Respiratory Syncytial Virus Persistence in Macrophages Upregulates Fcgamma Receptors Expression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
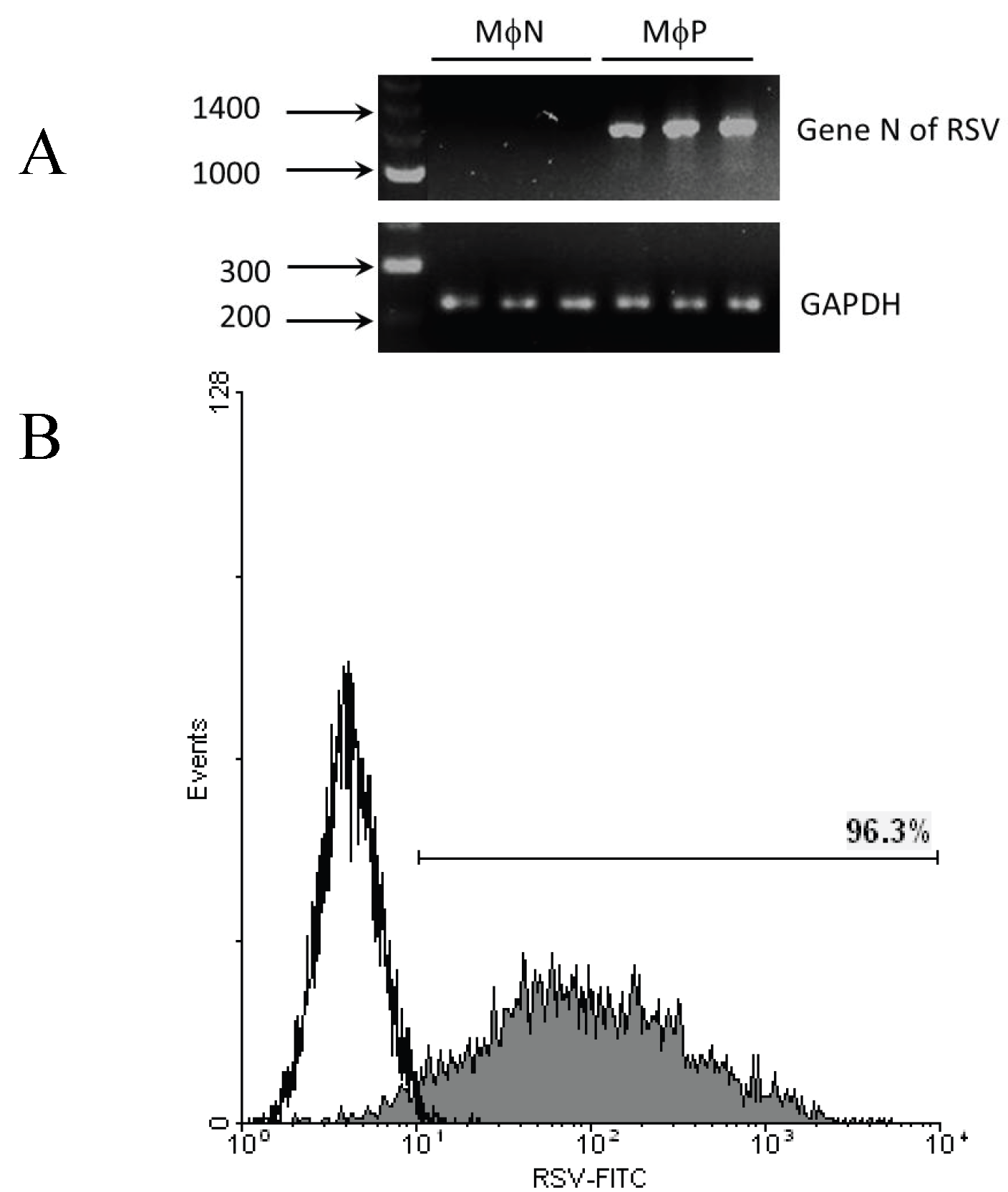
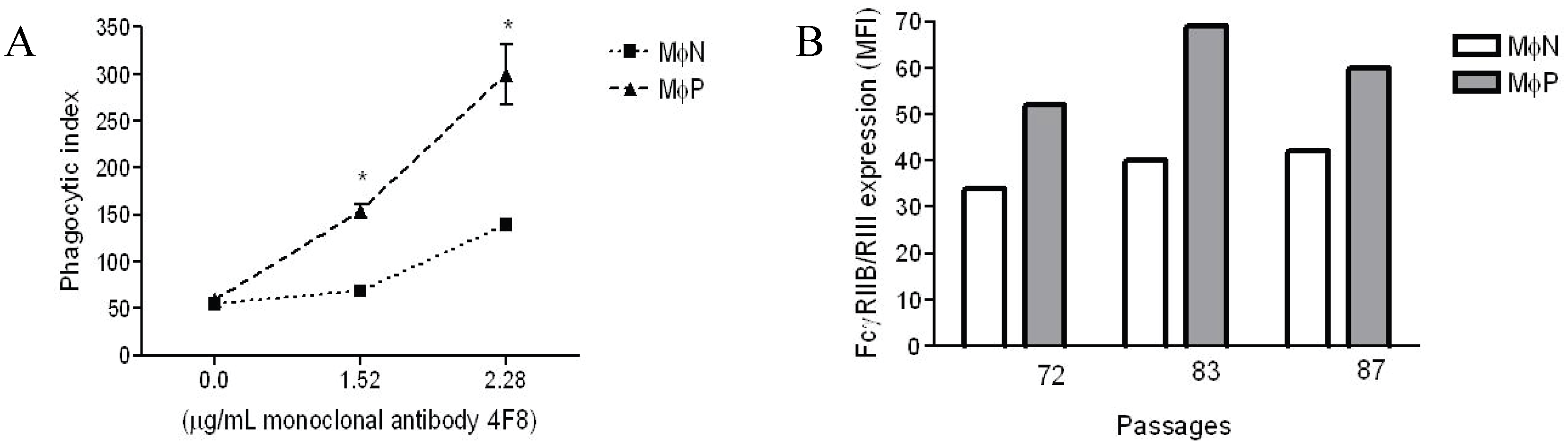
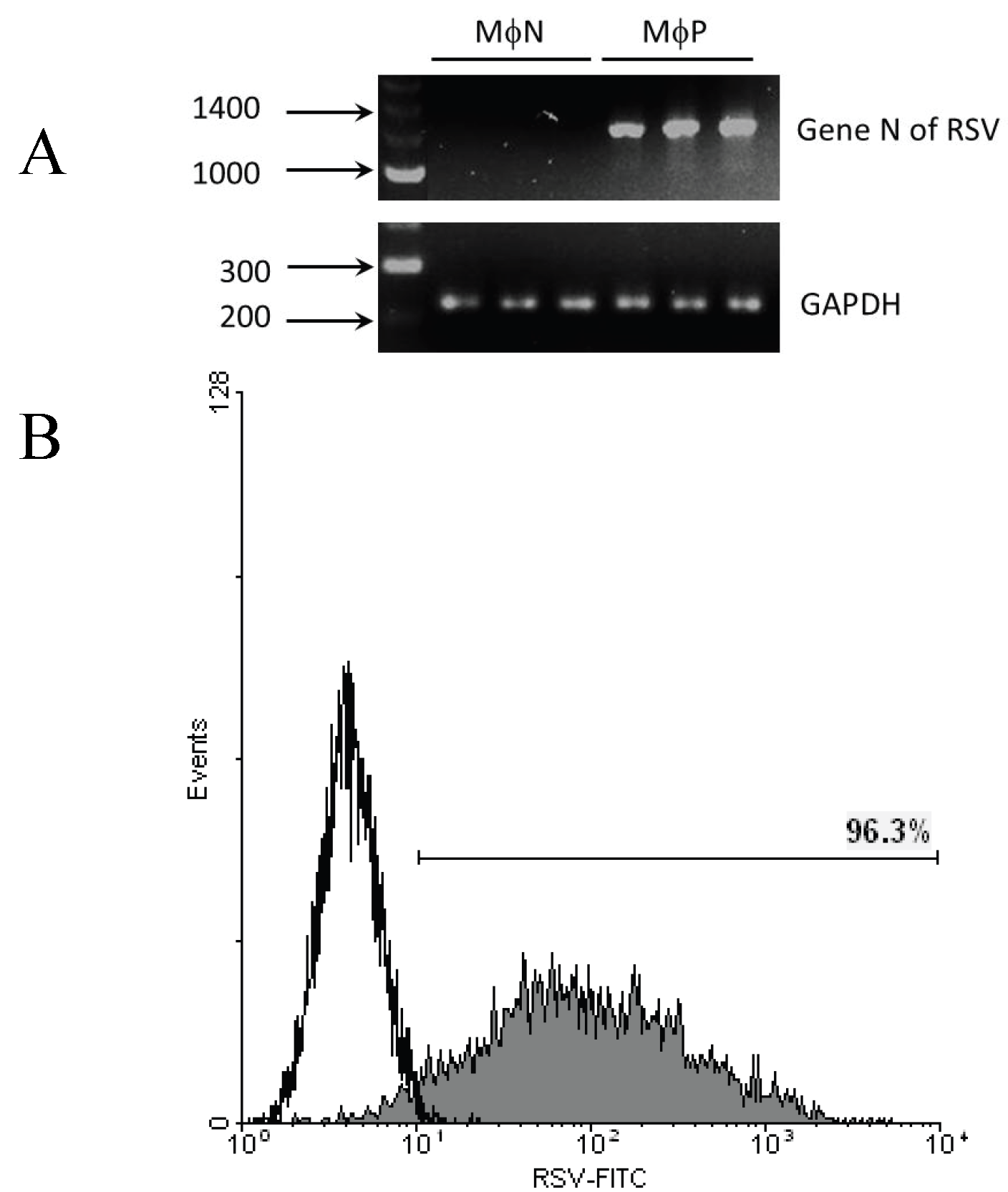
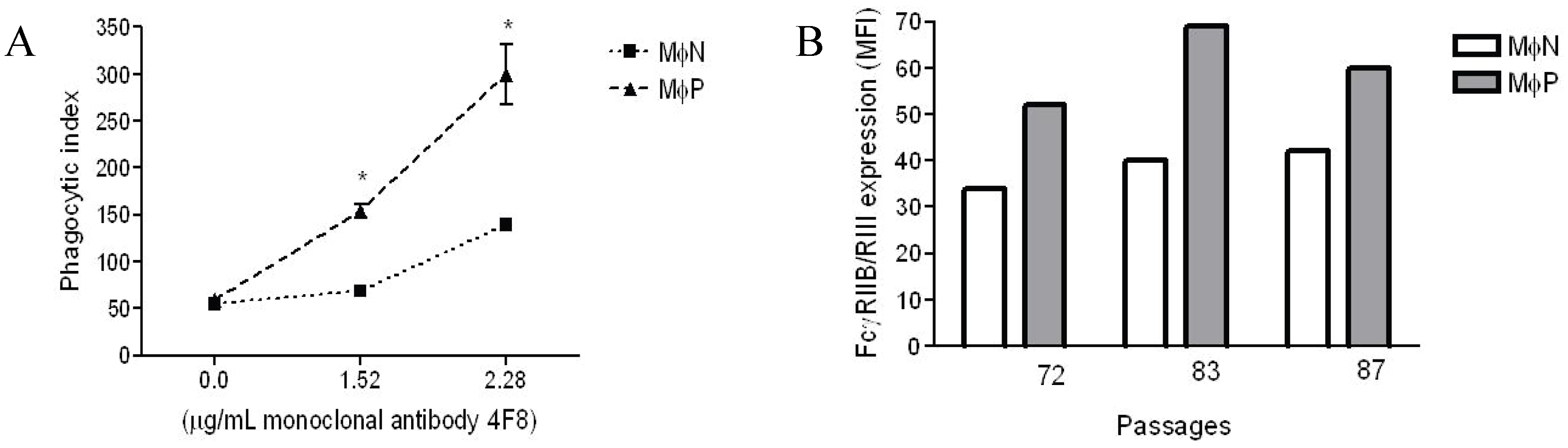
2.1.1. Persistent Infection by RSV Increases FcγR-mediated Phagocytosis and FcγRIIB/RIII Expression even at High Passage Numbers
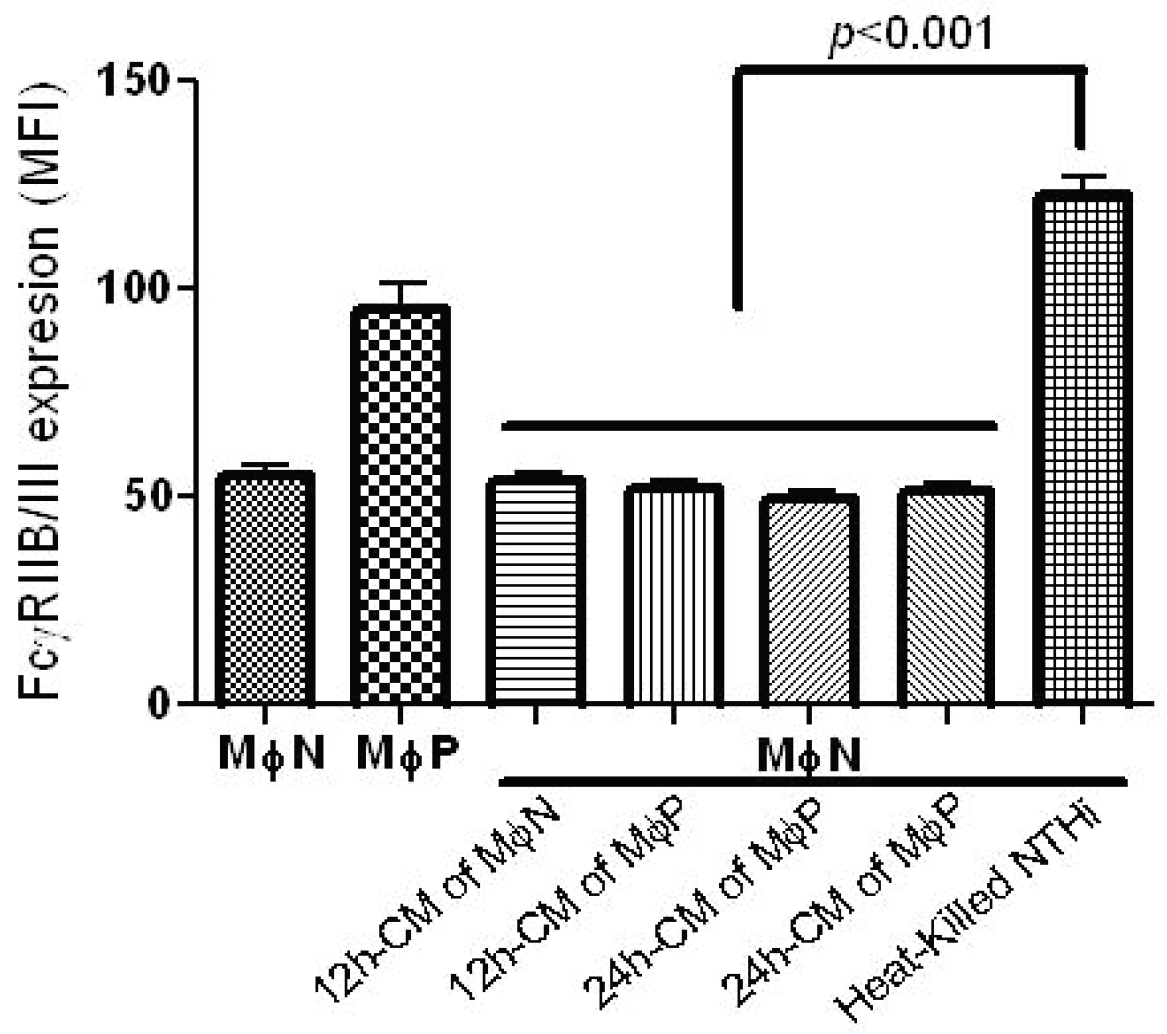
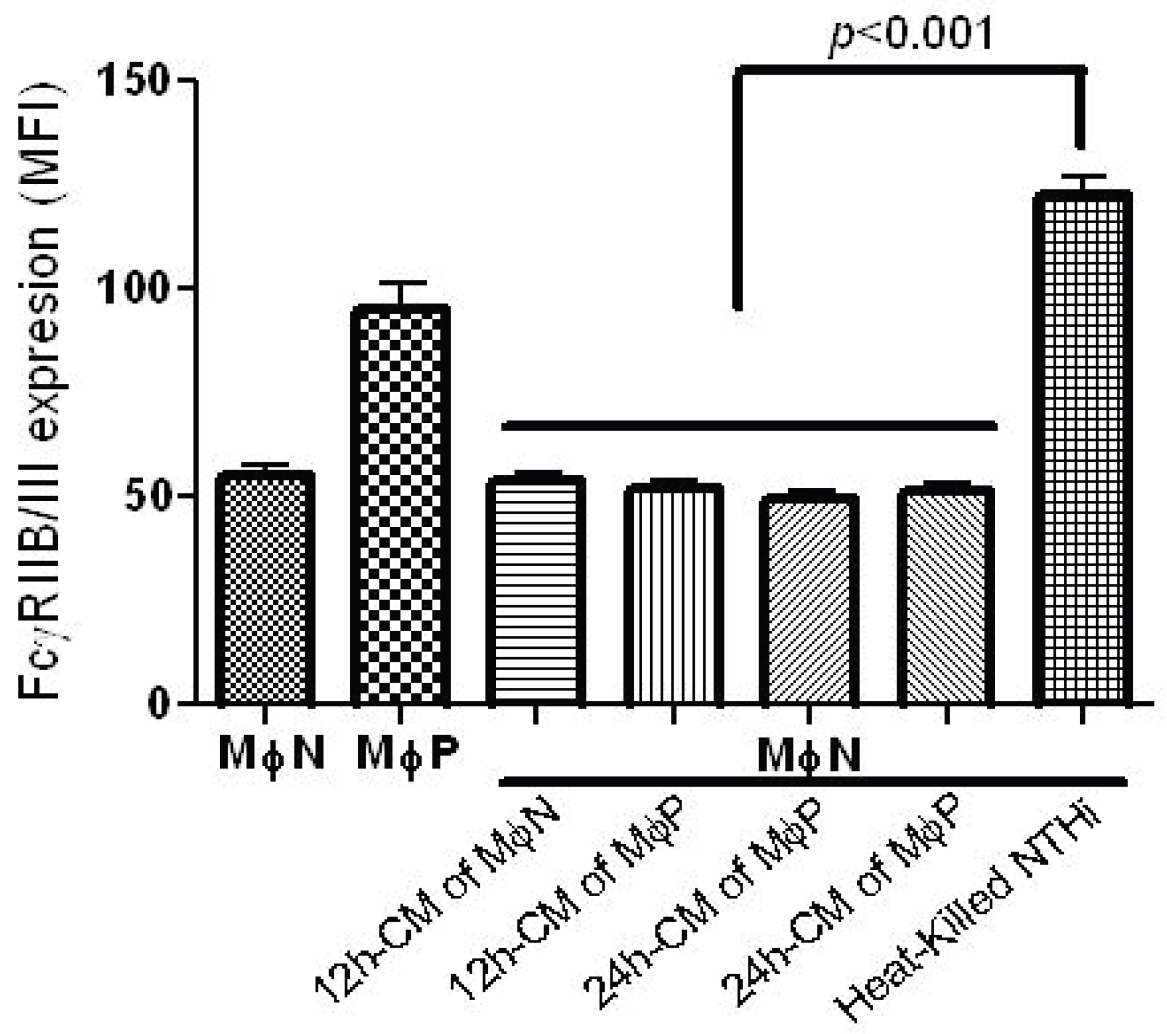
2.1.2. Increase in FcγRIIB/RIII Cell Membrane Expression Is Not Mediated by Soluble Factors
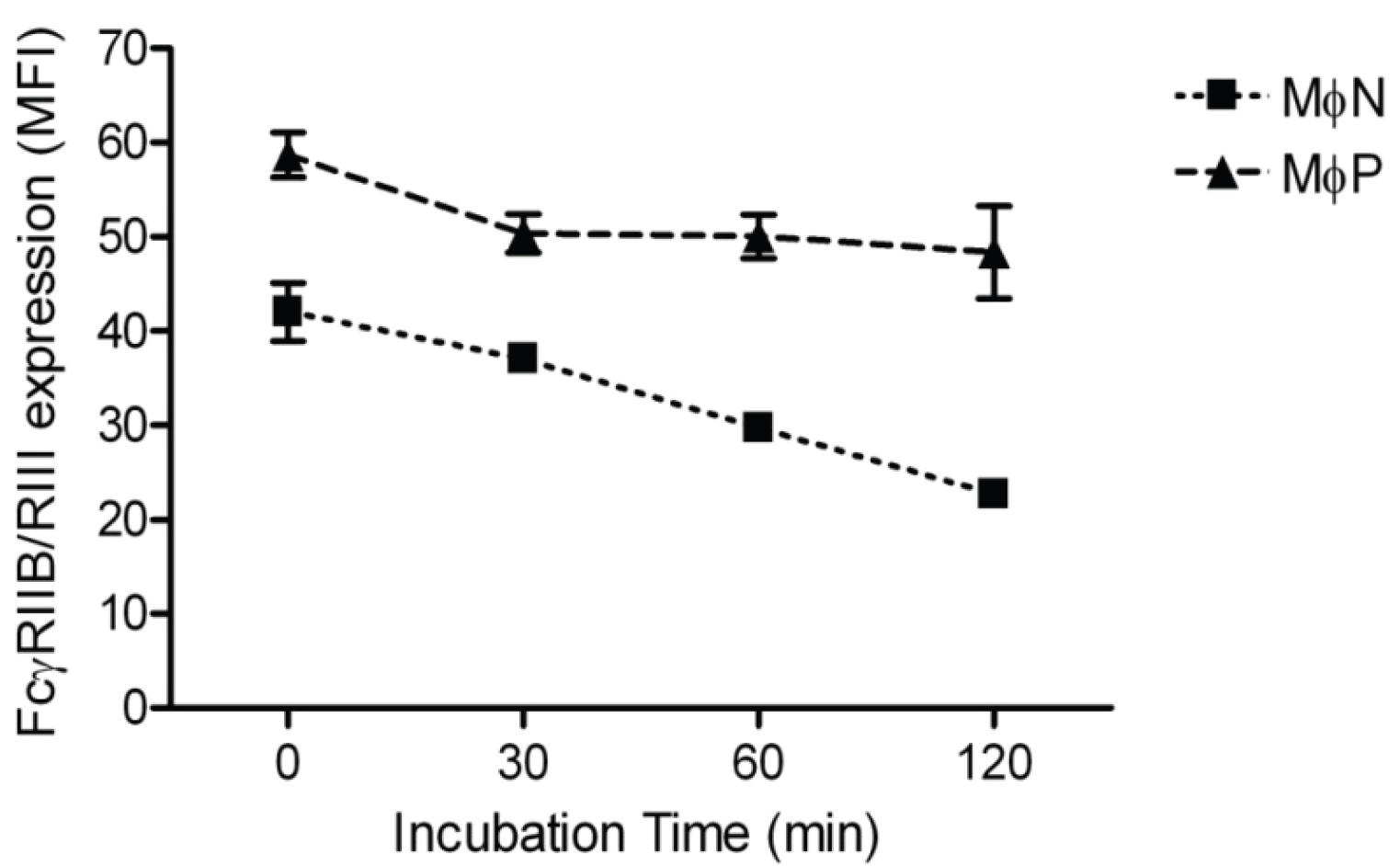
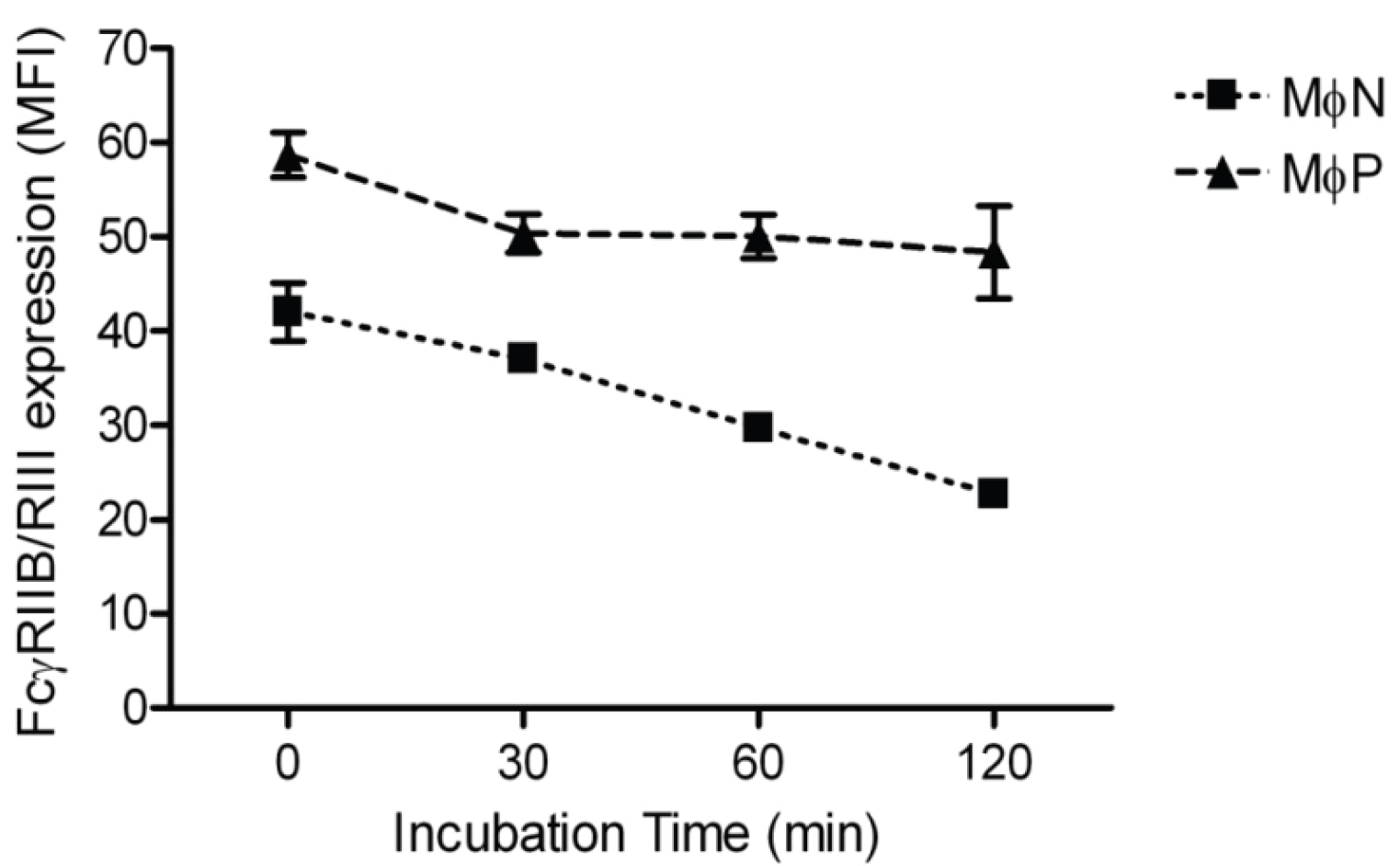
2.1.3. RSV Persistence does not Affect FcγRIIB/RIII Endocytosis
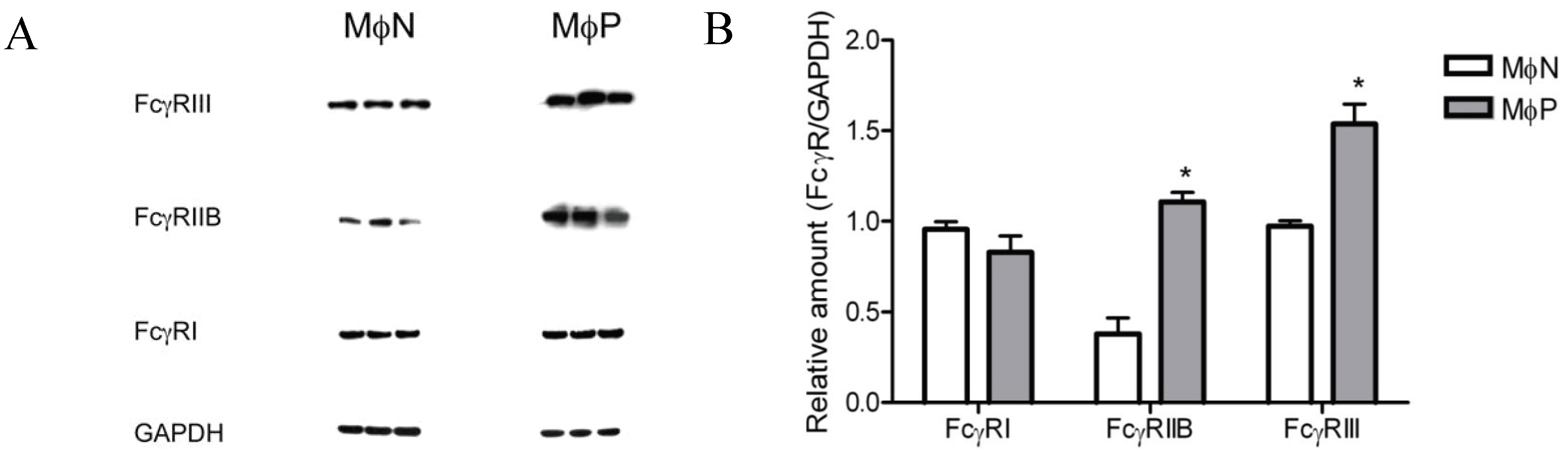
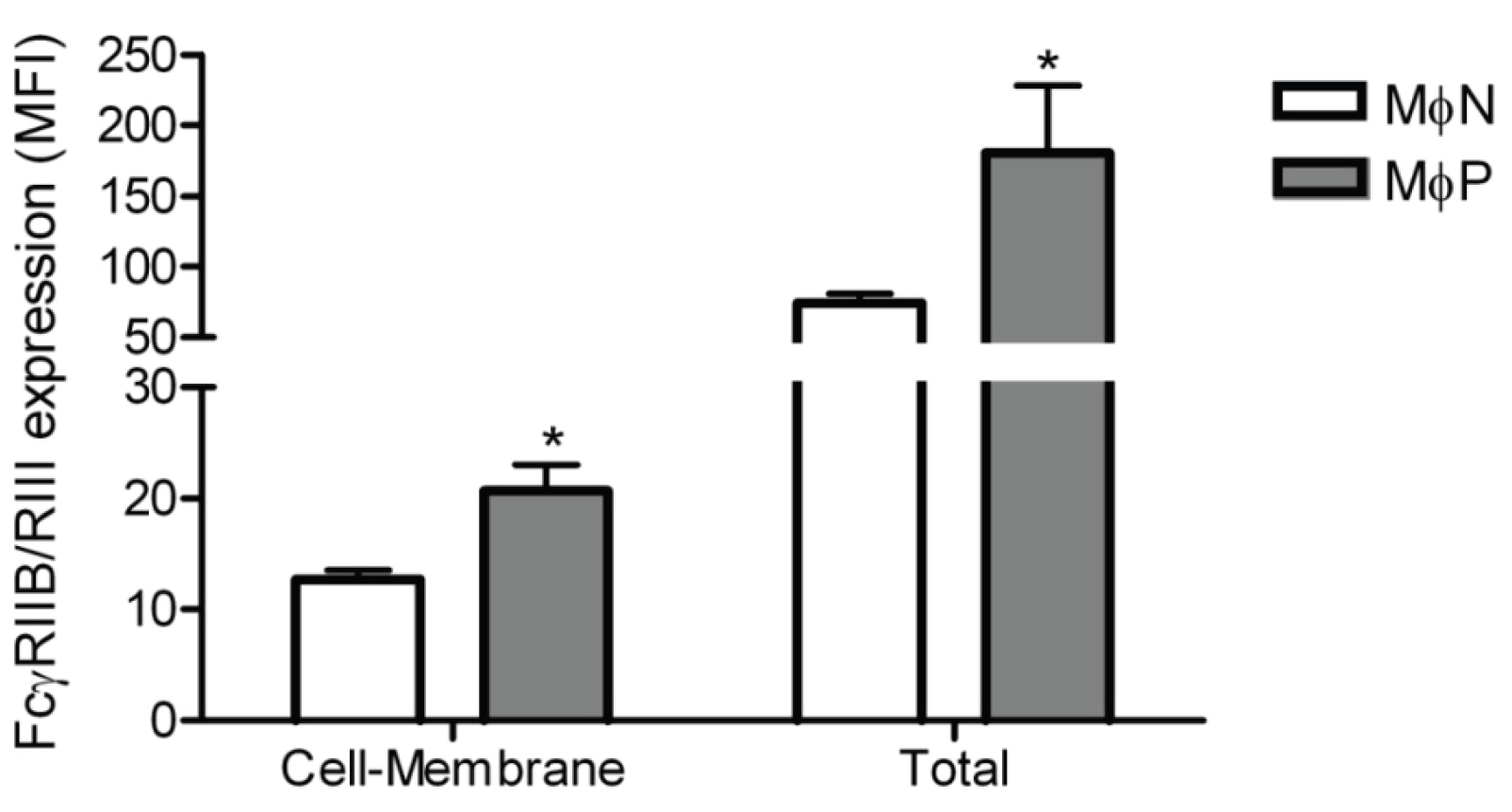
2.1.4. Intracellular Levels of FcγRIIB/RIII Proteins Are Increased in MɸP
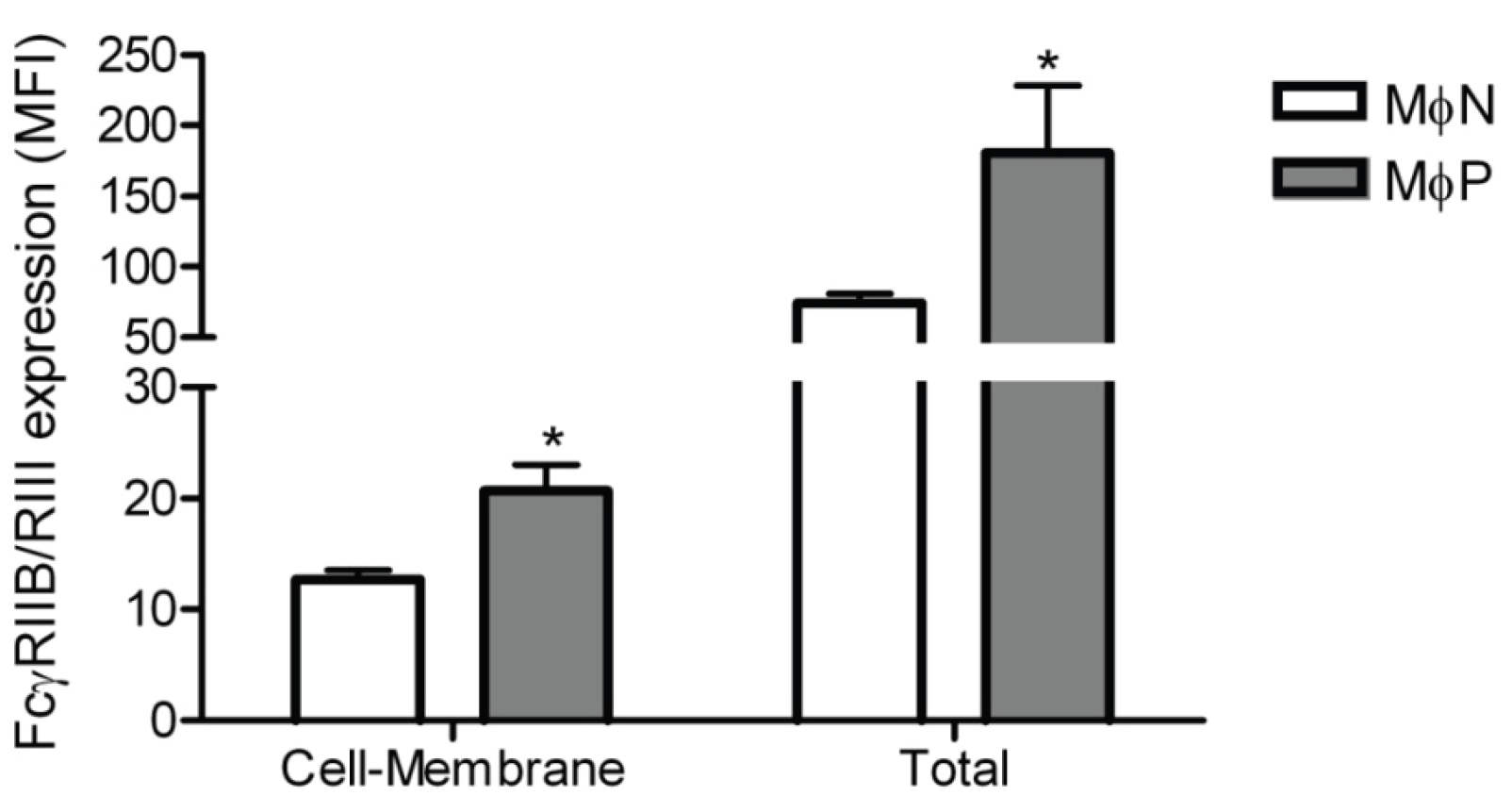
2.1.5. Membrane Expression of FcγRIIB and FcγRIII, but Not of FcγRI, Is Increased in RSV Persistently Infected Cells
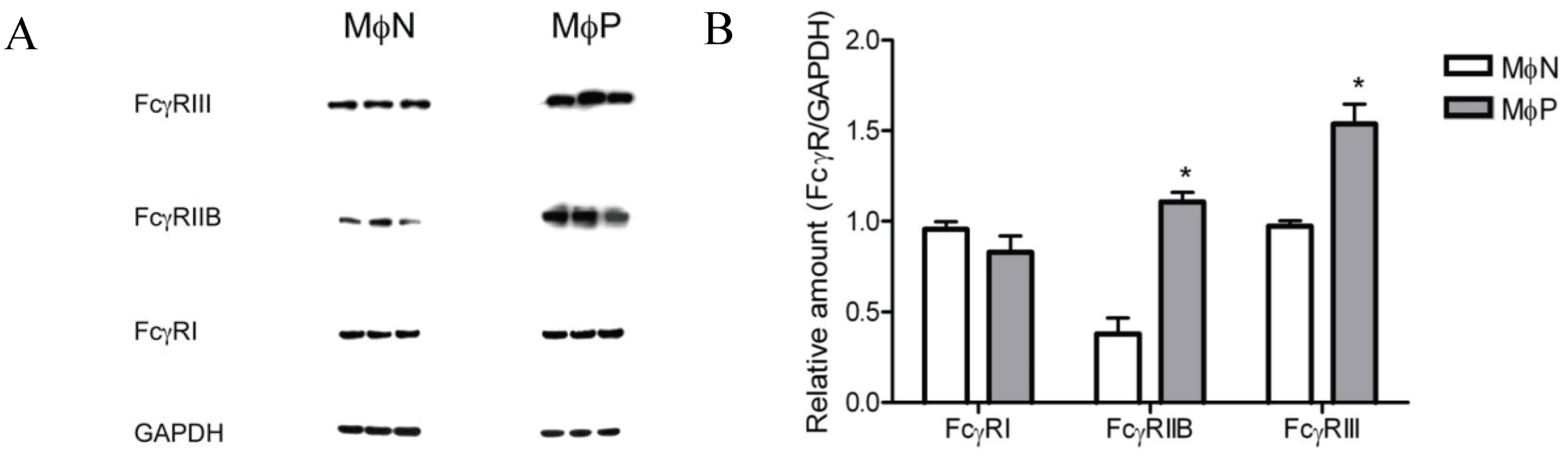
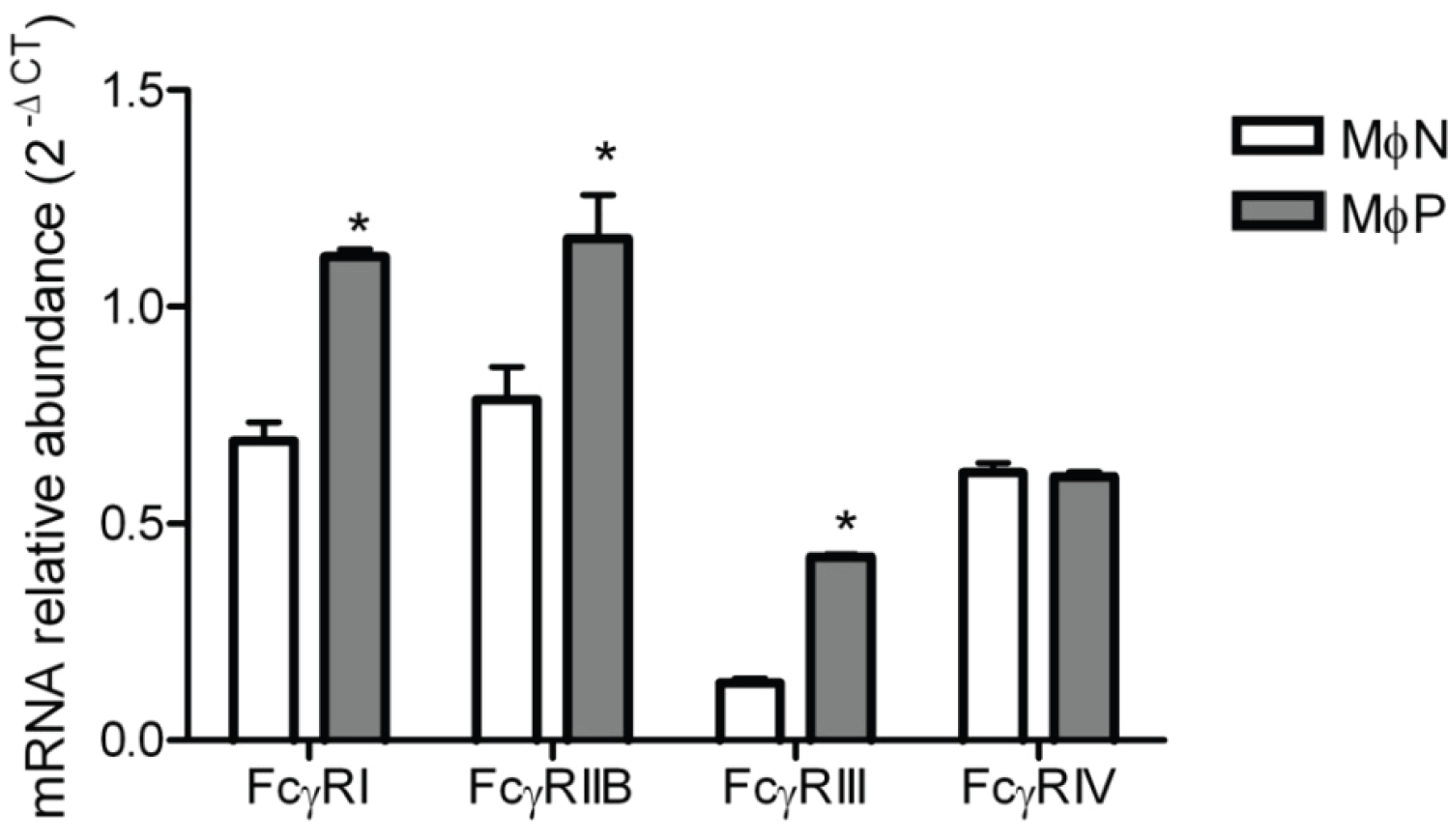
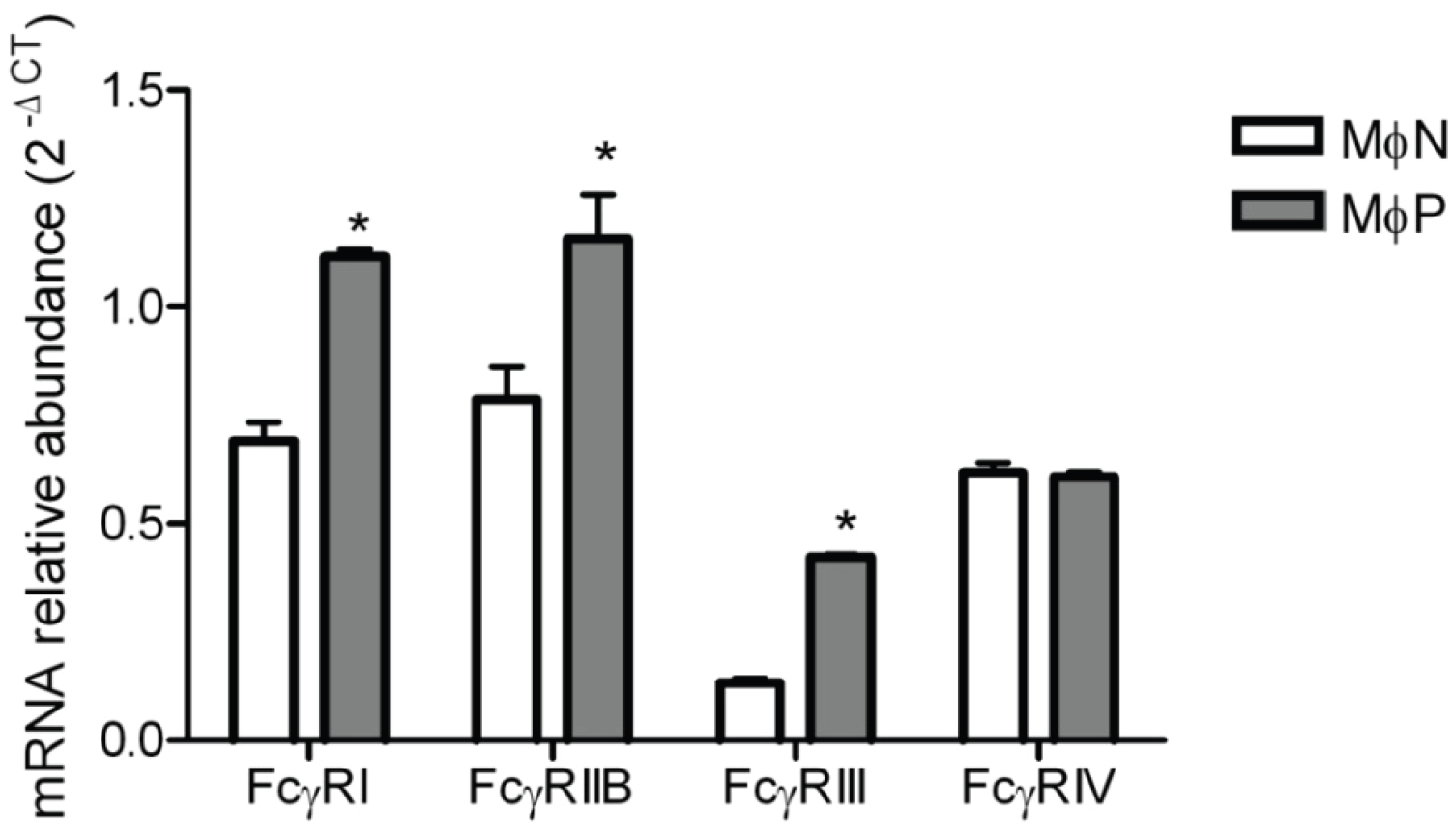
2.1.6. mRNA Expression of FcγRs Is Distinctively Affected by Viral Persistence
2.2. Discussion
3. Experimental Section
3.1. Virus and Cell Lines
3.2. Phagocytosis Assays
3.3. Flow Cytometry
3.4. Conditioned Medium
