Noncoding Subgenomic Flavivirus RNA: Multiple Functions in West Nile Virus Pathogenesis and Modulation of Host Responses



Abstract
:1. Conservation of sfRNA in the Genus Flavivirus
1.1. Genus Flavivirus
1.2. The 3′UTR of the Family Flaviviridae
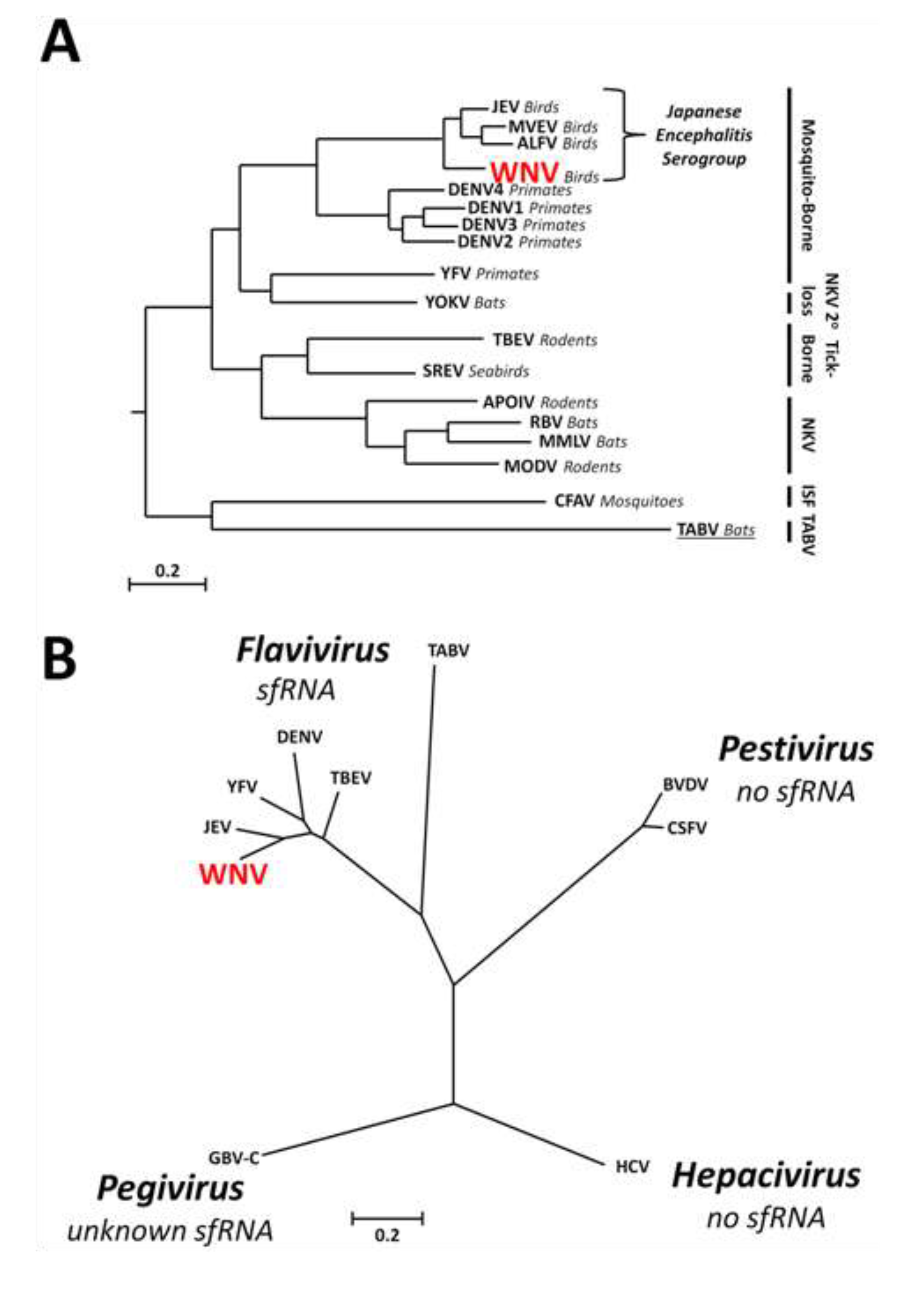
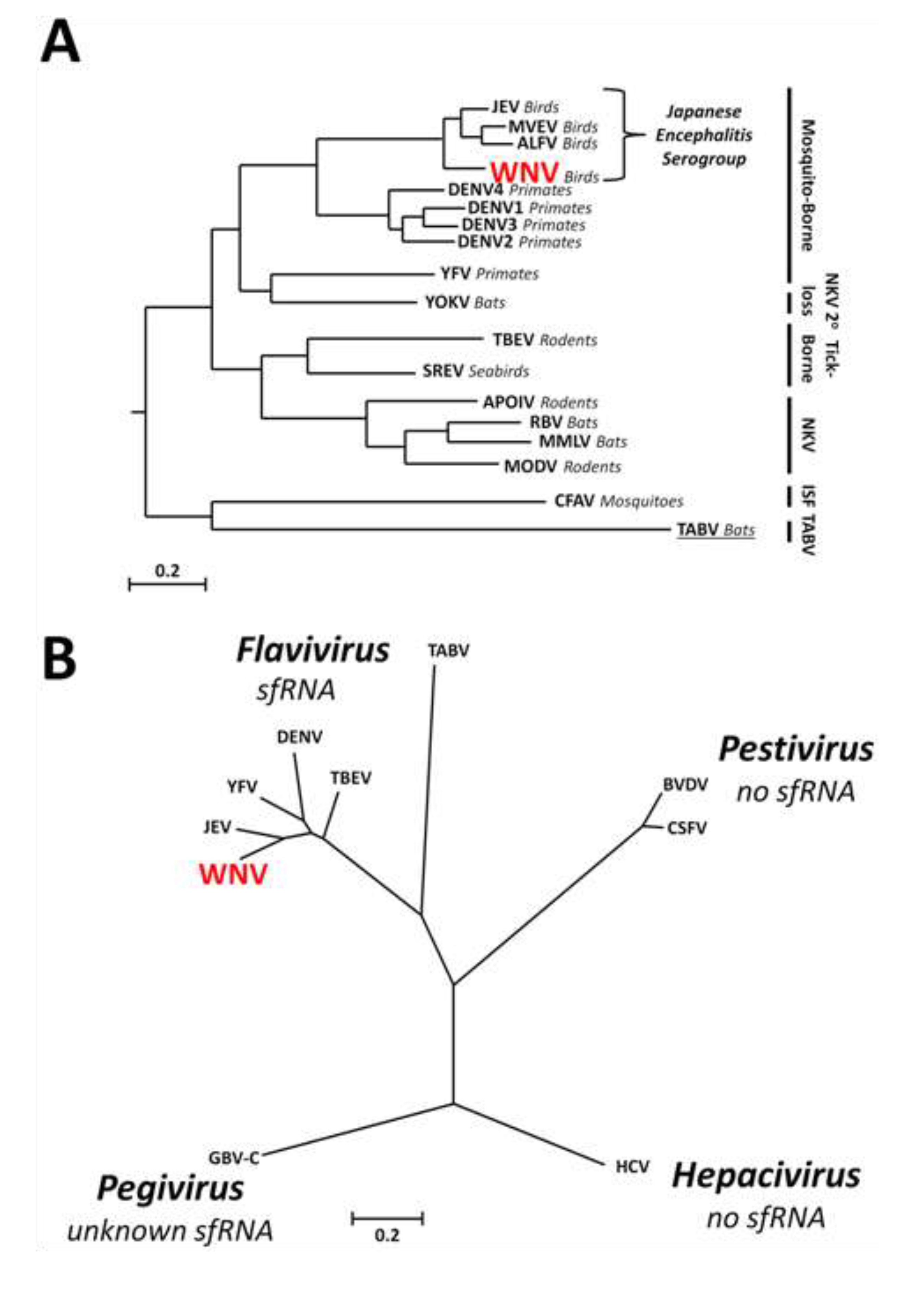
1.3. Discovery of sfRNA
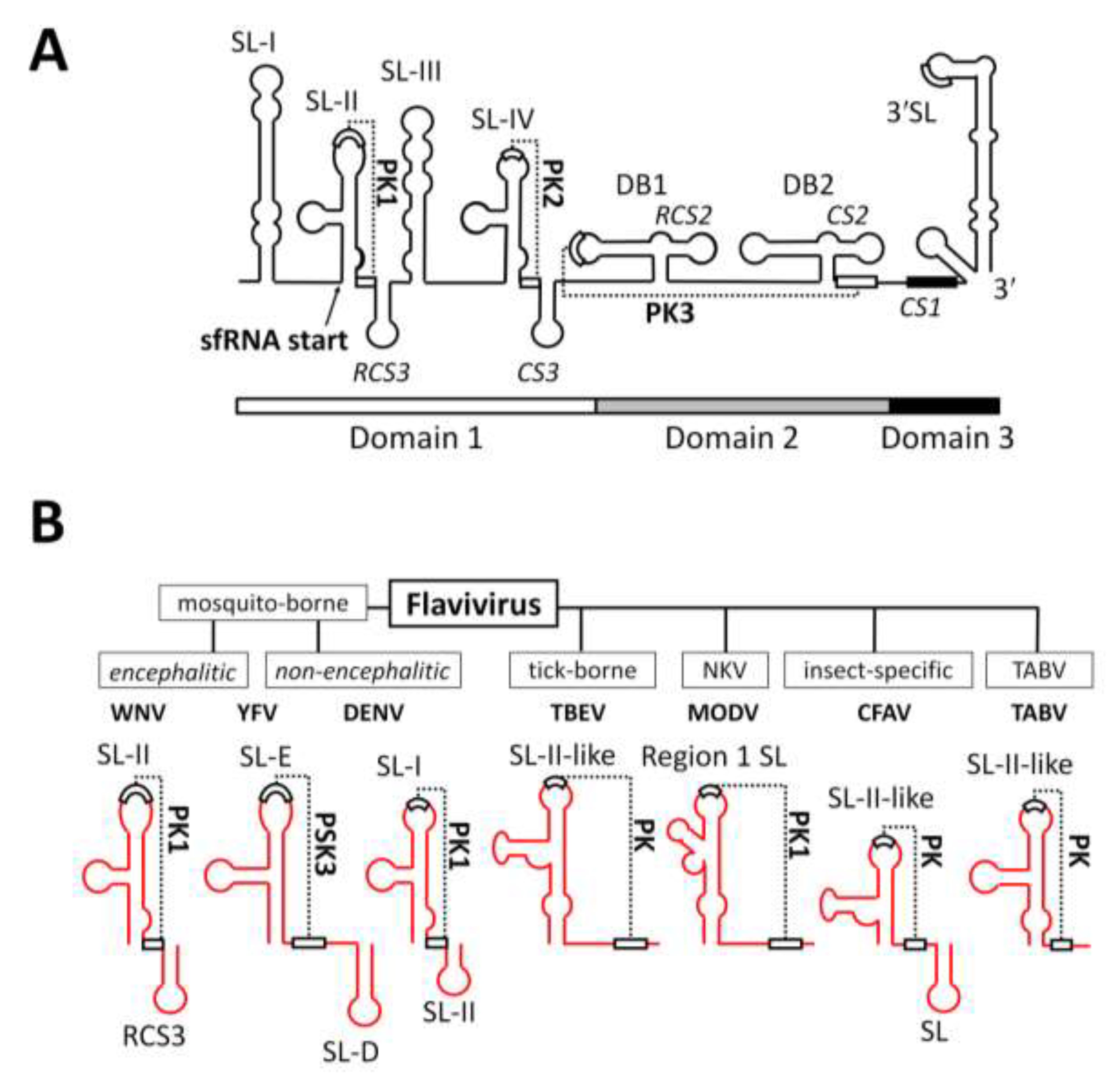
1.4. Conservation of sfRNA between Different Groups within the Genus Flavivirus
2. Mechanism of sfRNA Generation
2.1. Exoribonuclease Stalling
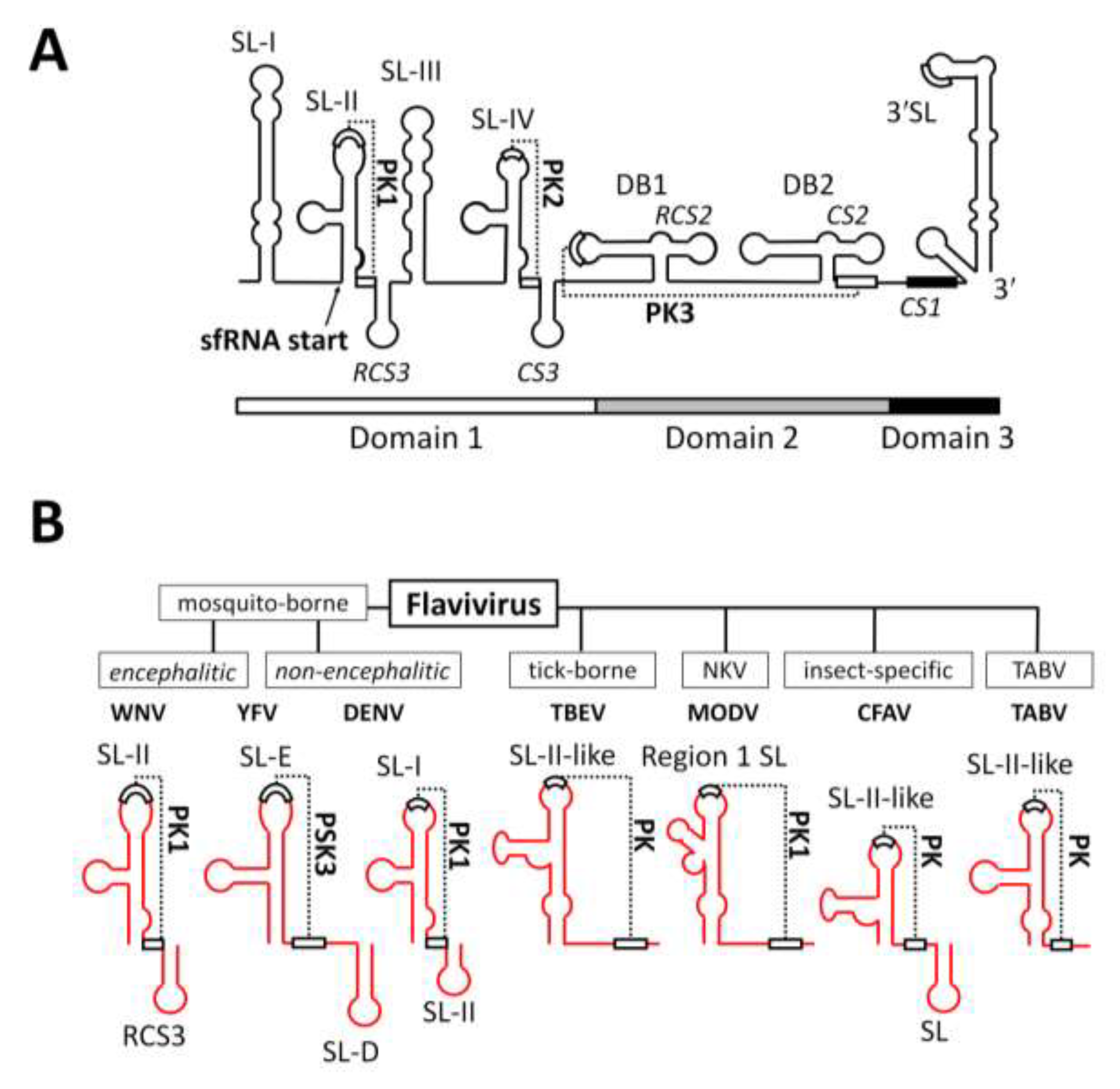
2.2. RNA Structures Required for sfRNA Production
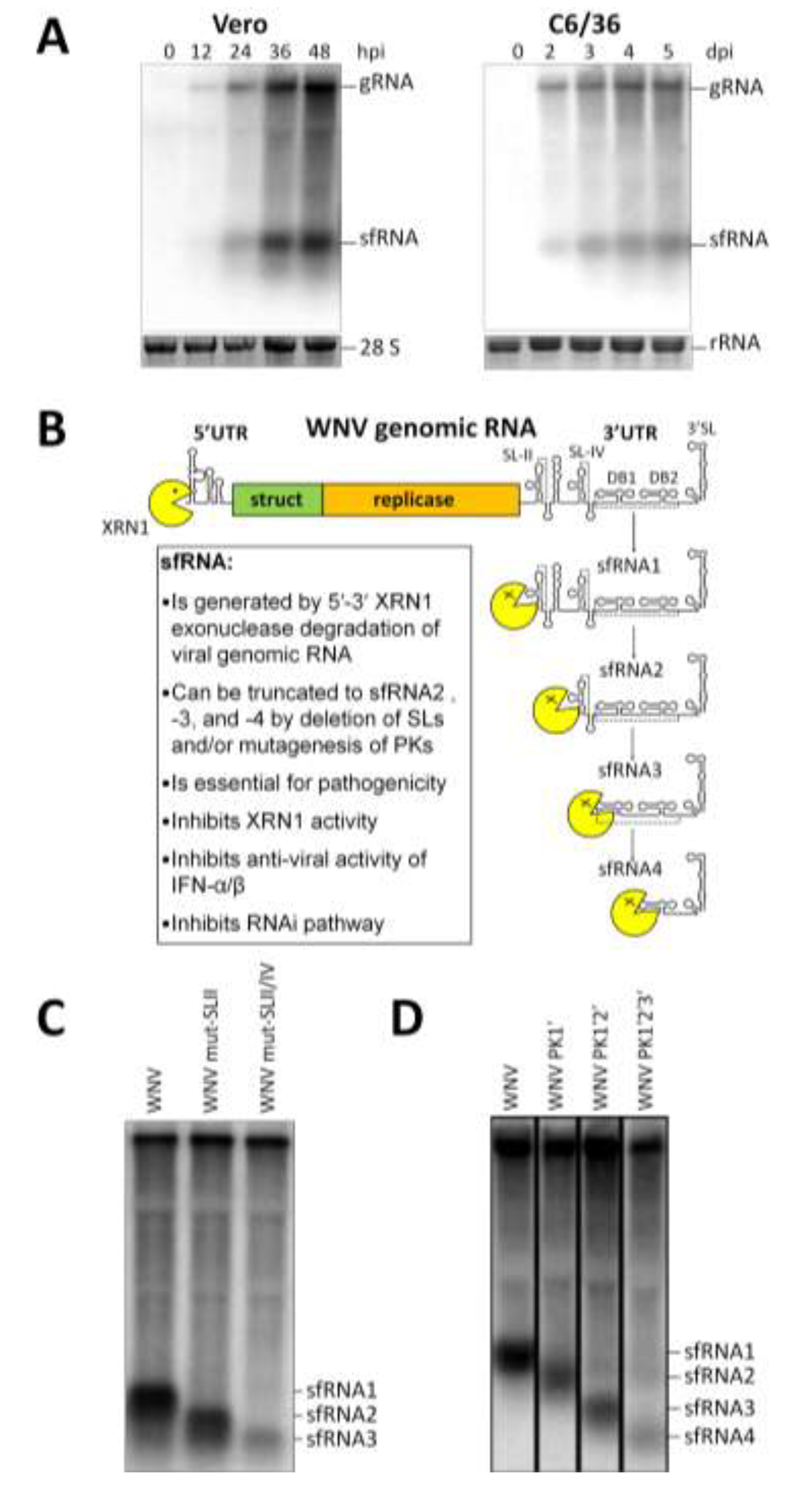
2.3. Endogenous Production of Truncated sfRNAs
3. Cellular Localisation of sfRNA
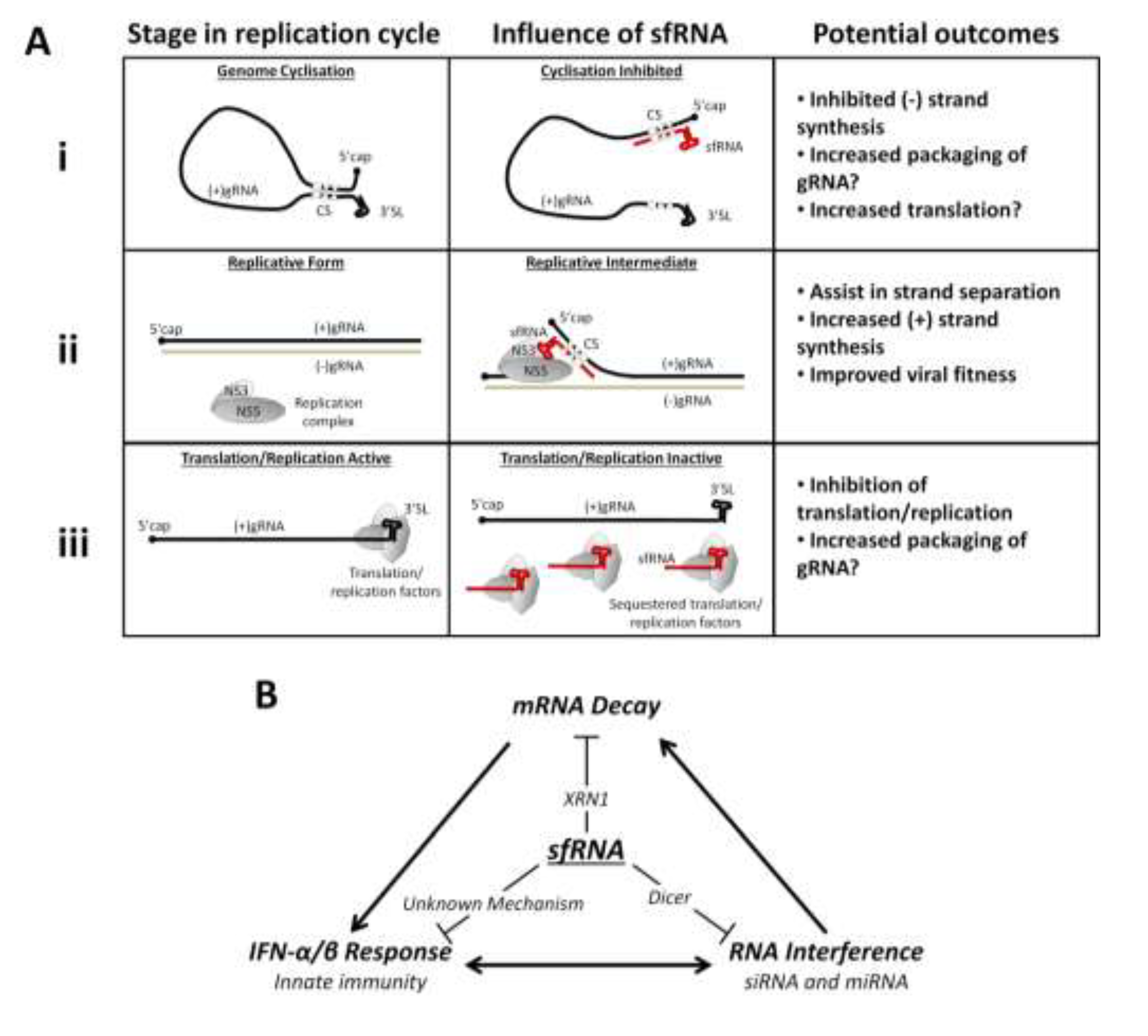
4. Flavivirus Replication and sfRNA
5. MicroRNA Production from sfRNA
6. Host Response and sfRNA
6.1. Cytopathicity in Cells and Pathogenicity in Mice are Dependent on sfRNA
6.2. The Interferon Response and sfRNA
6.3. Inhibition of Host mRNA Turnover Mediated by sfRNA
6.4. The RNAi Pathways and sfRNA
7. Host Binding Partners of the 3'UTR and/or sfRNA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Origin | Function | Binds 3'UTR? | Binds sfRNA? | Method of Identification | Ref. |
|---|---|---|---|---|---|---|
| NS5 | Virus | Polymerase 5' RNA cap | Yes, 3'SL | Likely | Infected cells Pull-down IVT 1 RNA | [71,79] |
| Capsid | Virus | Nucleocapsid | Yes | NK 3, Likely | Pull-down IVT RNA | [80] |
| NS2A | Virus | Viral RC Anti-IFN 2 | Yes, 3'SL | NK, Likely | IVT RNA | [81] |
| NS3 | Virus | Helicase, Protease, NTPase | Yes, 3'SL | NK, Likely | IVT RNA | [79,82] |
| EF1α 4 | Host | Translation elongation factor | Yes, middle of 3'SL | NK, Likely | Infected cells, Pull-down, IVT RNA | [24,25,83] |
| PABP 5 | Host | Translation initiation, SG component | Yes, A-rich regions flanking DBs | NK, Likely | IVT RNA | [23] |
| La autoantigen | Host | RNA chaperone, Protection from RNases | Yes, 3′SL | NK, Likely | Infected cells, Pull-down, IVT RNA | [25,84,85,86,87] |
| PTB 6 | Host | RNA splicing | Yes | NK, Likely | Pull-down, IVT RNA | [25,88] |
