Noncanonical Expression of a Murine Cytomegalovirus Early Protein CD8 T-Cell Epitope as an Immediate Early Epitope Based on Transcription from an Upstream Gene



Abstract
:1. Introduction
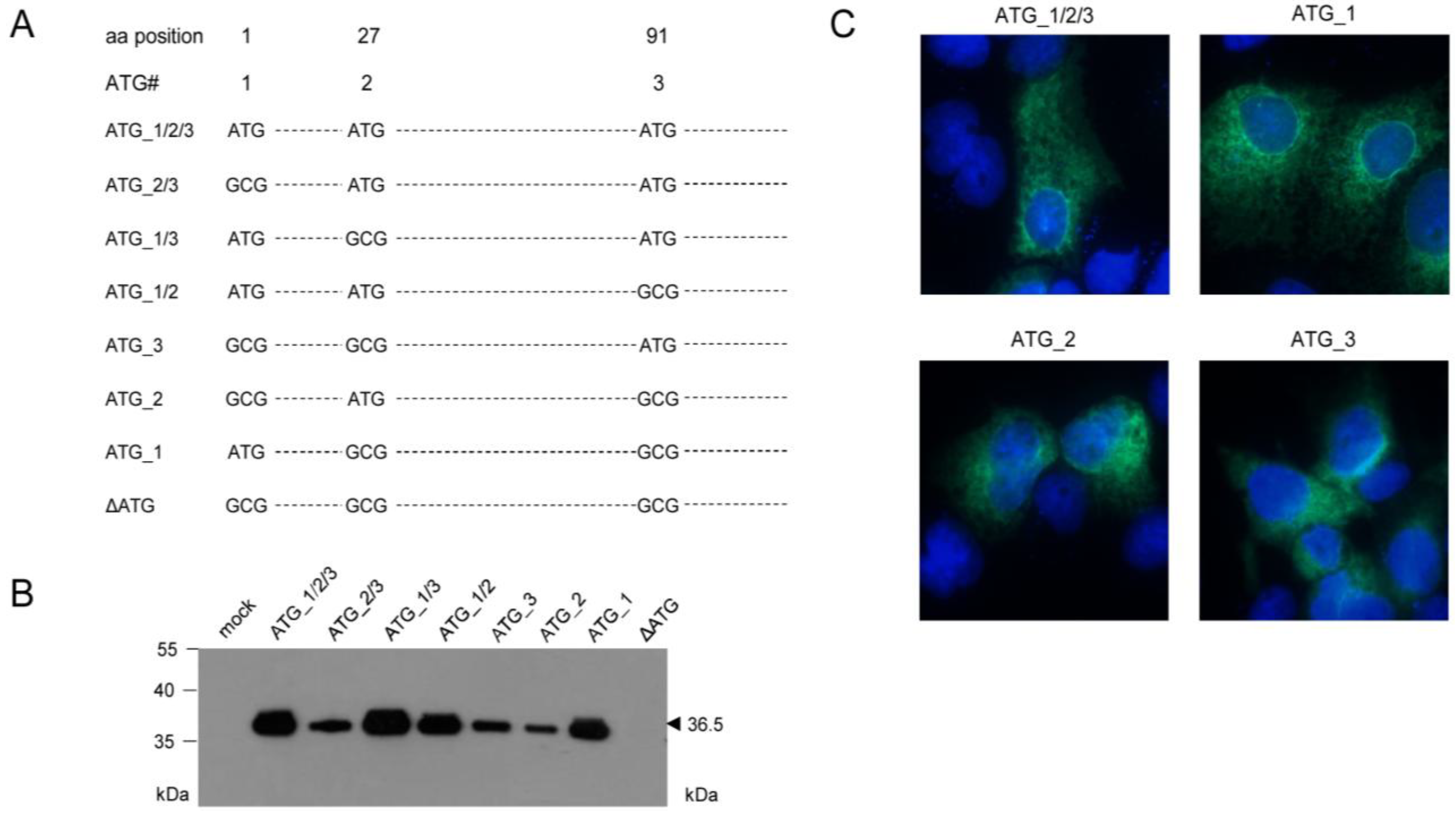
2. Results and Discussion
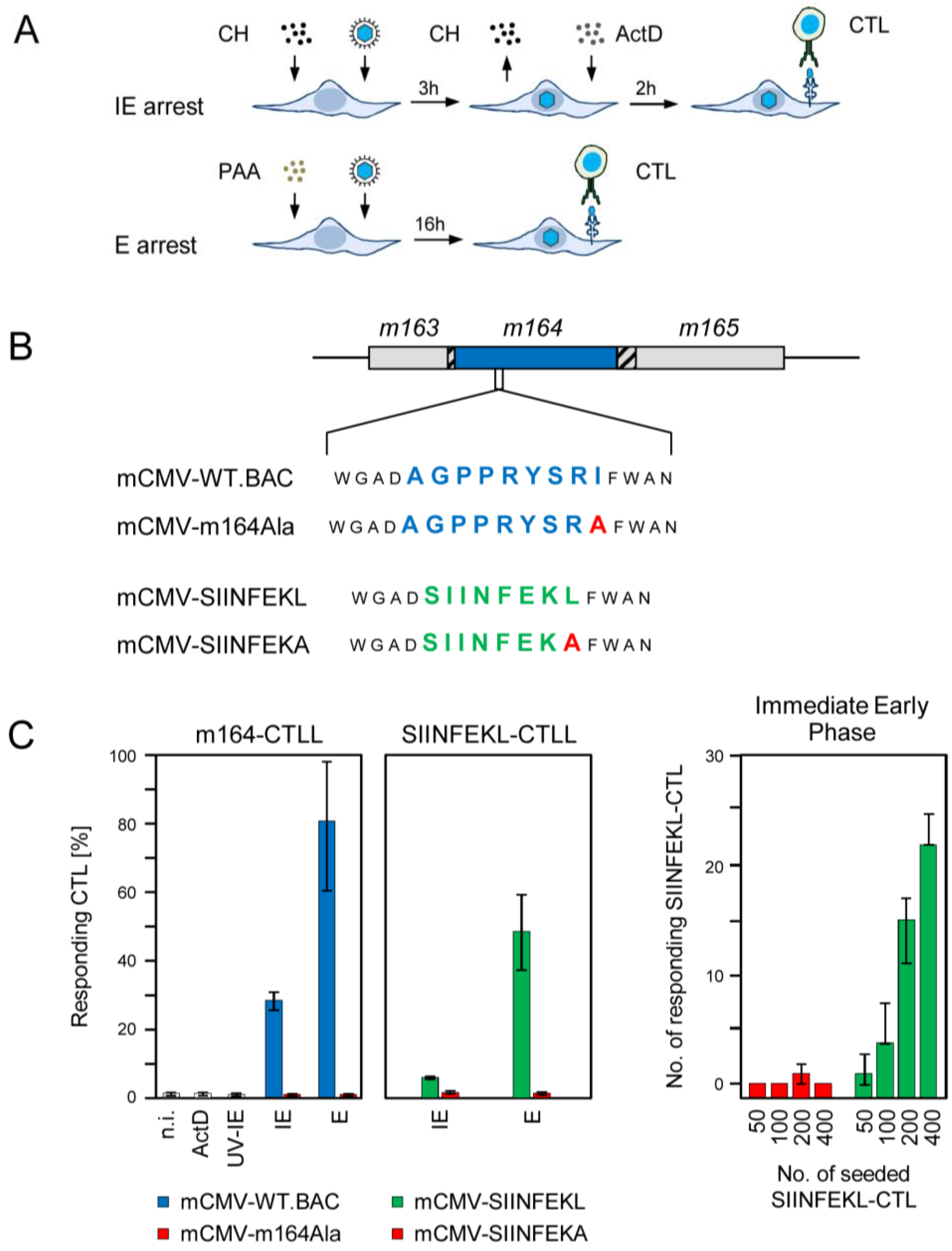
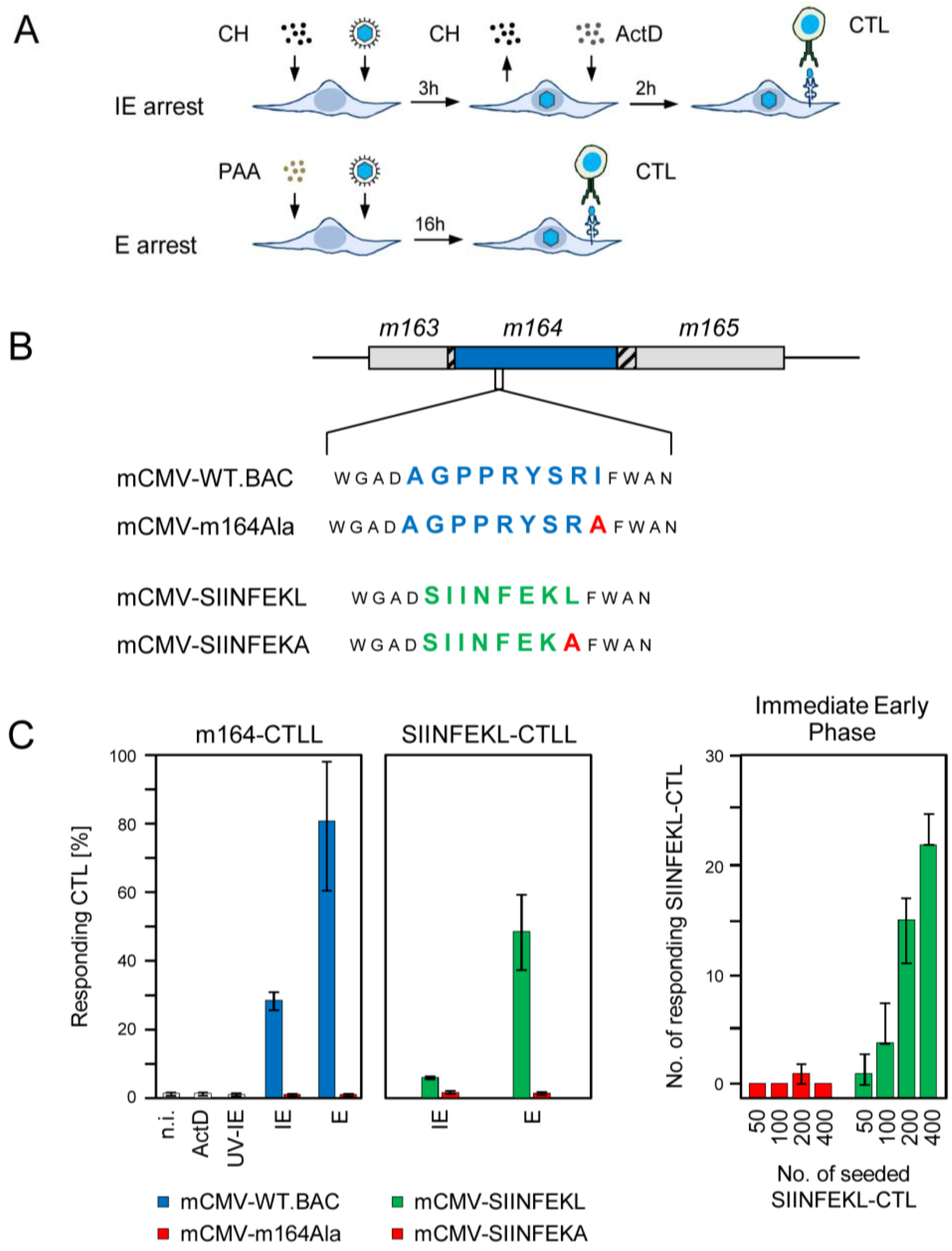
2.1. Antigenic Sequences Encoded by the Predicted ORFm164 Are Presented on Infected Cells Metabolically Arrested in the IE Phase as Well as in the E Phase
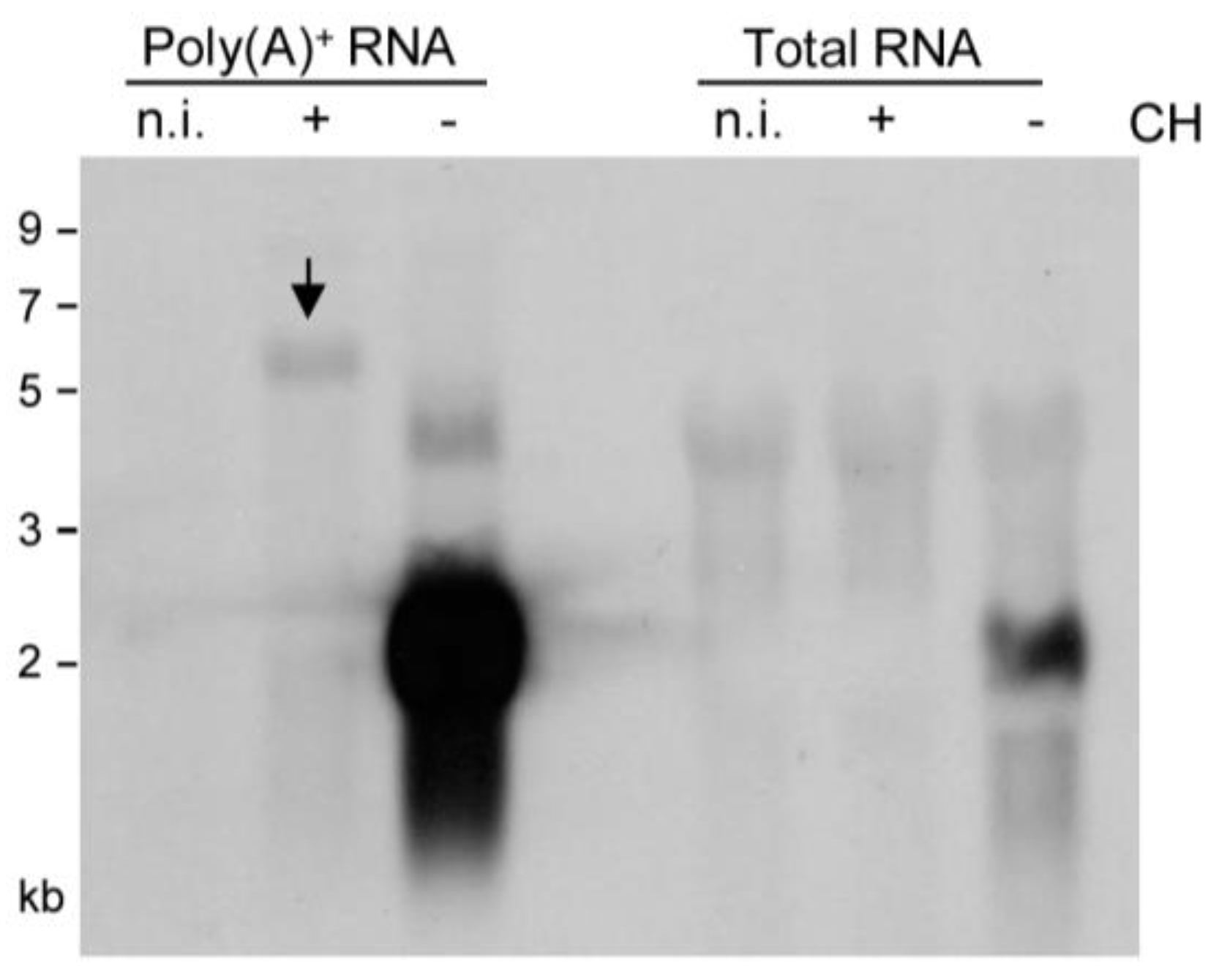
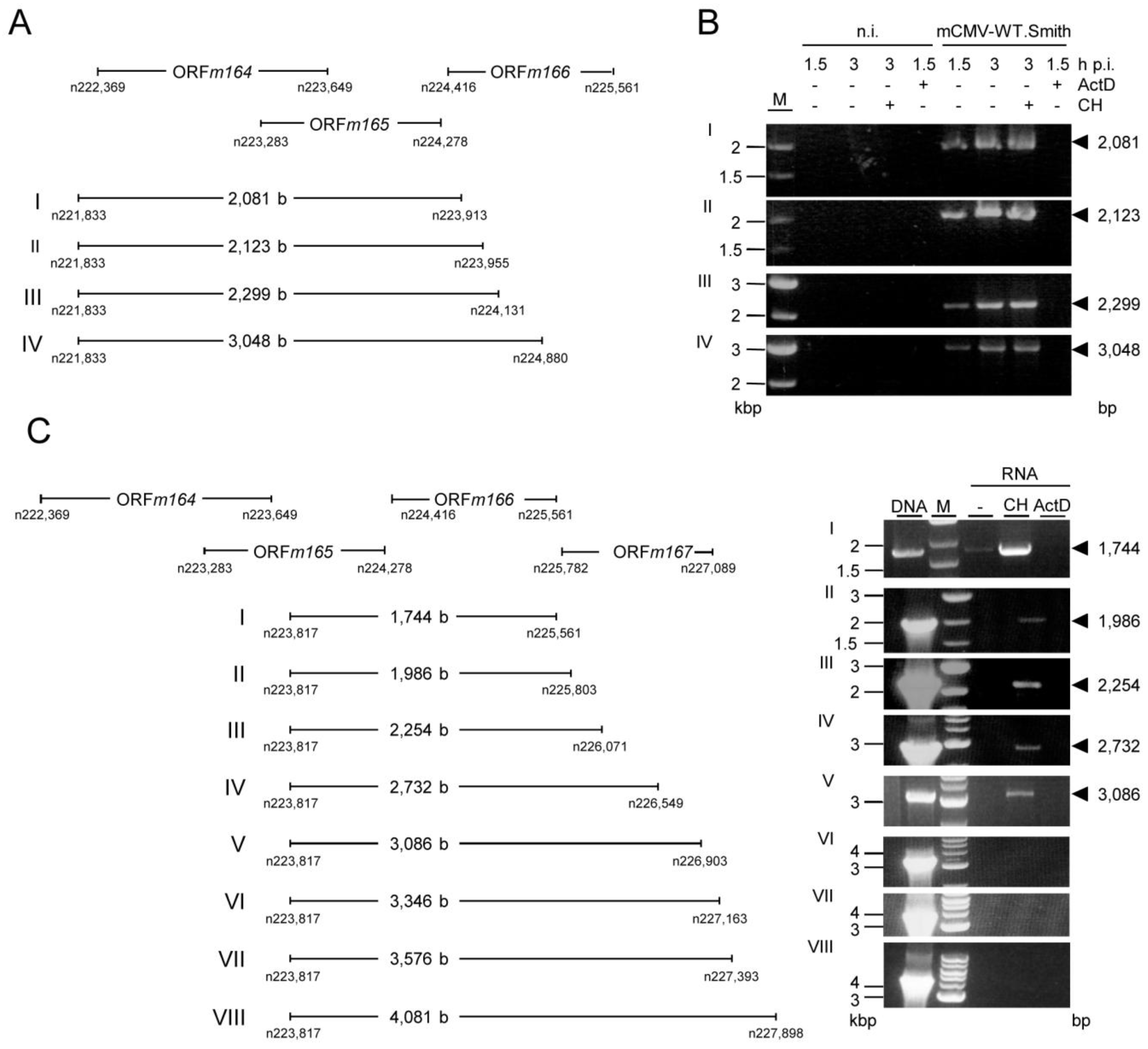
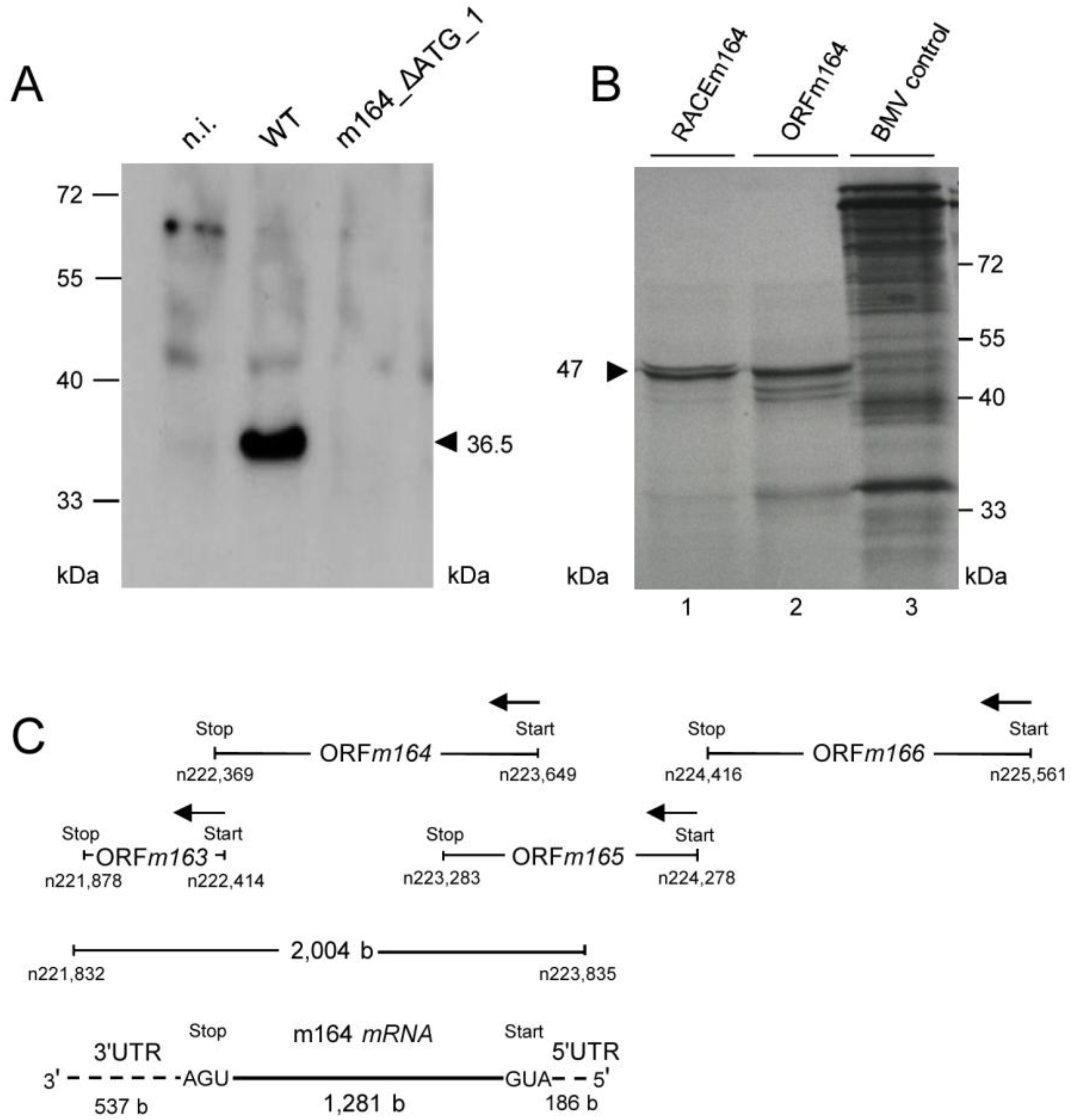
2.2. Detection of Distinct mRNA Species in the IE and E Phases by m164 Sequence-Specific Hybridization
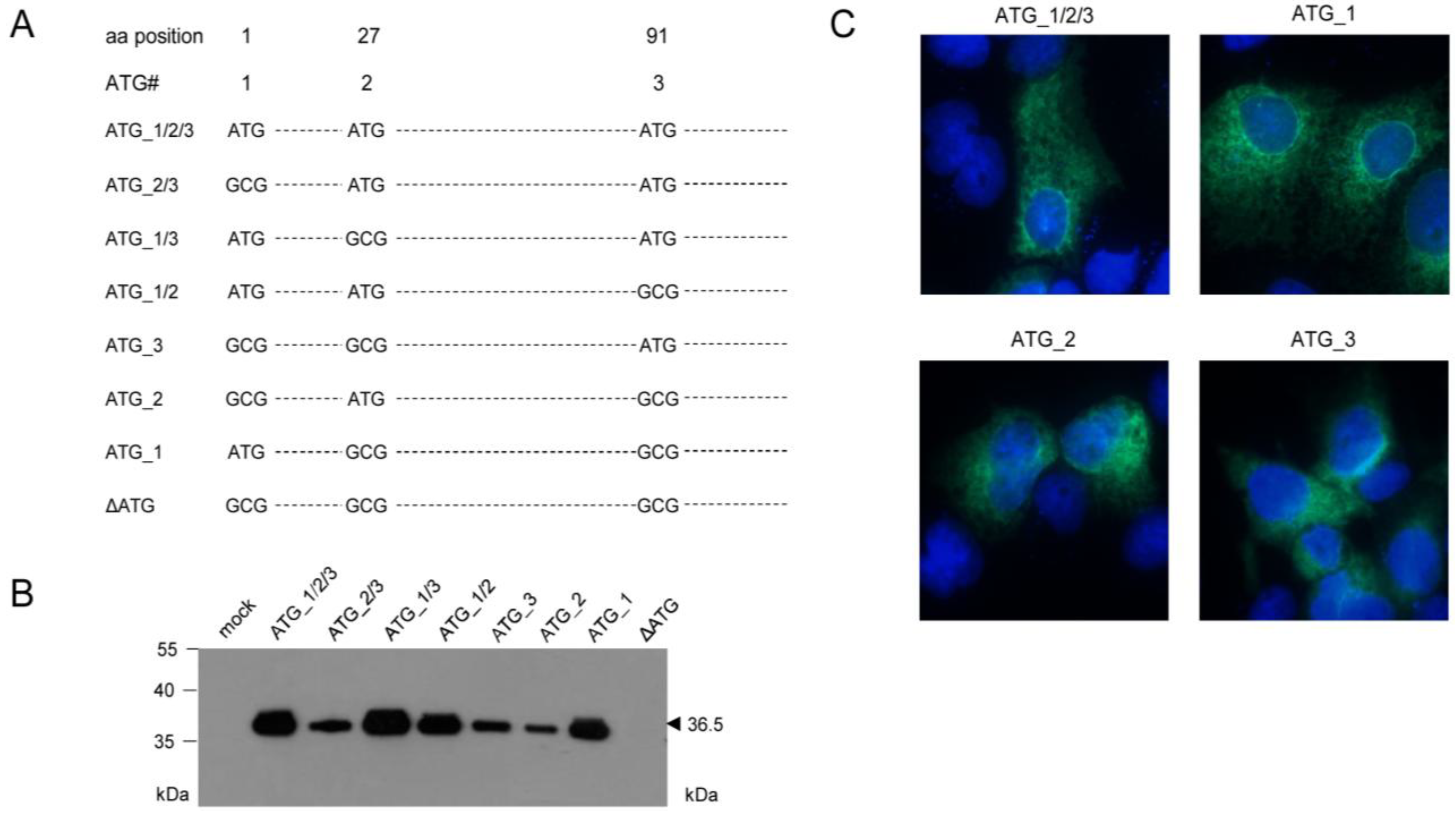
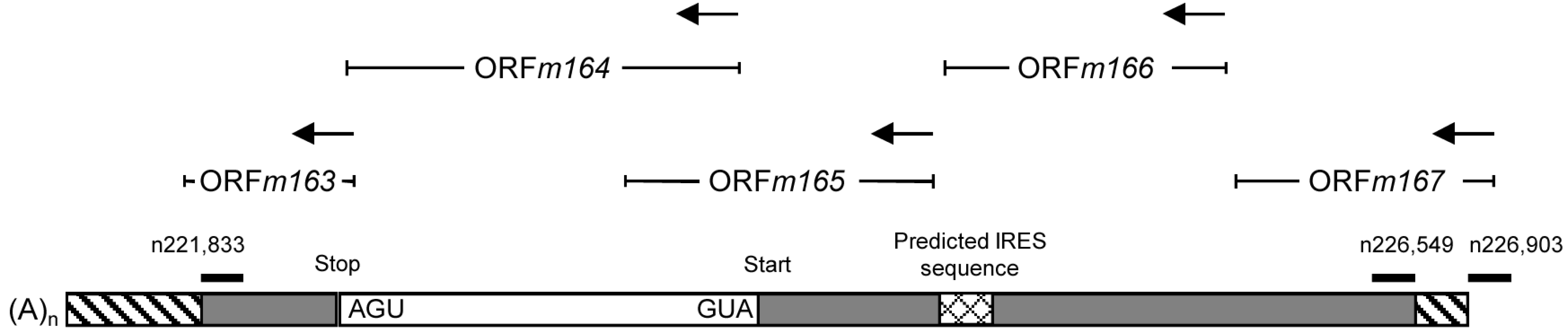
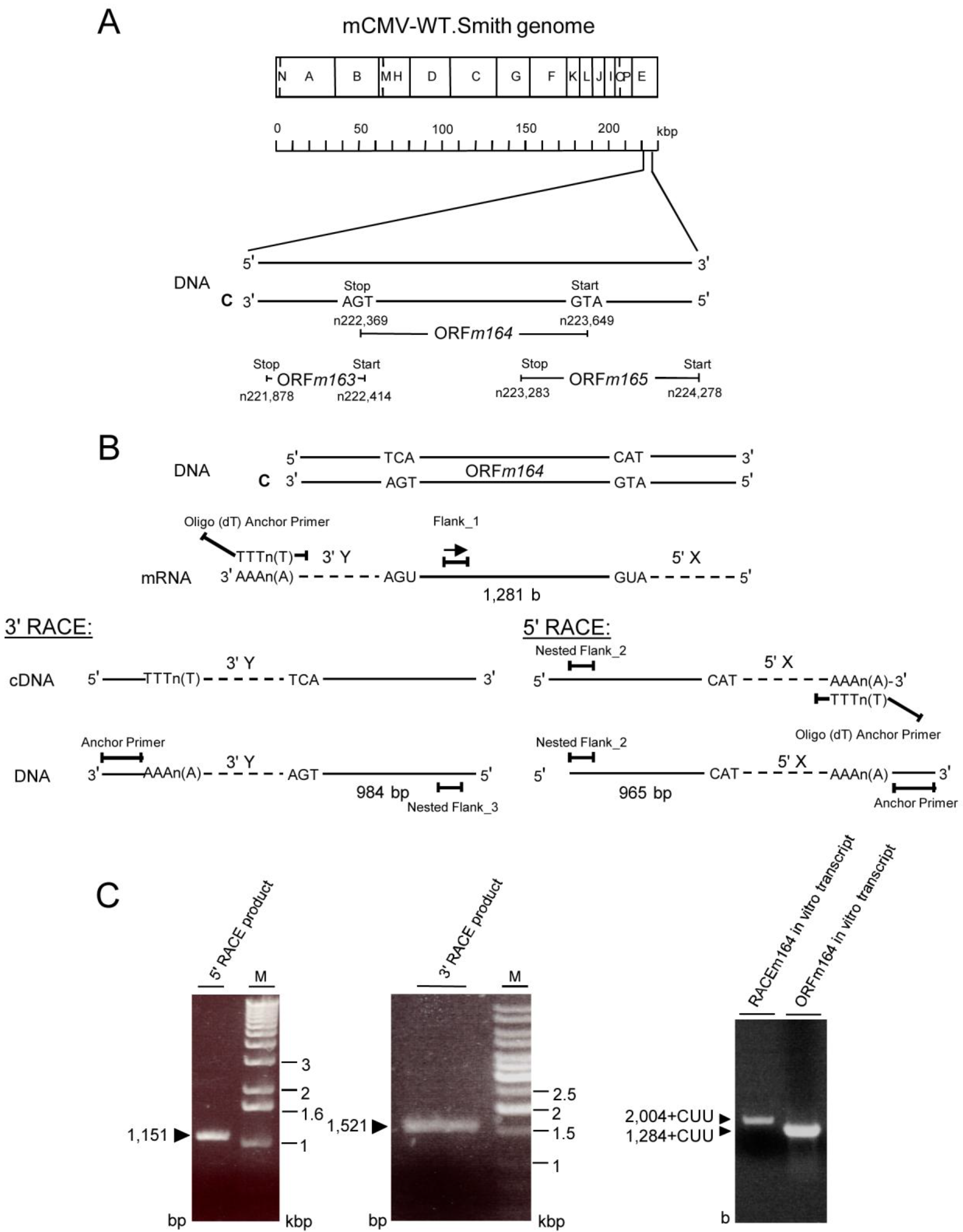
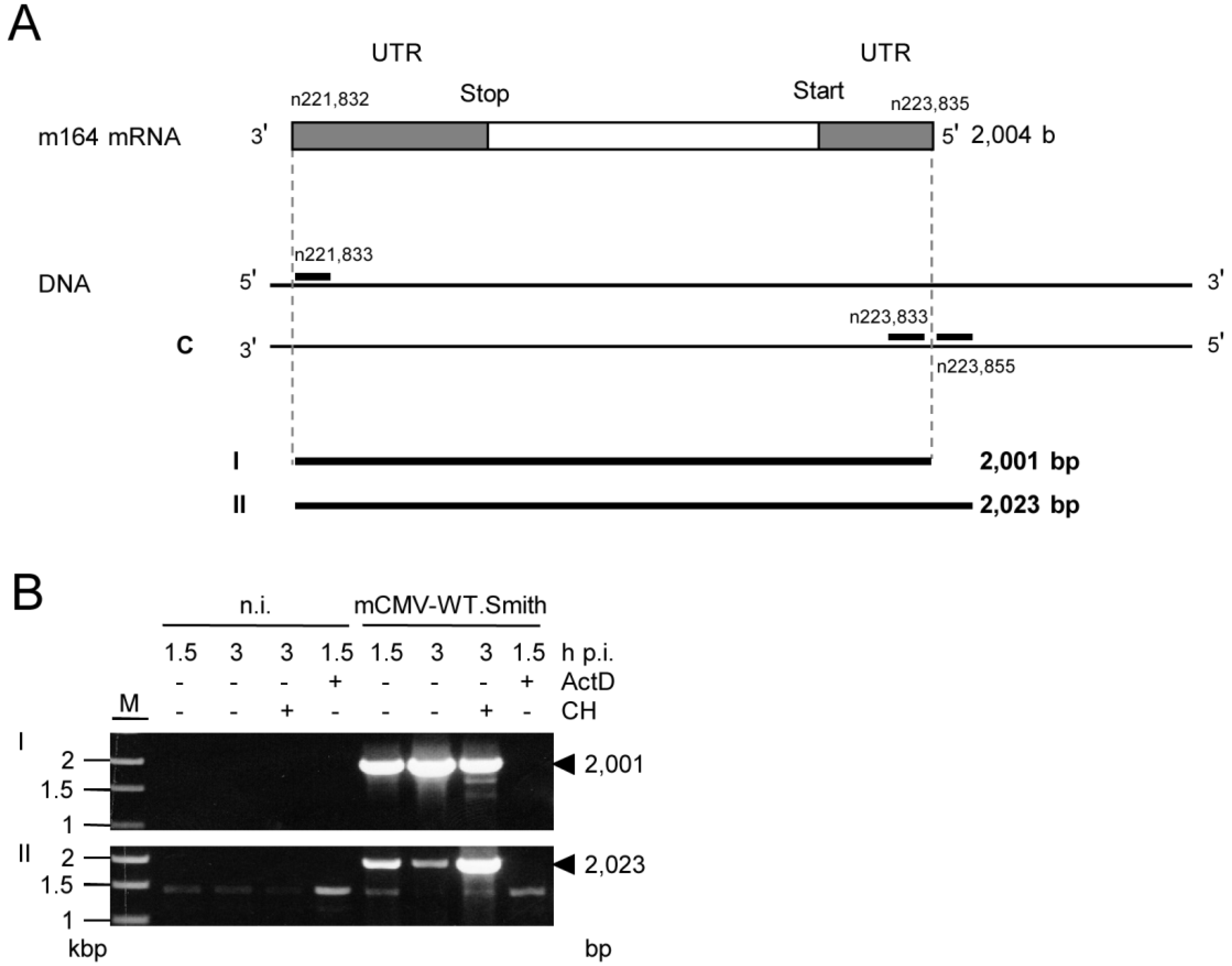
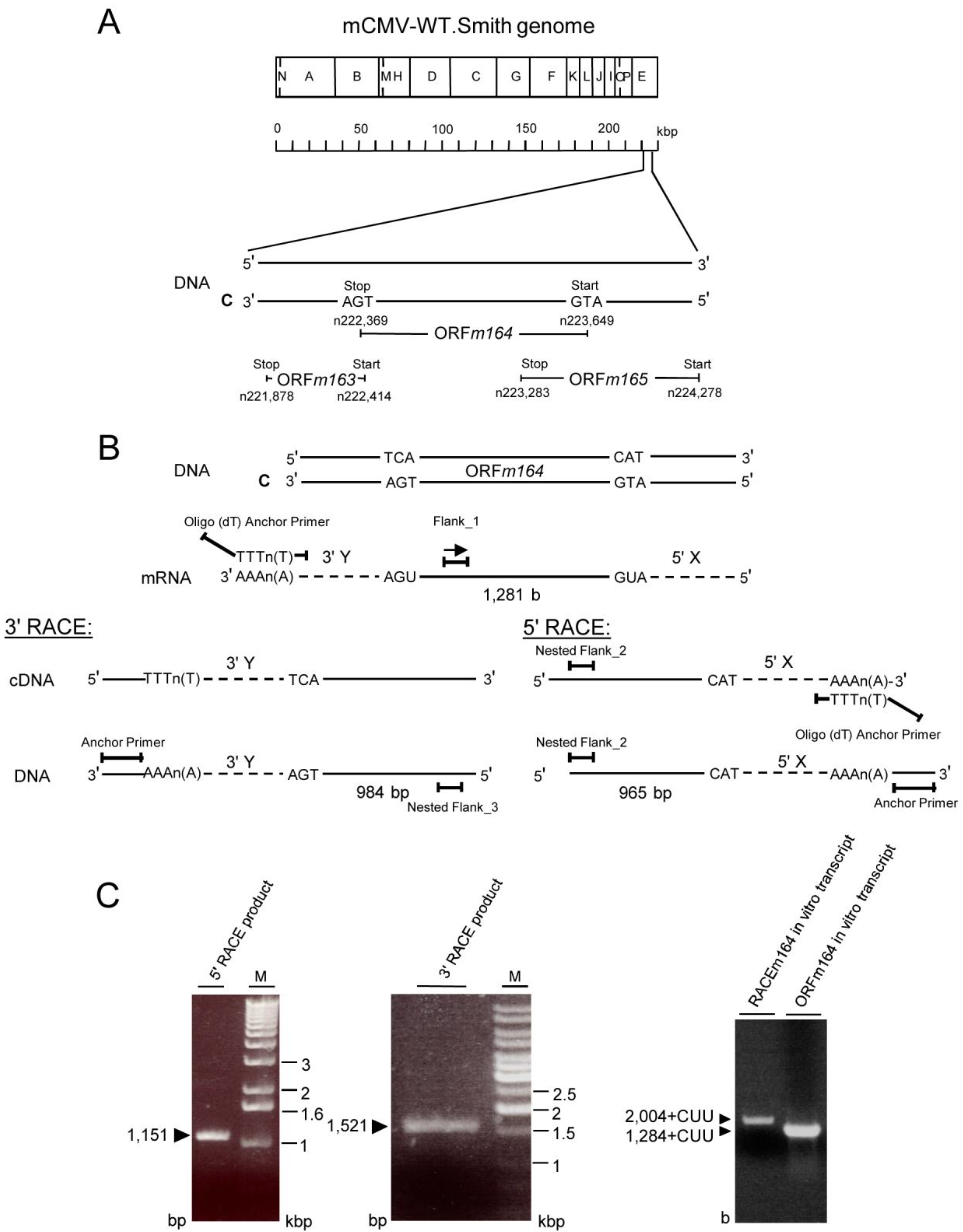
2.3. Mapping of the m164 E Phase Start Sites of Transcription and Translation
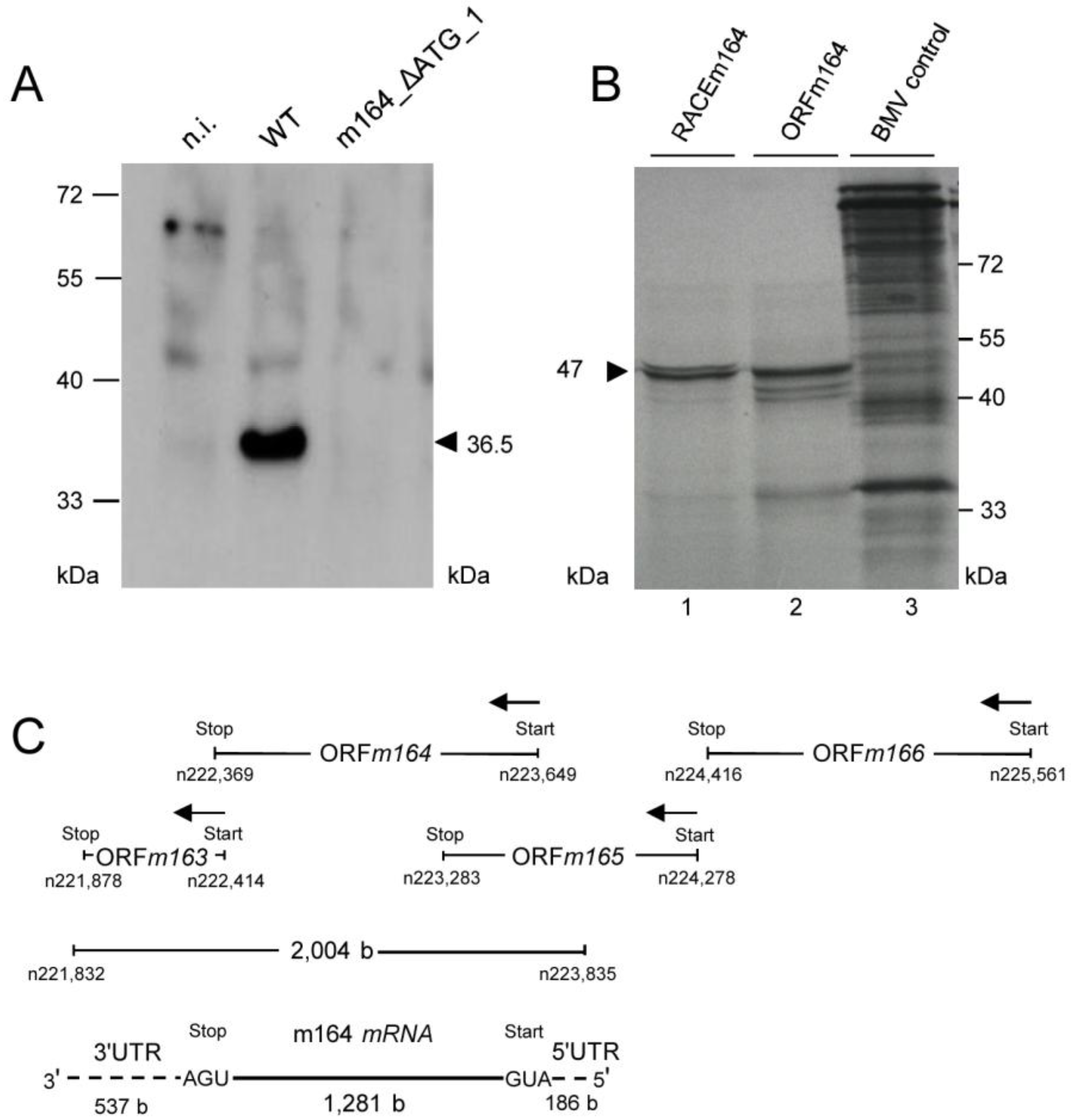
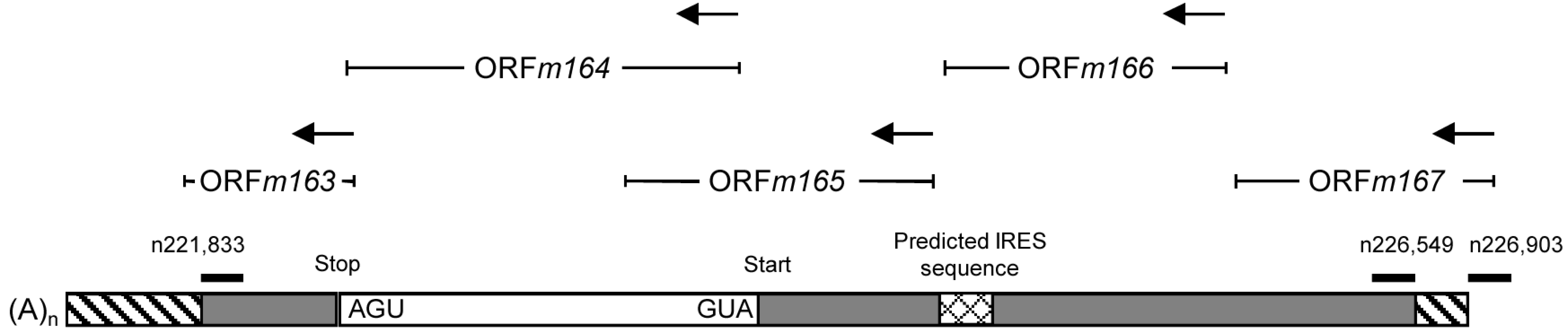
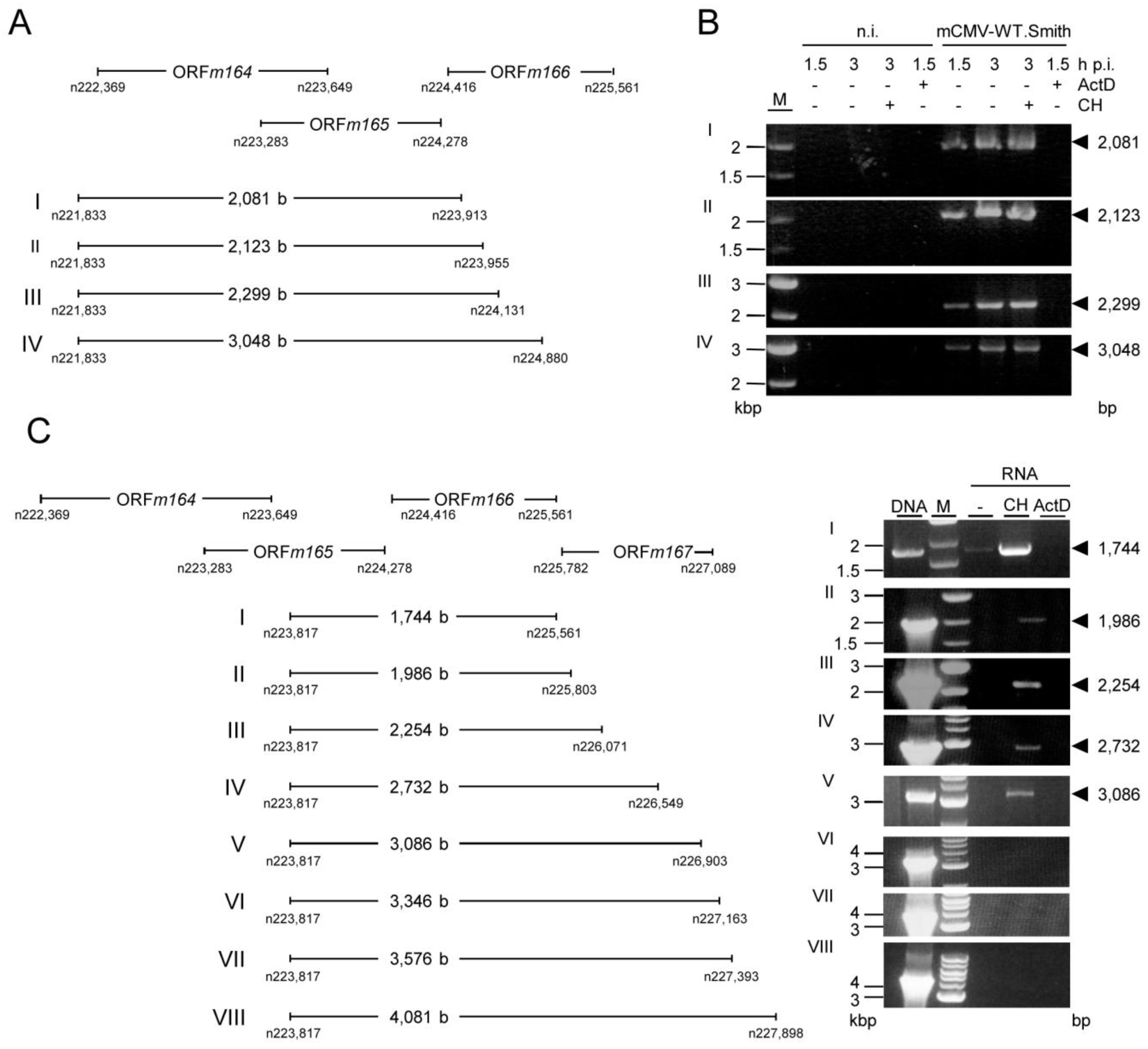
2.4. Confirmation of an Upstream IE Phase RNA Encompassing the Complete m164 E Phase RNA Sequence

3. Experimental Section
3.1. Cells, Viruses, and Mice
3.2. Infection Conditions
3.3. Construction of Expression Plasmids
3.4. Construction of Recombinant mCMV
3.5. Protein Extraction and Analysis
3.6. Transfection
3.7. Immunofluorescence Analysis
3.8. Northern Blot using RNA Probes
3.9. Mapping of mRNA by 5'/3'RACE
3.10. Primer Walking
3.11. In Vitro Transcription and Translation
3.12. ELISpot Assay
4. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Falk, K.; Rötzschke, O.; Stevanović, S.; Jung, G.; Rammensee, H.G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 1991, 351, 290–296. [Google Scholar] [CrossRef]