3.5. Endocytosis Assay
3.6. Western Blot
3.7. Real Time RT-PCR
3.8. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Oldstone, M.B. Anatomy of viral persistence. PLoS Pathog. 2009, 5, e1000523. [Google Scholar] [CrossRef]
- Anderson, C.F.; Gerber, J.S.; Mosser, D.M. Modulating macrophage function with IgG immune complexes. J. Endotoxin Res. 2002, 8, 477–481. [Google Scholar] [CrossRef]
- Biswas, S.K.; Chittezhath, M.; Shalova, I.N.; Lim, J.Y. Macrophage polarization and plasticity in health and disease. Immunol. Res. 2012, 53, 11–24. [Google Scholar] [CrossRef]
- Leeansyah, E.; Wines, B.D.; Crowe, S.M.; Jaworowski, A. The mechanism underlying defective Fcgamma receptor-mediated phagocytosis by HIV-1-infected human monocyte-derived macrophages. J. Immunol. 2007, 178, 1096–1104. [Google Scholar]
- Kedzierska, K.; Azzam, R.; Ellery, P.; Mak, J.; Jaworowski, A.; Crowe, S.M. Defective phagocytosis by human monocyte/macrophages following HIV-1 infection: Underlying mechanisms and modulation by adjunctive cytokine therapy. J. Clin. Virol. 2003, 26, 247–263. [Google Scholar] [CrossRef]
- Martinez, I.; Lombardia, L.; Herranz, C.; Garcia-Barreno, B.; Dominguez, O.; Melero, J.A. Cultures of HEp-2 cells persistently infected by human respiratory syncytial virus differ in chemokine expression and resistance to apoptosis as compared to lytic infections of the same cell type. Virology 2009, 388, 31–41. [Google Scholar] [CrossRef]