| DDX6 7 | Host | PB component, Promote RNA degradation | Yes, DB1 and DB2 | NK, Likely | Infected cells, Pull-down, IVT RNA, Quantitative mass-spec | [89] |
| Caprin1 8 | Host | Transport and translation of mRNAs, SG component | Yes, region SL-I to DB1 | NK, Possibly unless binds SL-I | Pull-down, IVT RNA, Quantitative mass-spec | [89] |
| G3BP1/2 9 | Host | dsDNA or dsRNA unwinding, SG components | Yes, region SL-I to DB1 | NK, Possibly unless binds SL-I | Pull-down, IVT RNA, Quantitative mass-spec | [89] |
| USP10 10 | Host | De-ubiquitination, SG component | Yes, region SL-I to DB1 | NK, Possibly unless binds SL-I | Pull-down, IVT RNA, Quantitative mass-spec | [89] |
| FBP1 11 | Host | ssDNA binding protein, Influence mRNA stability | Yes | NK, Likely | Pull-down, IVT RNA | [90] |
| p100 | Host | Transcription and RNA transport | Yes, 3'SL | NK, Likely | Pull-down, IVT RNA | [88] |
| IGF2BP1 12 | Host | Translation and mRNA stability | Yes | NK, Likely | Pull-down, IVT RNA | [88] |
| RBMX 13 | Host | Pre-mRNA splicing | Yes | NK, Likely | Pull-down, IVT RNA | [88] |
| YB-1 14 | Host | Transcription regulation, Translation regulation, mRNA stability | Yes, 3'SL | NK, Likely | Infected cells, Pull-down, IVT RNA, Mass-spec | [91] |
| hnRNP 15 Q | Host | Splicing, Translation regulation, mRNA stability | Yes | NK, Likely | Pull-down, IVT RNA, Mass-spec | [91] |
| hnRNP A1 | Host | Splicing and RNA synthesis | Yes | NK, Likely | Pull-down, IVT RNA, Mass-spec | [91] |
| hnRNP A2/B | Host | RNA trafficking | Yes | NK, Likely | Pull-down, IVT RNA, Mass-spec | [91] |
| Mov34 16 | Host | RNA transcription and translation, Proteasome | Yes, 3′SL | NK, Likely | IVT RNA | [92] |
| NF90 17 | Host | RNA export, RNA stabilization, Negative regulation of miRNA | Yes, 3'SL | NK, Likely | Pull-down, IVT RNA | [93] |
| RHA 18 | Host | Assist NF-κB signaling, Sense dsRNA, Unwind dsRNA | Yes, 3'SL, Maybe in vitro only | NK, Possibly | Pull-down, IVT RNA | [93] |
| XRN1 | Host | PB component, 5'–3' exoribonuclease | Yes | Yes | Infected cells, Pull-down | [35] |
8. Conclusions and Future Directions
Acknowledgements
Author Contributions
Conflicts of Interest
References and Notes