- Rammensee, H.; Bachmann, J.; Emmerich, N.P.; Bachor, O.A.; Stevanović, S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics 1999, 50, 213–219. [Google Scholar] [CrossRef]
- Tenzer, S.; Peters, B.; Bulik, S.; Schoor, O.; Lemmel, C.; Schatz, M.M.; Kloetzel, P.M.; Rammensee, H.G.; Schild, H.; Holzhütter, H.G. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol. Life Sci. 2005, 62, 1025–1037. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Keil, G.M.; Koszinowski, U.H. The cytolytic T lymphocyte response to the murine cytomegalovirus. II. Detection of virus replication stage-specific antigens by separate populations of in vivo active cytolytic T lymphocyte precursors. Eur. J. Immunol. 1984, 14, 56–61. [Google Scholar] [CrossRef]
- McLaughlin-Taylor, E.; Pande, H.; Forman, S.J.; Tanamachi, B.; Li, C.R.; Zaia, J.A.; Greenberg, P.D.; Riddell, S.R. Identification of the major late human cytomegalovirus matrix protein pp65 as a target antigen for CD8+ virus-specific cytotoxic T lymphocytes. J. Med. Virol. 1994, 43, 103–110. [Google Scholar] [CrossRef]
- Frankenberg, N.; Lischka, P.; Pepperl-Klindworth, S.; Stamminger, T.; Plachter, B. Nucleocytoplasmic shuttling and CRM1-dependent MHC class I peptide presentation of human cytomegalovirus pp65. Med. Microbiol. Immunol. 2012, 201, 567–579. [Google Scholar] [CrossRef]
- Bresnahan, W.A.; Shenk, T. A subset of viral transcripts packaged within human cytomegalovirus particles. Science 2000, 288, 2373–2376. [Google Scholar] [CrossRef]
- Reddehase, M.J. Antigens and immunoevasins: Opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2002, 2, 831–844. [Google Scholar] [CrossRef]
- Powers, C.; DeFilippis, V.; Malouli, D.; Früh, K. Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 2008, 325, 333–359. [Google Scholar]
- Hansen, T.H.; Bouvier, M. MHC class I antigen presentation: Learning from viral evasion strategies. Nat. Rev. Immunol. 2009, 9, 503–513. [Google Scholar] [CrossRef]