- Sarmiento, R.E.; Tirado, R.; Gomez, B. Characteristics of a respiratory syncytial virus persistently infected macrophage-like culture. Virus Res. 2002, 84, 45–58. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Ortega, E.; Gomez, B. Persistence of respiratory syncytial virus in macrophages alters phagocytosis and pro-inflammatory cytokine production. Viral Immunol. 2001, 14, 19–30. [Google Scholar] [CrossRef]
- Ravetch, J.V.; Bolland, S. IgG Fc receptors. Annu. Rev. Immunol. 2001, 19, 275–290. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 2005, 310, 1510–1512. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Analyzing antibody-Fc-receptor interactions. Methods Mol. Biol. 2008, 415, 151–162. [Google Scholar]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Antibody-mediated modulation of immune responses. Immunol. Rev. 2010, 236, 265–275. [Google Scholar] [CrossRef]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef]
- Bont, L.; Houben, M.L. The epidemiology of respiratory syncytial virus lower respiratory tract infection on young children. Pediatr. Infect. Dis. J. 2012, 30, 778–784. [Google Scholar]
- Van Drunen Littel-van den Hurk, S.; Watkiss, E.R. Pathogenesis of respiratory syncytial virus. Curr. Opin. Virol. 2012, 2, 300–305. [Google Scholar] [CrossRef]
- Habibi, M.S.; Openshaw, P.J. Benefit and harm from immunity to respiratory syncytial virus: Implications for treatment. Curr. Opin. Infect. Dis. 2012, 25, 687–694. [Google Scholar] [CrossRef]
