Google Scholar] [CrossRef]
- Jones, D.M.; Atoom, A.M.; Zhang, X.; Kottilil, S.; Russell, R.S. A genetic interaction between the core and NS3 proteins of hepatitis C virus is essential for production of infectious virus. J. Virol. 2011, 85, 12351–12361. [Google Scholar] [CrossRef]
- Mousseau, G.; Kota, S.; Takahashi, V.; Frick, D.N.; Strosberg, A.D. Dimerization-driven interaction of hepatitis C virus core protein with NS3 helicase. J. Gen. Virol. 2011, 92, 101–111. [Google Scholar] [CrossRef]
- Lo, S.Y.; Selby, M.J.; Ou, J.H. Interaction between hepatitis C virus core protein and E1 envelope protein. J. Virol. 1996, 70, 5177–5182. [Google Scholar]
- Ma, H.C.; Ke, C.H.; Hsieh, T.Y.; Lo, S.Y. The first hydrophobic domain of the hepatitis C virus E1 protein is important for interaction with the capsid protein. J. Gen. Virol. 2002, 83, 3085–3092. [Google Scholar]
- Nakai, K.; Okamoto, T.; Kimura-Someya, T.; Ishii, K.; Lim, C.K.; Tani, H.; Matsuo, E.; Abe, T.; Mori, Y.; Suzuki, T.; et al. Oligomerization of hepatitis C virus core protein is crucial for interaction with the cytoplasmic domain of E1 envelope protein. J. Virol. 2006, 80, 11265–11273. [Google Scholar] [CrossRef]
- Jirasko, V.; Montserret, R.; Lee, J.Y.; Gouttenoire, J.; Moradpour, D.; Penin, F.; Bartenschlager, R. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog. 2010, 6, e1001233. [Google Scholar] [CrossRef]
- Ma, Y.; Anantpadma, M.; Timpe, J.M.; Shanmugam, S.; Singh, S.M.; Lemon, S.M.; Yi, M. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J. Virol. 2011, 85, 86–97. [Google Scholar] [CrossRef]
- Murray, C.L.; Jones, C.T.; Tassello, J.; Rice, C.M. Alanine scanning of the hepatitis C virus core protein reveals numerous residues essential for production of infectious virus. J. Virol. 2007, 81, 10220–10231. [Google Scholar] [CrossRef]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.L.; Bartenschlager, R. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef]
- Gouklani, H.; Bull, R.A.; Beyer, C.; Coulibaly, F.; Gowans, E.J.; Drummer, H.E.; Netter, H.J.; White, P.A.; Haqshenas, G. Hepatitis C virus nonstructural protein 5B is involved in virus morphogenesis. J. Virol. 2012, 86, 5080–5088. [Google Scholar] [CrossRef]
- Han, Q.; Manna, D.; Belton, K.; Cole, R.; Konan, K.V. Modulation of hepatitis C virus genome encapsidation by nonstructural protein 4B. J. Virol. 2013, 87, 7409–7422. [Google Scholar] [CrossRef]
- McCormick, C.J.; Brown, D.; Griffin, S.; Challinor, L.; Rowlands, D.J.; Harris, M. A link between translation of the hepatitis C virus polyprotein and polymerase function; possible consequences for hyperphosphorylation of NS5A. J. Gen. Virol. 2006, 87, 93–102. [Google Scholar] [CrossRef]
- Gentzsch, J.; Brohm, C.; Steinmann, E.; Friesland, M.; Menzel, N.; Vieyres, G.; Perin, P.M.; Frentzen, A.; Kaderali, L.; Pietschmann, T. hepatitis c Virus p7 is critical for capsid assembly and envelopment. PLoS Pathog. 2013, 9, e1003355. [Google Scholar] [CrossRef]
- Santolini, E.; Migliaccio, G.; La Monica, N. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 1994, 68, 3631–3641. [Google Scholar]
- Kunkel, M.; Lorinczi, M.; Rijnbrand, R.; Lemon, S.M.; Watowich, S.J. Self-assembly of nucleocapsid-like particles from recombinant hepatitis C virus core protein. J. Virol. 2001, 75, 2119–2129. [Google Scholar] [CrossRef]
- Targett-Adams, P.; Boulant, S.; McLauchlan, J. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 2008, 82, 2182–2195. [Google Scholar] [CrossRef]