- Hayes, E.B.; Gubler, D.J. West Nile virus: Epidemiology and clinical features of an emerging epidemic in the United States. In Annual Review of Medicine; Annual Reviews: Palo Alto, SA, USA, 2006; Volume 57, pp. 181–194. [Google Scholar]
- Gyure, K.A. West Nile Virus Infections. J. Neuropathol. Exp. Neurol. 2009, 68, 1053–1060. [Google Scholar] [CrossRef]
- Davis, L.E.; DeBiasi, R.; Goade, D.E.; Haaland, K.Y.; Harrington, J.A.; Harnar, J.B.; Pergam, S.A.; King, M.K.; DeMasters, B.K.; Tyler, K.L. West Nile virus neuroinvasive disease. Annu. Neurol. 2006, 60, 286–300. [Google Scholar] [CrossRef]
- Cook, S.; Holmes, E.C. A multigene analysis of the phylogenetic relationships among the flaviviruses (Family: Flaviviridae) and the evolution of vector transmission. Arch. Virol. 2006, 151, 309–325. [Google Scholar] [CrossRef]
- Cook, S.; Moureau, G.; Kitchen, A.; Gould, E.A.; de Lamballerie, X.; Holmes, E.C.; Harbachl, R.E. Molecular evolution of the insect-specific flaviviruses. J. Gen. Virol. 2012, 93, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, A.; Shackelton, L.A.; Holmes, E.C. Family level phylogenies reveal modes of macroevolution in RNA viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 238–243. [Google Scholar] [CrossRef]
- Kuno, G.; Chang, G.J.J.; Tsuchiya, K.R.; Karabatsos, N.; Cropp, C.B. Phylogeny of the genus Flavivirus. J. Virol. 1998, 72, 73–83. [Google Scholar]
- Gubler, D.J. Flaviviruses: Past, present and future. In Molecular Virology and Control of Flaviviruses; Shi, P.Y., Ed.; Caister Academic Press: Wymondham, UK, 2012; pp. 1–7. [Google Scholar]
- Varelas-Wesley, I.; Calisher, C.H. Antigenic relationships of flaviviruses with undetermined arthropod-borne status. Am. J. Trop. Med. Hyg. 1982, 31, 1273–1284. [Google Scholar]
- De Lamballerie, X.; Crochu, S.; Billoir, F.; Neyts, J.; de Micco, P.; Holmes, E.C.; Gould, E.A. Genome sequence analysis of Tamana bat virus and its relationship with the genus Flavivirus. J. Gen. Virol. 2002, 83, 2443–2454. [Google Scholar]
- Stapleton, J.T.; Foung, S.; Muerhoff, A.S.; Bukh, J.; Simmonds, P. The GB viruses: A review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J. Gen. Virol. 2011, 92, 233–246. [Google Scholar] [CrossRef]