- Walsh, E.E. Respiratory syncytial virus infection in adults. Semin. Respir. Crit. Care Med. 2011, 32, 423–432. [Google Scholar] [CrossRef]
- Welliver, R.C. Review of epidemiology and clinical risk factors for severe respiratory syncytial virus (RSV) infection. J. Pediatr. 2003, 143, S112–S117. [Google Scholar] [CrossRef]
- Zeng, R.; Li, C.; Li, N.; Wei, L.; Cui, Y. The role of cytokines and chemokines in severe respiratory syncytial virus infection and subsequent asthma. Cytokine 2011, 53, 1–7. [Google Scholar] [CrossRef]
- Hall, C.B.; Douglas, R.G.; Geiman, J.M. Respiratory syncytial virus infections in infants: Quantitation and duration of shedding. J. Pediatr. 1976, 89, 11–15. [Google Scholar] [CrossRef]
- Szabo, S.M.; Levy, A.R.; Gooch, K.L.; Bradt, P.; Wijaya, H.; Mitchell, I. Elevated risk of asthma after hospitalization for respiratory syncytial virus infection in infancy. Paediatr. Respir. Rev. 2013, 13, S9–S15. [Google Scholar] [CrossRef]
- Piedimonte, G. Respiratory syncytial virus and asthma: Speed-dating or long-term relationship? Curr. Opin. Pediatr. 2013, 25, 344–349. [Google Scholar] [CrossRef]
- Openshaw, P.J.; Dean, G.S.; Culley, F.J. Links between respiratory syncytial virus bronchiolitis and childhood asthma: Clinical and research approaches. Pediatr. Infect. Dis. J. 2003, 22, S58–S64. [Google Scholar]