- Alsaleh, K.; Delavalle, P.Y.; Pillez, A.; Duverlie, G.; Descamps, V.; Rouille, Y.; Dubuisson, J.; Wychowski, C. Identification of basic amino acids at the N-terminal end of the core protein that are crucial for hepatitis C virus infectivity. J. Virol. 2010, 84, 12515–12528. [Google Scholar] [CrossRef]
- Steinmann, E.; Doerrbecker, J.; Friesland, M.; Riebesehl, N.; Ginkel, C.; Hillung, J.; Gentzsch, J.; Lauber, C.; Brown, R.; Frentzen, A.; Pietschmann, T. Characterization of hepatitis C virus intra- and inter-genotypic chimeras reveals a role of the glycoproteins in virus envelopment. J. Virol. 2013, 87, 13297–12306. [Google Scholar] [CrossRef]
- Corless, L.; Crump, C.M.; Griffin, S.D.; Harris, M. Vps4 and the ESCRT-III complex are required for the release of infectious hepatitis C virus particles. J. Gen. Virol. 2010, 91, 362–372. [Google Scholar] [CrossRef]
- Steinmann, E.; Whitfield, T.; Kallis, S.; Dwek, R.A.; Zitzmann, N.; Pietschmann, T.; Bartenschlager, R. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology 2007, 46, 330–338. [Google Scholar]
- Ai, L.S.; Lee, Y.W.; Chen, S.S. Characterization of hepatitis C virus core protein multimerization and membrane envelopment: Revelation of a cascade of core-membrane interactions. J. Virol. 2009, 83, 9923–9939. [Google Scholar] [CrossRef]
- Blanchard, E.; Hourioux, C.; Brand, D.; Ait-Goughoulte, M.; Moreau, A.; Trassard, S.; Sizaret, P.Y.; Dubois, F.; Roingeard, P. Hepatitis C virus-like particle budding: Role of the core protein and importance of its Asp111. J. Virol. 2003, 77, 10131–10138. [Google Scholar] [CrossRef]
- Riedel, C.; Lamp, B.; Heimann, M.; Konig, M.; Blome, S.; Moennig, V.; Schuttler, C.; Thiel, H.J.; Rumenapf, T. The core protein of classical Swine Fever virus is dispensable for virus propagation in vitro. PLoS Pathog. 2012, 8, e1002598. [Google Scholar] [CrossRef]
- Stapleton, J.T.; Foung, S.; Muerhoff, A.S.; Bukh, J.; Simmonds, P. The GB viruses: A review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J. Gen. Virol. 2011, 92, 233–246. [Google Scholar] [CrossRef]
- Gubler, D.J.; Kuno, G.; Markoff, L. Flaviviruses. In Fields Virology, 5th ed.; Knipe, D.M., Howley, M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 1153–1252. [Google Scholar]
- Schalich, J.; Allison, S.L.; Stiasny, K.; Mandl, C.W.; Kunz, C.; Heinz, F.X. Recombinant subviral particles from tick-borne encephalitis virus are fusogenic and provide a model system for studying flavivirus envelope glycoprotein functions. J. Virol. 1996, 70, 4549–4557. [Google Scholar]
- Icard, V.; Diaz, O.; Scholtes, C.; Perrin-Cocon, L.; Ramiere, C.; Bartenschlager, R.; Penin, F.; Lotteau, V.; Andre, P. Secretion of hepatitis C virus envelope glycoproteins depends on assembly of apolipoprotein B positive lipoproteins. PLoS One 2009, 4, e4233. [Google Scholar] [CrossRef]
- Scholtes, C.; Ramiere, C.; Rainteau, D.; Perrin-Cocon, L.; Wolf, C.; Humbert, L.; Carreras, M.; Guironnet-Paquet, A.; Zoulim, F.; Bartenschlager, R.; Lotteau, V.; Andre, P.; Diaz, O. High plasma level of nucleocapsid-free envelope glycoprotein-positive lipoproteins in hepatitis C patients. Hepatology 2012, 56, 39–48. [Google Scholar] [CrossRef]
- Menzel, N.; Fischl, W.; Hueging, K.; Bankwitz, D.; Frentzen, A.; Haid, S.; Gentzsch, J.; Kaderali, L.; Bartenschlager, R.; Pietschmann, T. MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS Pathog. 2012, 8, e1002829. [Google Scholar] [CrossRef]
- Ariumi, Y.; Kuroki, M.; Maki, M.; Ikeda, M.; Dansako, H.; Wakita, T.; Kato, N. The ESCRT system is required for hepatitis C virus production. PLoS One 2011, 6, e14517. [Google Scholar]