- Thurner, C.; Witwer, C.; Hofacker, I.L.; Stadler, P.F. Conserved RNA secondary structures in Flaviviridae genomes. J. Gen. Virol. 2004, 85, 1113–1124. [Google Scholar] [CrossRef]
- Liu, Y.; Wimmer, E.; Paul, A.V. cis-Acting RNA elements in human and animal plus-strand RNA viruses. Biochim. Biophys. Acta 2009, 1789, 495–517. [Google Scholar]
- Lodeiro, M.F.; Filomatori, C.V.; Gamarnik, A.V. Structural and functional studies of the promoter element for Dengue virus RNA replication. J. Virol. 2009, 83, 993–1008. [Google Scholar] [CrossRef]
- Roby, J.A.; Funk, A.; Khromykh, A.A. Flavivirus replication and assembly. In Molecular Virology and Control of Flaviviruses; Shi, P.Y., Ed.; Caister Academic Press: Wymondham, UK, 2012. [Google Scholar]
- Olsthoorn, R.C.; Bol, J.F. Sequence comparison and secondary structure analysis of the 3' noncoding region of flavivirus genomes reveals multiple pseudoknots. RNA 2001, 7, 1370–1377. [Google Scholar]
- Proutski, V.; Gould, E.A.; Holmes, E.C. Secondary structure of the 3' untranslated region of flaviviruses: Similarities and differences. Nucleic Acids Res. 1997, 25, 1194–1202. [Google Scholar] [CrossRef]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef]
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Melian, E.B.; Edmonds, J.; Dong, H.; Shi, P.Y.; Khromykh, A.A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010, 84, 11407–11417. [Google Scholar] [CrossRef]
- Filomatori, C.V.; Lodeiro, M.F.; Alvarez, D.E.; Samsa, M.M.; Pietrasanta, L.; Gamarnik, A.V. A 5' RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev. 2006, 20, 2238–2249. [Google Scholar] [CrossRef]
- Dong, H.P.; Zhang, B.; Shi, P.Y. Flavivirus methyltransferase: A novel antiviral target. Antivir. Res. 2008, 80, 1–10. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, H.; Stein, D.A.; Iversen, P.L.; Shi, P.Y. West Nile virus genome cyclization and RNA replication require two pairs of long-distance RNA interactions. Virology 2008, 373, 1–13. [Google Scholar] [CrossRef]
- Polacek, C.; Friebe, P.; Harris, E. Poly(A)-binding protein binds to the non-polyadenylated 3' untranslated region of dengue virus and modulates translation efficiency. J. Gen. Virol. 2009, 90, 687–692. [Google Scholar] [CrossRef]