- Holtappels, R.; Thomas, D.; Podlech, J.; Reddehase, M.J. Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J. Virol. 2002, 76, 151–164. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Rothbard, J.B.; Koszinowski, U.H. A pentapeptide as minimal antigenic determinant for MHC class I-restricted T lymphocytes. Nature 1989, 337, 651–653. [Google Scholar]
- Holtappels, R.; Simon, C.O.; Munks, M.W.; Thomas, D.; Deegen, P.; Kühnapfel, B.; Däubner, T.; Emde, S.; Podlech, J.; Grzimek, N.K.; et al. Subdominant CD8 T-cell epitopes account for protection against cytomegalovirus independent of immunodomination. J. Virol. 2008, 82, 5781–5796. [Google Scholar] [CrossRef]
- Holtappels, R.; Pahl-Seibert, M.F.; Thomas, D.; Reddehase, M.J. Enrichment of immediate-early 1 (m123/pp89) peptide-specific CD8 T cells in a pulmonary CD62L(lo) memory-effector cell pool during latent murine cytomegalovirus infection of the lungs. J. Virol. 2000, 74, 11495–11503. [Google Scholar] [CrossRef]
- Karrer, U.; Sierro, S.; Wagner, M.; Oxenius, A.; Hengel, H.; Koszinowski, U.H.; Phillips, R.E.; Klenerman, P. Memory inflation: Continuous accumulation of antiviral CD8+ T cells over time. J. Immunol. 2003, 170, 2022–2029. [Google Scholar]
- Klenerman, P.; Dunbar, P.R. CMV and the art of memory maintenance. Immunity 2008, 29, 520–522. [Google Scholar] [CrossRef]
- Snyder, C.M. Buffered memory: A hypothesis for the maintenance of functional, virus-specific CD8+ T cells during cytomegalovirus infection. Immunol. Res. 2011, 51, 195–204. [Google Scholar] [CrossRef]
- O’Hara, G.A.; Welten, S.P.; Klenerman, P.; Arens, R. Memory T cell inflation: Understanding cause and effect. Trends Immunol. 2012, 33, 84–90. [Google Scholar] [CrossRef]
- Munks, M.W.; Cho, K.S.; Pinto, A.K.; Sierro, S.; Klenerman, P.; Hill, A.B. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J. Immunol. 2006, 177, 450–458. [Google Scholar]