- Tamai, K.; Shiina, M.; Tanaka, N.; Nakano, T.; Yamamoto, A.; Kondo, Y.; Kakazu, E.; Inoue, J.; Fukushima, K.; Sano, K.; Ueno, Y.; Shimosegawa, T.; Sugamura, K. Regulation of hepatitis C virus secretion by the Hrs-dependent exosomal pathway. Virology 2012, 422, 377–385. [Google Scholar] [CrossRef]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M., Jr.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar] [CrossRef]
- Long, G.; Hiet, M.S.; Windisch, M.P.; Lee, J.Y.; Lohmann, V.; Bartenschlager, R. Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology 2011, 141, 1057–1066. [Google Scholar] [CrossRef]
- Olofsson, S.O.; Stillemark-Billton, P.; Asp, L. Intracellular assembly of VLDL: Two major steps in separate cell compartments. Trends Cardiovasc. Med. 2000, 10, 338–345. [Google Scholar] [CrossRef]
- Monazahian, M.; Bohme, I.; Bonk, S.; Koch, A.; Scholz, C.; Grethe, S.; Thomssen, R. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J. Med. Virol. 1999, 57, 223–229. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Hiet, M.-S.; Long, G.; Romero-Brey, I.; Bartenschlager, R. Apolipoprotein E interacts with Hepatitis C virus E2 envelope glycoprotein and critically determines virion assembly. In Proceedings of the 19th International Symposium on Hepatitis C Virus and Related Viruses, Venice, Italy, 5–9 October 2012.
- Benga, W.J.; Krieger, S.E.; Dimitrova, M.; Zeisel, M.B.; Parnot, M.; Lupberger, J.; Hildt, E.; Luo, G.; McLauchlan, J.; Baumert, T.F.; Schuster, C. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 2010, 51, 43–53. [Google Scholar] [CrossRef]
- Evans, M.J.; Rice, C.M.; Goff, S.P. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. USA 2004, 101, 13038–13043. [Google Scholar] [CrossRef]
- Cun, W.; Jiang, J.; Luo, G. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J. Virol. 2010, 84, 11532–11541. [Google Scholar] [CrossRef]
- Albecka, A.; Belouzard, S.; Op de Beeck, A.; Descamps, V.; Goueslain, L.; Bertrand-Michel, J.; Terce, F.; Duverlie, G.; Rouille, Y.; Dubuisson, J. Role of LDL receptor in the hepatitis c virus life cycle. Hepatology 2012, 55, 998–1007. [Google Scholar] [CrossRef]
- Vieyres, G.; Dubuisson, J.; Patel, A.H. Characterization of antibody-mediated neutralization directed against the hypervariable region 1 of hepatitis C virus E2 glycoprotein. J. Gen. Virol. 2011, 92, 494–506. [Google Scholar] [CrossRef]
- Dreux, M.; Boson, B.; Ricard-Blum, S.; Molle, J.; Lavillette, D.; Bartosch, B.; Pecheur, E.I.; Cosset, F.L. The exchangeable apolipoprotein ApoC-I promotes membrane fusion of hepatitis C virus. J. Biol. Chem. 2007, 282, 32357–32369. [Google Scholar] [CrossRef]
- Bankwitz, D.; Steinmann, E.; Bitzegeio, J.; Ciesek, S.; Friesland, M.; Herrmann, E.; Zeisel, M.B.; Baumert, T.F.; Keck, Z.Y.; Foung, S.K.; Pecheur, E.I.; Pietschmann, T. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J. Virol. 2010, 84, 5751–5763. [Google Scholar] [CrossRef]
- Prentoe, J.; Jensen, T.B.; Meuleman, P.; Serre, S.B.; Scheel, T.K.; Leroux-Roels, G.; Gottwein, J.M.; Bukh, J. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J. Virol. 2011, 85, 2224–2234. [Google Scholar] [CrossRef]
- Bartosch, B.; Verney, G.; Dreux, M.; Donot, P.; Morice, Y.; Penin, F.; Pawlotsky, J.M.; Lavillette, D.; Cosset, F.L. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 2005, 79, 8217–8229. [Google Scholar] [CrossRef]
- Grove, J.; Nielsen, S.; Zhong, J.; Bassendine, M.F.; Drummer, H.E.; Balfe, P.; McKeating, J.A. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J. Virol. 2008, 82, 12020–12029. [Google Scholar] [CrossRef]