- Blackwell, J.L.; Brinton, M.A. Translation elongation factor-1 alpha interacts with the 3' stem-loop region of West Nile virus genomic RNA. J. Virol. 1997, 71, 6433–6444. [Google Scholar]
- De Nova-Ocampo, M.; Villegas-Sepuveda, N.; del Angel, R.M. Translation elongation factor-1 alpha, La, and PTB interact with the 3' untranslated region of dengue 4 virus RNA. Virology 2002, 295, 337–347. [Google Scholar] [CrossRef]
- Urosevic, N.; van Maanen, M.; Mansfield, J.P.; Mackenzie, J.S.; Shellam, G.R. Molecular characterization of virus-specific RNA produced in the brains of flavivirus-susceptible and -resistant mice after challenge with Murray Valley encephalitis virus. J. Gen. Virol. 1997, 78, 23–29. [Google Scholar]
- Lin, K.C.; Chang, H.L.; Chang, R.Y. Accumulation of a 3'-terminal genome fragment in Japanese encephalitis virus-infected mammalian and mosquito cells. J. Virol. 2004, 78, 5133–5138. [Google Scholar] [CrossRef]
- Scherbik, S.V.; Paranjape, J.M.; Stockman, B.M.; Silverman, R.H.; Brinton, M.A. RNase L plays a role in the antiviral response to West Nile virus. J. Virol. 2006, 80, 2987–2999. [Google Scholar] [CrossRef]
- Fan, Y.H.; Nadar, M.; Chen, C.C.; Weng, C.C.; Lin, Y.T.; Chang, R.Y. Small noncoding RNA modulates Japanese encephalitis virus replication and translation in trans. Virol. J. 2011, 8. [Google Scholar] [CrossRef]
- Silva, P.A.; Pereira, C.F.; Dalebout, T.J.; Spaan, W.J.; Bredenbeek, P.J. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. J. Virol. 2010, 84, 11395–11406. [Google Scholar] [CrossRef]
- Liu, R.; Yue, L.; Li, X.; Yu, X.; Zhao, H.; Jiang, Z.; Qin, E.; Qin, C. Identification and characterization of small sub-genomic RNAs in dengue 1–4 virus-infected cell cultures and tissues. Biochem. Biophys. Res. Commun. 2010, 391, 1099–1103. [Google Scholar] [CrossRef]
- Silva, P.; Jiang, X.; Pereira, C.F.; Dalebout, T.J.; Bredenbeek, P.J. Characterization of the RNA structures required for sfRNA production in flaviviruses with no known vector. In Proceedings of the American Society of Virology 29th Annual Meeting, Montana State University, Bozeman, MT, USA, 17–21 July 2010.