- Seckert, C.K.; Griessl, M.; Büttner, J.K.; Scheller, S.; Simon, C.O.; Kropp, K.A.; Renzaho, A.; Kühnapfel, B.; Grzimek, N.K.; Reddehase, M.J. Viral latency drives “memory inflation”: A unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 551–566. [Google Scholar] [CrossRef]
- Seckert, C.K.; Grießl, M.; Büttner, J.K.; Freitag, K.; Lemmermann, N.A.W.; Hummel, M.A.; Liu, X.-F.; Abecassis, M.I.; Angulo, A.; Messerle, M.; et al. Immune surveillance of cytomegalovirus latency and reactivation in murine models: Link to “memory inflation”. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; pp. 374–416. [Google Scholar]
- Däubner, T.; Fink, A.; Seitz, A.; Tenzer, S.; Müller, J.; Strand, D.; Seckert, C.K.; Janssen, C.; Renzaho, A.; Grzimek, N.K.; et al. A novel transmembrane domain mediating retention of a highly motile herpesvirus glycoprotein in the endoplasmic reticulum. J. Gen. Virol. 2010, 91, 1524–1534. [Google Scholar] [CrossRef]
- Holtappels, R.; Grzimek, N.K.; Simon, C.O.; Thomas, D.; Dreis, D.; Reddehase, M.J. Processing and presentation of murine cytomegalovirus pORFm164-derived peptide in fibroblasts in the face of all viral immunosubversive early gene functions. J. Virol. 2002, 76, 6044–6053. [Google Scholar] [CrossRef]
- Holtappels, R.; Böhm, V.; Podlech, J.; Reddehase, M.J. CD8 T-cell-based immunotherapy of cytomegalovirus infection: “Proof of concept” provided by the murine model. Med. Microbiol. Immunol. 2008, 197, 125–134. [Google Scholar] [CrossRef]
- Ebert, S.; Podlech, J.; Gillert-Marien, D.; Gergely, K.M.; Büttner, J.K.; Fink, A.; Freitag, K.; Thomas, D.; Reddehase, M.J.; Holtappels, R. Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 527–539. [Google Scholar] [CrossRef]
- Lemmermann, N.A.; Kropp, K.A.; Seckert, C.K.; Grzimek, N.K.; Reddehase, M.J. Reverse genetics modification of cytomegalovirus antigenicity and immunogenicity by CD8 T-cell epitope deletion and insertion. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef]