- Keck, Z.Y.; Xia, J.; Cai, Z.; Li, T.K.; Owsianka, A.M.; Patel, A.H.; Luo, G.; Foung, S.K. Immunogenic and functional organization of hepatitis C virus (HCV) glycoprotein E2 on infectious HCV virions. J. Virol. 2007, 81, 1043–1047. [Google Scholar] [CrossRef]
- Owsianka, A.M.; Tarr, A.W.; Keck, Z.Y.; Li, T.K.; Witteveldt, J.; Adair, R.; Foung, S.K.; Ball, J.K.; Patel, A.H. Broadly neutralizing human monoclonal antibodies to the hepatitis C virus E2 glycoprotein. J. Gen. Virol. 2008, 89, 653–659. [Google Scholar] [CrossRef]
- Stamataki, Z.; Coates, S.; Evans, M.J.; Wininger, M.; Crawford, K.; Dong, C.; Fong, Y.L.; Chien, D.; Abrignani, S.; Balfe, P.; Rice, C.M.; McKeating, J.A.; Houghton, M. Hepatitis C virus envelope glycoprotein immunization of rodents elicits cross-reactive neutralizing antibodies. Vaccine 2007, 25, 7773–7784. [Google Scholar] [CrossRef]
- Meunier, J.C.; Gottwein, J.M.; Houghton, M.; Russell, R.S.; Emerson, S.U.; Bukh, J.; Purcell, R.H. Vaccine-induced cross-genotype reactive neutralizing antibodies against hepatitis C virus. J. Infect. Dis. 2011, 204, 1186–1190. [Google Scholar] [CrossRef]
- Keck, Z.Y.; Li, T.K.; Xia, J.; Gal-Tanamy, M.; Olson, O.; Li, S.H.; Patel, A.H.; Ball, J.K.; Lemon, S.M.; Foung, S.K. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J. Virol. 2008, 82, 6061–6066. [Google Scholar] [CrossRef]
- Krey, T.; Thiel, H.J.; Rumenapf, T. Acid-resistant bovine pestivirus requires activation for pH-triggered fusion during entry. J. Virol. 2005, 79, 4191–4200. [Google Scholar] [CrossRef]
- Sharma, N.R.; Mateu, G.; Dreux, M.; Grakoui, A.; Cosset, F.L.; Melikyan, G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011, 286, 30361–30376. [Google Scholar]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef]
- Mothes, W.; Boerger, A.L.; Narayan, S.; Cunningham, J.M.; Young, J.A. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 2000, 103, 679–689. [Google Scholar] [CrossRef]
- Wozniak, A.L.; Griffin, S.; Rowlands, D.; Harris, M.; Yi, M.; Lemon, S.M.; Weinman, S.A. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 2010, 6, e1001087. [Google Scholar] [CrossRef]
- Meunier, J.C.; Russell, R.S.; Goossens, V.; Priem, S.; Walter, H.; Depla, E.; Union, A.; Faulk, K.N.; Bukh, J.; Emerson, S.U.; Purcell, R.H. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J. Virol. 2008, 82, 966–973. [Google Scholar] [CrossRef]
- Haberstroh, A.; Schnober, E.K.; Zeisel, M.B.; Carolla, P.; Barth, H.; Blum, H.E.; Cosset, F.L.; Koutsoudakis, G.; Bartenschlager, R.; Union, A.; et al. Neutralizing host responses in hepatitis C virus infection target viral entry at postbinding steps and membrane fusion. Gastroenterology 2008, 135, 1719–1728.e1. [Google Scholar] [CrossRef]
- Dreux, M.; Pietschmann, T.; Granier, C.; Voisset, C.; Ricard-Blum, S.; Mangeot, P.E.; Keck, Z.; Foung, S.; Vu-Dac, N.; Dubuisson, J.; Bartenschlager, R.; Lavillette, D.; Cosset, F.L. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J. Biol. Chem. 2006, 281, 18285–18295. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vieyres, G.; Dubuisson, J.; Pietschmann, T. Incorporation of Hepatitis C Virus E1 and E2 Glycoproteins: The Keystones on a Peculiar Virion. Viruses 2014, 6, 1149-1187. https://doi.org/10.3390/v6031149
Vieyres G, Dubuisson J, Pietschmann T. Incorporation of Hepatitis C Virus E1 and E2 Glycoproteins: The Keystones on a Peculiar Virion. Viruses. 2014; 6(3):1149-1187. https://doi.org/10.3390/v6031149
Chicago/Turabian StyleVieyres, Gabrielle, Jean Dubuisson, and Thomas Pietschmann. 2014. "Incorporation of Hepatitis C Virus E1 and E2 Glycoproteins: The Keystones on a Peculiar Virion" Viruses 6, no. 3: 1149-1187. https://doi.org/10.3390/v6031149