- Sikkel, M.B.; Quint, J.K.; Mallia, P.; Wedzicha, J.A.; Johnston, S.L. Respiratory syncytial virus persistence in chronic obstructive pulmonary disease. Pediatr. Infect. Dis. J. 2008, 27, S63–S70. [Google Scholar] [CrossRef]
- Tripp, R.A. The brume surrounding respiratory syncytial virus persistence. Am. J. Respir. Crit. Care Med. 2004, 169, 778–779. [Google Scholar] [CrossRef]
- McNamara, P.S.; Smyth, R.L. The pathogenesis of respiratory syncytial virus disease in childhood. Br. Med. Bull. 2002, 61, 13–28. [Google Scholar] [CrossRef]
- Mejias, A.; Chavez-Bueno, S.; Gomez, A.M.; Somers, C.; Estripeaut, D.; Torres, J.P.; Jafri, H.S.; Ramilo, O. Respiratory syncytial virus persistence: Evidence in the mouse model. Pediatr. infect. Dis. J. 2008, 27, S60–S62. [Google Scholar] [CrossRef]
- Sutton, T.C.; Tayyari, F.; Khan, M.A.; Manson, H.E.; Hegele, R.G. T helper 1 background protects against airway hyperresponsiveness and inflammation in guinea pigs with persistent respiratory syncytial virus infection. Pediatr. Res. 2007, 61, 525–529. [Google Scholar] [CrossRef]
- Schwarze, J.; O’Donnell, D.R.; Rohwedder, A.; Openshaw, P.J. Latency and persistence of respiratory syncytial virus despite T cell immunity. Am. J. Respir. Crit. Care Med. 2004, 169, 801–805. [Google Scholar] [CrossRef]
- Bramley, A.M.; Vitalis, T.Z.; Wiggs, B.R.; Hegele, R.G. Effects of respiratory syncytial virus persistence on airway responsiveness and inflammation in guinea-pigs. Eur. Respir. J. 1999, 14, 1061–1067. [Google Scholar] [CrossRef]