- Bredenbeek, P. Leiden University Medical Centre, Leiden, The Netherlands. Personal Communication, 2013. [Google Scholar]
- Schnettler, E. MRC—University of Glasgow CVR, Glasgow, United Kingdom. Personal Communication, 2013. [Google Scholar]
- Moon, S.L.; Anderson, J.R.; Kumagai, Y.; Wilusz, C.J.; Akira, S.; Khromykh, A.A.; Wilusz, J. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 2012, 18, 2029–2040. [Google Scholar] [Green Version]
- Sheth, U.; Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003, 300, 805–808. [Google Scholar] [CrossRef]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 2007, 8, 9–22. [Google Scholar] [CrossRef]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar]
- Covarrubias, S.; Gaglia, M.M.; Kumar, G.R.; Wong, W.; Jackson, A.O.; Glaunsinger, B.A. Coordinated destruction of cellular messages in translation complexes by the gammaherpesvirus host shutoff factor and the mammalian exonuclease Xrn1. PLoS Pathog. 2011, 7, e1002339. [Google Scholar] [CrossRef]
- Markoff, L. 5'- And 3'-noncoding regions in flavivirus RNA. Adv. Virus Res. 2003, 59, 177–228. [Google Scholar] [CrossRef]
- Brinton, M.A.; Fernandez, A.V.; Dispoto, J.H. The 3'-nucleotides of flavivirus genomic RNA form a conserved secondary structure. Virology 1986, 153, 113–121. [Google Scholar] [CrossRef]
- Shi, P.Y.; Brinton, M.A.; Veal, J.M.; Zhong, Y.Y.; Wilson, W.D. Evidence for the existence of a pseudoknot structure at the 3'-terminus of the flavivirus genomic RNA. Biochemistry 1996, 35, 4222–4430. [Google Scholar] [CrossRef]
- Schuessler, A.; Funk, A.; Lazear, H.M.; Cooper, D.A.; Torres, S.; Daffis, S.; Jha, B.K.; Kumagai, Y.; Takeuchi, O.; Hertzog, P.; et al. West Nile Virus noncoding subgenomic RNA contributes to Viral evasion of the type I interferon-mediated antiviral response. J. Virol. 2012, 86, 5708–5718. [Google Scholar] [CrossRef]
- Zhu, W.; Qin, C.; Chen, S.; Jiang, T.; Yu, M.; Yu, X.; Qin, E. Attenuated dengue 2 viruses with deletions in capsid protein derived from an infectious full-length cDNA clone. Virus Res. 2007, 126, 226–232. [Google Scholar] [CrossRef]
- Pahl, J.; Funk, A.; Khromykh, A. University of Queensland, Brisbane, Australia, Unpublished work. 2008.