- Simon, C.O.; Holtappels, R.; Tervo, H.-M.; Böhm, V.; Däubner, T.; Oehrlein-Karpi, S.A.; Kühnapfel, B.; Renzaho, A.; Strand, D.; Podlech, J.; et al. CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J. Virol. 2006, 80, 10436–10456. [Google Scholar] [CrossRef]
- Böhm, V.; Podlech, J.; Thomas, D.; Deegen, P.; Pahl-Seibert, M.; Lemmermann, N.A.; Grzimek, N.K.; Oehrlein-Karpi, S.A.; Reddehase, M.J.; Holtappels, R. Epitope-specific in vivo protection against cytomegalovirus disease by CD8 T cells in the murine model of preemptive immunotherapy. Med. Microbiol. Immunol. 2008, 197, 135–144. [Google Scholar] [CrossRef]
- Lemmermann, N.A.; Gergely, K.M.; Böhm, V.; Deegen, P.; Däubner, T.; Reddehase, M.J. Immune evasion proteins of murine cytomegalovirus preferentially affect cell surface display of recently generated peptide presentation complexes. J. Virol. 2010, 84, 1221–1236. [Google Scholar] [CrossRef]
- Ebert, S.; Lemmermann, N.A.; Thomas, D.; Renzaho, A.; Reddehase, M.J.; Holtappels, R. Immune control in the absence of immunodominant epitopes: Implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med. Microbiol. Immunol. 2012, 201, 541–550. [Google Scholar] [CrossRef]
- Nauerth, M.; Weißbrich, B.; Knall, R.; Franz, T.; Dössinger, G.; Bet, J.; Paszkiewicz, P.J.; Pfeifer, L.; Bunse, M.; Uckert, W.; et al. TCR-ligand koff rate correlates with the protective capacity of antigen-specific CD8+ T cells for adoptive transfer. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Caposio, P.; Streblow, D.; Nelson, J.A. Cytomegalovirus proteomics. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume I, pp. 86–108. [Google Scholar]
- Redwood, A.J.; Shellam, G.R.; Smith, L.M. Molecular evolution of murine cytomegalovirus genomes. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume I, pp. 23–37. [Google Scholar]