- Hegele, R.G.; Hayashi, S.; Bramley, A.M.; Hogg, J.C. Persistence of respiratory syncytial virus genome and protein after acute bronchiolitis in guinea pigs. Chest 1994, 105, 1848–1854. [Google Scholar] [CrossRef]
- Riedel, F.; Oberdieck, B.; Streckert, H.J.; Philippou, S.; Krusat, T.; Marek, W. Persistence of airway hyperresponsiveness and viral antigen following respiratory syncytial virus bronchiolitis in young guinea-pigs. Eur. Respir. J. 1997, 10, 639–645. [Google Scholar]
- Valarcher, J.F.; Bourhy, H.; Lavenu, A.; Bourges-Abella, N.; Roth, M.; Andreoletti, O.; Ave, P.; Schelcher, F. Persistent infection of b lymphocytes by bovine respiratory syncytial virus. Virology 2001, 291, 55–67. [Google Scholar] [CrossRef]
- Mills, B.G.; Frausto, A.; Singer, F.R.; Ohsaki, Y.; Demulder, A.; Roodman, G.D. Multinucleated cells formed in vitro from paget’s bone marrow express viral antigens. Bone 1994, 15, 443–448. [Google Scholar] [CrossRef]
- Cubie, H.A.; Duncan, L.A.; Marshall, L.A.; Smith, N.M. Detection of respiratory syncytial virus nucleic acid in archival postmortem tissue from infants. Pediatr. Pathol. Lab. Med. 1997, 17, 927–938. [Google Scholar]
- Mohapatra, S.S.; Boyapalle, S. Epidemiologic, experimental, and clinical links between respiratory syncytial virus infection and asthma. Clin. Microbiol. Rev. 2008, 21, 495–504. [Google Scholar] [CrossRef]