- Chahar, H.S.; Chen, S.; Manjunath, N. P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 2013, 436, 1–7. [Google Scholar] [CrossRef]
- Reineke, L.C.; Lloyd, R.E. Diversion of stress granules and P-bodies during viral infection. Virology 2013, 436, 255–267. [Google Scholar] [CrossRef]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef]
- Courtney, S.C.; Scherbik, S.V.; Stockman, B.M.; Brinton, M.A. West Nile virus infections suppress early viral RNA synthesis and avoid inducing the cell stress granule response. J. Virol. 2012, 86, 3647–3657. [Google Scholar] [CrossRef]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996, 220, 232–240. [Google Scholar]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.E.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and Three-Dimensional Architecture of the Dengue Virus Replication and Assembly Sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The Endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef]
- Ding, S.W. RNA-based antiviral immunity. Nat. Rev. Immunol. 2010, 10, 632–644. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Morris, K.V.; Chan, S.W.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar] [CrossRef]
- Brackney, D.E.; Beane, J.E.; Ebel, G.D. RNAi targeting of West Nile virus in mosquito midguts promotes virus diversification. PLoS Pathog. 2009, 5, e1000502. [Google Scholar] [CrossRef]
- Jinek, M.; Doudna, J.A. A three-dimensional view of the molecular machinery of RNA interference. Nature 2009, 457, 405–412. [Google Scholar] [CrossRef]
- Yates, L.A.; Norbury, C.J.; Gilbert, R.J. The long and short of microRNA. Cell 2013, 153, 516–519. [Google Scholar] [CrossRef]
- Rouha, H.; Thurner, C.; Mandl, C.W. Functional microRNA generated from a cytoplasmic RNA virus. Nucleic Acids Res. 2010, 38, 8328–8337. [Google Scholar] [CrossRef]
- Varble, A.; ten Oever, B.R. Implications of RNA virus-produced miRNAs. RNA Biol. 2011, 8, 190–194. [Google Scholar] [CrossRef]
- Shapiro, J.S.; Varble, A.; Pham, A.M.; Tenoever, B.R. Noncanonical cytoplasmic processing of viral microRNAs. RNA 2010, 16, 2068–2074. [Google Scholar] [CrossRef]
- Hussain, M.; Torres, S.; Schnettler, E.; Funk, A.; Grundhoff, A.; Pijlman, G.P.; Khromykh, A.A.; Asgari, S. West Nile virus encodes a microRNA-like small RNA in the 3' untranslated region which up-regulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res. 2012, 40, 2210–2223. [Google Scholar] [CrossRef]
- Diamond, M.S.; Gale, M., Jr. Cell-intrinsic innate immune control of West Nile virus infection. Trends Immunol. 2012, 33, 522–530. [Google Scholar] [CrossRef]
- Suthar, M.S.; Diamond, M.S.; Gale, M. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Gilfoy, F.D.; Mason, P.W. West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J. Virol. 2007, 81, 11148–11158. [Google Scholar] [CrossRef]
- Samuel, M.A.; Whitby, K.; Keller, B.C.; Marri, A.; Barchet, W.; Williams, B.R.G.; Silverman, R.H.; Gale, M.; Diamond, M.S. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 2006, 80, 7009–7019. [Google Scholar] [CrossRef]
- Elbahesh, H.; Scherbik, S.V.; Brinton, M.A. West Nile virus infection does not induce PKR activation in rodent cells. Virology 2011, 421, 51–60. [Google Scholar] [CrossRef]
- Tu, Y.C.; Yu, C.Y.; Liang, J.J.; Lin, E.L.; Liao, C.L.; Lin, Y.L. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J. Virol. 2012, 86, 10347–10358. [Google Scholar] [CrossRef]
- Diamond, M.S.; Harris, E. Interferon inhibits dengue virus infection by preventing translation of viral RNA through a PKR-independent mechanism. Virology 2001, 289, 297–311. [Google Scholar] [CrossRef]
- Li, X.L.; Ezelle, H.J.; Hsi, T.Y.; Hassel, B.A. A central role for RNA in the induction and biological activities of type 1 interferons. Wiley Interdiscip Rev. RNA 2011, 2, 58–78. [Google Scholar] [CrossRef]
- Chang, R.Y.; Hsu, T.W.; Chen, Y.L.; Liu, S.F.; Tsai, Y.J.; Lin, Y.T.; Chen, Y.S.; Fan, Y.H. Japanese encephalitis virus non-coding RNA inhibits activation of interferon by blocking nuclear translocation of interferon regulatory factor 3. Vet. Microbiol. 2013, 166, 11–21. [Google Scholar] [CrossRef]
- Braun, J.E.; Truffault, V.; Boland, A.; Huntzinger, E.; Chang, C.T.; Haas, G.; Weichenrieder, O.; Coles, M.; Izaurralde, E. A direct interaction between DCP1 and XRN1 couples mRNA decapping to 5' exonucleolytic degradation. Nat. Struct. Mol. Biol. 2012, 19, 1324–1331. [Google Scholar]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with west nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar] [CrossRef]
- Fragkoudis, R.; Attarzadeh-Yazdi, G.; Nash, A.A.; Fazakerley, J.K.; Kohl, A. Advances in dissecting mosquito innate immune responses to arbovirus infection. J. Gen. Virol. 2009, 90, 2061–2072. [Google Scholar] [CrossRef]