- Rawlinson, W.; Farrell, H.; Barrell, B. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996, 70, 8833–8849. [Google Scholar]
- Bendtsen, J.D.; Nielsen, H.; von Heijne, G.; Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef]
- Dölken, L.; Department of Medicine, University of Cambridge, Cambridge, UK. Personal communication, 2013.
- Erhard, F.; Institut für Informatik, Ludwigs-Maximilians-Universität München, München, Germany. Personal communication, 2013.
- Zhu, J.; Chen, J.; Hai, R.; Tong, T.; Xiao, J.; Zhan, X.; Lu, S.; Liu, F. In vitro and in vivo characterization of a murine cytomegalovirus with a mutation at open reading frame m166. J. Virol. 2003, 77, 2882–2891. [Google Scholar] [CrossRef]
- Mercer, J.A.; Marks, J.R.; Spector, D.H. Molecular cloning and restriction endonuclease mapping of the murine cytomegalovirus genome (Smith Strain). Virology 1983, 129, 94–106. [Google Scholar] [CrossRef]
- Wing, B.A.; Browne, E.P.; Shenk, T.E. Modulation of cellular gene expression by MCMV evaluated by DNA microarray analysis. In Proceedings of the 26th International Herpesvirus Workshop, Regensburg, Germany, 14–17 May 2001. Abstract Number 1.08.
- Shenk, T.E.; Department of Molecular Biology, Princeton University, Princeton, NJ, USA. Personal communication, 2007.
- Hong, J.-J.; Wu, T.-Y.; Chang, T.-Y.; Chen, C.-Y. Viral IRES prediction system—A web server for prediction of the IRES secondary structure in silico. PLoS One 2013, 8, e79288. [Google Scholar]
- Baird, S.D.; Turcotte, M.; Robert, G.; Korneluk, R.G.; Holcik, M. Searching for IRES. RNA 2006, 12, 1755–1785. [Google Scholar] [CrossRef]
- Podlech, J.; Holtappels, R.; Grzimek, N.K.; Reddehase, M.J. Animal models: Murine cytomegalovirus. In Methods in Microbiology, Immunology of Infection, 2nd ed.; Kaufmann, S., Kabelitz, D., Eds.; Academic Press: London, UK, 2002; pp. 493–525. [Google Scholar]