- Herra, C.M.; Keane, C.T.; Whelan, A. Increased expression of Fc gamma receptors on neutrophils and monocytes may reflect ongoing bacterial infection. J. Med. Microbiol. 1996, 44, 135–140. [Google Scholar] [CrossRef]
- Unkeless, J.C. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte fc receptors. J. Exp. Med. 1979, 150, 580–596. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors: Old friends and new family members. Immunity 2006, 24, 19–28. [Google Scholar] [CrossRef]
- Arrevillaga, G.; Gaona, J.; Sanchez, C.; Rosales, V.; Gomez, B. Respiratory syncytial virus persistence in macrophages downregulates intercellular adhesion molecule-1 expression and reduces adhesion of non-typeable haemophilus influenzae. Intervirology 2012, 55, 442–450. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Ortega, E.; Ortiz-Navarrete, V.; Gomez, B. Antigen presentation by a macrophage-like cell line persistently infected with respiratory syncytial virus. Virus Res. 2004, 99, 95–100. [Google Scholar] [CrossRef]
- Tirado, R.; Gómez, B. National Autonomous University of Mexico (UNAM): Ciudad Universitaria, D.F., México, 2013; Unpublished work.
- Nakamura-Lopez, Y.; Villegas-Sepulveda, N.; Sarmiento-Silva, R.E.; Gomez, B. Intrinsic apoptotic pathway is subverted in mouse macrophages persistently infected by RSV. Virus Res. 2011, 158, 98–107. [Google Scholar] [CrossRef]
- Frausto-Del-Río, D.; Soto-Cruz, I.; Garay-Canales, C.; Ambriz, X.; Soldevila, G.; Carretero-Ortega, J.; Vázquez-Prado, J.; Ortega, E. Interferon gamma induces actin polymerization, rac1 activation and down regulates phagocytosis in human monocytic cells. Cytokine 2012, 57, 158–168. [Google Scholar] [CrossRef]
- Berclaz, P.Y.; Shibata, Y.; Whitsett, J.A.; Trapnell, B.C. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood 2002, 100, 4193–4200. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar]
- Jungi, T.W. A rapid and sensitive method allowing photometric determination of erythrophagocytosis by mononuclear phagocytes. J. Immunol. Methods 1985, 82, 141–153. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gaona, J.; Santiago-Olivares, C.; Ortega, E.; Gómez, B. Respiratory Syncytial Virus Persistence in Macrophages Upregulates Fcgamma Receptors Expression. Viruses 2014, 6, 624-639. https://doi.org/10.3390/v6020624
Gaona J, Santiago-Olivares C, Ortega E, Gómez B. Respiratory Syncytial Virus Persistence in Macrophages Upregulates Fcgamma Receptors Expression. Viruses. 2014; 6(2):624-639. https://doi.org/10.3390/v6020624
Chicago/Turabian StyleGaona, Jorge, Carlos Santiago-Olivares, Enrique Ortega, and Beatriz Gómez. 2014. "Respiratory Syncytial Virus Persistence in Macrophages Upregulates Fcgamma Receptors Expression" Viruses 6, no. 2: 624-639. https://doi.org/10.3390/v6020624