- Lemmermann, N.A.; Podlech, J.; Seckert, C.K.; Kropp, K.A.; Grzimek, N.K.; Reddehase, M.J.; Holtappels, R. CD8 T-cell immunotherapy of cytomegalovirus disease in the murine model. In Methods in Microbiology; Kabelitz, D., Kaufmann, S., Eds.; Academic Press: Oxford. UK, 2010; Volume 37, pp. 369–420. [Google Scholar]
- Wagner, M.; Jonjić, S.; Koszinowski, U.H.; Messerle, M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 1999, 73, 7056–7060. [Google Scholar]
- CIOMS and ICLAS release the new International Guiding Principles for Biomedical Research Involving Animals. Available online: http://www.cioms.ch/images/stories/CIOMS/IGP2012.pdf (accessed on 1 December 2013).
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [CrossRef]
- Holtappels, R.; Gillert-Marien, D.; Thomas, D.; Podlech, J.; Deegen, P.; Herter, S.; Oehrlein-Karpi, S.A.; Strand, D.; Wagner, M.; Reddehase, M.J. Cytomegalovirus encodes a positive regulator of antigen presentation. J. Virol. 2006, 80, 7613–7624. [Google Scholar] [CrossRef]
- Frohman, M.A. On beyond classic RACE (rapid amplification of cDNA ends). PCR Methods Appl. 1994, 4, 40–58. [Google Scholar] [CrossRef]
- Krieg, P.A.; Melton, D.A. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 1984, 12, 7057–7070. [Google Scholar] [CrossRef]
- Pahl-Seibert, M.F.; Juelch, M.; Podlech, J.; Thomas, D.; Deegen, P.; Reddehase, M.J.; Holtappels, R. Highly protective in vivo function of cytomegalovirus IE1 epitope-specific memory CD8 829 T cells purified by T-cell receptor-based cell sorting. J. Virol. 2005, 79, 5400–5413. [Google Scholar]
- Böhm, V.; Simon, C.O.; Podlech, J.; Seckert, C.K.; Gendig, D.; Deegen, P.; Gillert-Marien, D.; Lemmermann, N.A.; Holtappels, R.; Reddehase, M.J. The immune evasion paradox: Immunoevasins of murine cytomegalovirus enhance priming of CD8 T cells by preventing negative feedback regulation. J. Virol. 2008, 82, 11637–11650. [Google Scholar] [CrossRef]
- Wolfram Mathematica, version 9; Wolfram Research: Champaign, IL, USA, 2012.
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar]
- Fortunato, E.A.; Spector, D.H. Regulation of human cytomegalovirus gene expression. Adv. Virus Res. 1999, 54, 61–128. [Google Scholar] [CrossRef]
- Davison, A.J.; Holton, M.; Dolan, A.; Dargan, D.J.; Gatherer, D.; Hayward, G.S. Comparative genomics of primate cytomegaloviruses. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume I, pp. 1–22. [Google Scholar]
- Kurz, S.K.; Rapp, M.; Steffens, H.P.; Grzimek, N.K.; Schmalz, S.; Reddehase, M.J. Focal transcriptional activity of murine cytomegalovirus during latency in the lungs. J. Virol. 1999, 73, 482–494. [Google Scholar]
- Grzimek, N.K.; Dreis, D.; Schmalz, S.; Reddehase, M.J. Random, asynchronous, and asymmetric transcriptional activity of enhancer-flanking major immediate-early genes ie1/3 and ie2 during murine cytomegalovirus latency in the lungs. J. Virol. 2001, 75, 2692–2705. [Google Scholar] [CrossRef]
- Simon, C.O.; Seckert, C.K.; Dreis, D.; Reddehase, M.J.; Grzimek, N.K. Role for tumor necrosis factor alpha in murine cytomegalovirus transcriptional reactivation in latently infected lungs. J. Virol. 2005, 79, 326–340. [Google Scholar] [CrossRef]
- Ebeling, A.; Keil, G.M.; Knust, E.; Koszinowski, U.H. Molecular cloning and physical mapping of murine cytomegalovirus DNA. J. Virol. 1983, 47, 421–433. [Google Scholar]
Appendices

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| pEPKan-S_m164_1stATG_for | 5'-CCTGCCAGATCTTGGGGCCTCGGGTCGCCACACACGTGGGAAGGCG  TTTCTCCGCGGCTGTCGGCAACCAATTAACCAATTCTGATTAG-3' TTTCTCCGCGGCTGTCGGCAACCAATTAACCAATTCTGATTAG-3' |
| pEPKan-S_m164_1stATG_rev | 5'-GCTGCGCGTCTCCCCCTCCGACAGCCGCGGAGAAA  CGCCTTCCCACGTGTGTGGCGAGGATGACGACGATAAGTAGGG-3' CGCCTTCCCACGTGTGTGGCGAGGATGACGACGATAAGTAGGG-3' |
| m164_1stATG_short | 5'-CCTGCCAGATCTTGGGGCCTCGGG-3' |
| pEPKan-S_rev | 5'-AGGATGACGACGATAAGTAGGG-3' |
| mRNAint_for | 5'-GGCTTCTCGGGCCTCGTCGTGTGC-3' |
| mRNAint_rev | 5'-GCCACGTGGACGGGTCGACATTCG-3' |
| m164mRNA_rev | 5'-ACGCACATCACCAGTG-3' |
| mRNAin_for | 5'-GTTTAGTCGGGAGCGTG-3' |
| m164mRNAout_for | 5'-GTGAGGTGGTGGATAGATAC-3' |
| m164mRNAout50_for | 5'-GTGTGAGCAGTATCTGGG-3' |
| m164mRNAout100_for | 5'-GAGACGGCTAGCGAATC-3' |
| m164mRNAout300_for | 5'-GCGGACTACATCTCGATC-3' |
| m164mRNAout1000_for | 5'-GAGTGGAGCGAGAACAAC-3' |
| m164mRNAin_rev | 5'-CACGCTCCCGACTAAAC-3' |
| m166_ORF_end_for | 5'-ATGGCGCTTCCAGCAATTCC-3' |
| m166_ORF_end250_for | 5'-CAGGTTCCGCAACAGTC-3' |
| m166_ORF_end500_for | 5'-ATCGGATGGTGCTTCAG-3' |
| m166_ORF_end1000_for | 5'-AAGCCTCCGAGGAAGAC-3' |
| m166.5_ORF_end_for | 5'-GGCGGTAGAACAGTTGC-3' |
| m166.5_ORF_end250_for | 5'-CCTTTGAGTGTGGAAAGTTTG-3' |
| m166.5_ORF_end500_for | 5'-TTACCCGCGTCACATGG-3' |
| m166.5_ORF_end1000_for | 5'-CCGTCCTTAGGTTTCGTTTC-3' |
| Flank_1 | 5'-CACGAAGGCGGCGAGGAAAC-3' |
| Nested Flank_2 | 5'-GACGACGCACGATACGCAGAC-3' |
| Nested Flank_3 | 5'-CTCATCTACGCGGCGGCTTCAG-3' |
| m164_rev_BamHI | 5'-ATATGGATCCGGAGAAGAGTCAGAATCATGACGAACG-3' |
| m164_full_HindIII | 5'-ATATAAGCTTCTCCTGCCAGATCTTG-3' |
| m164_3rdATG_rev | 5'-GCGTAGATGAGGGCGAG CCCGGCCTTGAAGAGTCG-3' |
| m164_3rdATG_for | 5'-CGACTCTTCAAGGCCGGG CTCGCCCTCATCTACGC-3' |
| m164_2ndATG_rev | 5'-GCAACGCGTCGTCGGCA CGGCAACTCGGGTTCCCCG-3' |
| m164_2ndATG_for | 5'-CGGGGAACCCGAGTTGCCG TGCCGACGACGCGTTGC-3' |
| m164_1stATG_rev | 5'-CCGACAGCCGCGGAGAAA CGCCTTCCCACGTGTGTG-3'G |
| m164_1stATG_for | 5'-CCACACACGTGGGAAGGCGGCGTTTCTCCGCGGCTGTCG-3' |
| Product | Primer rev | Primer for | Annealing | Elongation | Size [bp] |
|---|---|---|---|---|---|
| Figure 4 | |||||
| I | m164mRNA_rev | m164mRNAin_for | 51.7 °C | 1 min 30 s; 72 °C | 2,001 |
| II | m164mRNA_rev | m164mRNAout_for | 51.7 °C | 90 s; 72 °C | 2,023 |
| Figure A2A | |||||
| I | m164mRNA_rev | m164mRNAout50_for | 51.7 °C | 2 min10 s; 68 °C | 2,081 |
| II | m164mRNA_rev | m164mRNAout100_for | 51.7 °C | 2 min10 s; 68 °C | 2,123 |
| III | m164mRNA_rev | m164mRNAout300_for | 51.7 °C | 2 min 18 s; 68 °C | 2,299 |
| IV | m164mRNA_rev | m164mRNAout1000_for | 51.7 °C | 3 min; 68 °C | 3,048 |
| Figure A2C | |||||
| I | m164mRNAin_rev | m166_ORF_end_for | 55 °C | 1 min 30 s; 72 °C | 1,744 |
| II | m164mRNAin_rev | m166_ORF_end250_for | 55 °C | 1 min 30 s; 72 °C | 1,986 |
| III | m164mRNAin_rev | m166_ORF_end500_for | 55 °C | 2 min 18 s; 68 °C | 2,254 |
| IV | m164mRNAin_rev | m166_ORF_end1000_for | 55 °C | 3 min; 68 °C | 2,732 |
| V | m164mRNAin_rev | m166.5_ORF_end_for | 55 °C | 3 min; 68 °C | 3,086 |
| VI | m164mRNAin_rev | m166.5_ORF_end250_for | 55 °C | 3 min 20 s; 68 °C | 3,346 |
| VII | m164mRNAin_rev | m166.5_ORF_end500_for | 55 °C | 3 min 30 s; 68 °C | 3,576 |
| VIII | m164mRNAin_rev | m166.5_ORF_end1000_for | 55 °C | 4 min; 68 °C | 4,081 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fink, A.; Büttner, J.K.; Thomas, D.; Holtappels, R.; Reddehase, M.J.; Lemmermann, N.A.W. Noncanonical Expression of a Murine Cytomegalovirus Early Protein CD8 T-Cell Epitope as an Immediate Early Epitope Based on Transcription from an Upstream Gene. Viruses 2014, 6, 808-831. https://doi.org/10.3390/v6020808
Fink A, Büttner JK, Thomas D, Holtappels R, Reddehase MJ, Lemmermann NAW. Noncanonical Expression of a Murine Cytomegalovirus Early Protein CD8 T-Cell Epitope as an Immediate Early Epitope Based on Transcription from an Upstream Gene. Viruses. 2014; 6(2):808-831. https://doi.org/10.3390/v6020808
Chicago/Turabian StyleFink, Annette, Julia K. Büttner, Doris Thomas, Rafaela Holtappels, Matthias J. Reddehase, and Niels A. W. Lemmermann. 2014. "Noncanonical Expression of a Murine Cytomegalovirus Early Protein CD8 T-Cell Epitope as an Immediate Early Epitope Based on Transcription from an Upstream Gene" Viruses 6, no. 2: 808-831. https://doi.org/10.3390/v6020808