- Blair, C.D. Mosquito RNAi is the major innate immune pathway controlling arbovirus infection and transmission. Future Microbiol. 2011, 6, 265–277. [Google Scholar] [CrossRef]
- Siu, R.W.; Fragkoudis, R.; Simmonds, P.; Donald, C.L.; Chase-Topping, M.E.; Barry, G.; Attarzadeh-Yazdi, G.; Rodriguez-Andres, J.; Nash, A.A.; Merits, A.; et al. Antiviral RNA interference responses induced by Semliki Forest virus infection of mosquito cells: Characterization, origin, and frequency-dependent functions of virus-derived small interfering RNAs. J. Virol 2011, 85, 2907–2917. [Google Scholar] [CrossRef]
- Schnettler, E.; Sterken, M.G.; Leung, J.Y.; Metz, S.W.; Geertsema, C.; Goldbach, R.W.; Vlak, J.M.; Kohl, A.; Khromykh, A.A.; Pijlman, G.P. Noncoding flavivirus RNA displays RNA interference suppressor activity in insect and Mammalian cells. J. Virol. 2012, 86, 13486–13500. [Google Scholar] [CrossRef]
- Schnettler, E.; de Vries, W.; Hemmes, H.; Haasnoot, J.; Kormelink, R.; Goldbach, R.; Berkhout, B. The NS3 protein of rice hoja blanca virus complements the RNAi suppressor function of HIV-1 Tat. EMBO Rep. 2009, 10, 258–263. [Google Scholar] [CrossRef]
- Chen, C.J.; Kuo, M.D.; Chien, L.J.; Hsu, S.L.; Wang, Y.M.; Lin, J.H. RNA-protein interactions: Involvement of NS3, NS5, and 3' noncoding regions of Japanese encephalitis virus genomic RNA. J. Virol. 1997, 71, 3466–3473. [Google Scholar]
- Khromykh, A.A.; Westaway, E.G. RNA binding properties of core protein of the flavivirus Kunjin. Arch. Virol 1996, 141, 685–699. [Google Scholar] [CrossRef]
- Mackenzie, J.M.; Khromykh, A.A.; Jones, M.K.; Westaway, E.G. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 1998, 245, 203–215. [Google Scholar] [CrossRef]
- Cui, T.; Sugrue, R.J.; Xu, Q.; Lee, A.K.; Chan, Y.C.; Fu, J. Recombinant dengue virus type 1 NS3 protein exhibits specific viral RNA binding and NTPase activity regulated by the NS5 protein. Virology 1998, 246, 409–417. [Google Scholar] [CrossRef]
- Davis, W.G.; Blackwell, J.L.; Shi, P.Y.; Brinton, M.A. Interaction between the cellular protein eEF1A and the 3'-terminal stem-loop of West Nile virus genomic RNA facilitates viral minus-strand RNA synthesis. J. Virol. 2007, 81, 10172–10187. [Google Scholar] [CrossRef]
- Vashist, S.; Bhullar, D.; Vrati, S. La protein can simultaneously bind to both 3'- and 5'-noncoding regions of Japanese encephalitis virus genome. DNA Cell Biol. 2011, 30, 339–346. [Google Scholar] [CrossRef]
- Vashist, S.; Anantpadma, M.; Sharma, H.; Vrati, S. La protein binds the predicted loop structures in the 3' non-coding region of Japanese encephalitis virus genome: Role in virus replication. J. Gen. Virol. 2009, 90, 1343–1352. [Google Scholar] [CrossRef]
- Yocupicio-Monroy, M.; Padmanabhan, R.; Medina, F.; del Angel, R.M. Mosquito La protein binds to the 3' untranslated region of the positive and negative polarity dengue virus RNAs and relocates to the cytoplasm of infected cells. Virology 2007, 357, 29–40. [Google Scholar] [CrossRef]
- Garcia-Montalvo, B.M.; Medina, F.; del Angel, R.M. La protein binds to NS5 and NS3 and to the 5' and 3' ends of Dengue 4 virus RNA. Virus Res. 2004, 102, 141–150. [Google Scholar] [CrossRef]
- Lei, Y.; Huang, Y.; Zhang, H.; Yu, L.; Zhang, M.; Dayton, A. Functional interaction between cellular p100 and the dengue virus 3' UTR. J. Gen. Virol. 2011, 92, 796–806. [Google Scholar] [CrossRef]
- Ward, A.M.; Bidet, K.; Yinglin, A.; Ler, S.G.; Hogue, K.; Blackstock, W.; Gunaratne, J.; Garcia-Blanco, M.A. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3' UTR structures. RNA Biol. 2011, 8, 1173–1186. [Google Scholar] [CrossRef]
- Chien, H.L.; Liao, C.L.; Lin, Y.L. FUSE binding protein 1 interacts with untranslated regions of Japanese encephalitis virus RNA and negatively regulates viral replication. J. Virol. 2011, 85, 4698–4706. [Google Scholar] [CrossRef]
- Paranjape, S.M.; Harris, E. Y box-binding protein-1 binds to the dengue virus 3'-untranslated region and mediates antiviral effects. J. Biol. Chem. 2007, 282, 30497–30508. [Google Scholar] [CrossRef]
- Ta, M.; Vrati, S. Mov34 protein from mouse brain interacts with the 3' noncoding region of Japanese encephalitis virus. J. Virol. 2000, 74, 5108–5115. [Google Scholar] [CrossRef]
- Gomila, R.C.; Martin, G.W.; Gehrke, L. NF90 binds the dengue virus RNA 3' terminus and is a positive regulator of dengue virus replication. PLoS One 2011, 6, e16687. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Padmanabhan, R. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J. Biol.Chem. 2001, 276, 39926–39937. [Google Scholar] [CrossRef]
- Chu, P.W.; Westaway, E.G. Replication strategy of Kunjin virus: Evidence for recycling role of replicative form RNA as template in semiconservative and asymmetric replication. Virology 1985, 140, 68–79. [Google Scholar] [CrossRef]
- Blackwell, J.L.; Brinton, M.A. BHK cell proteins that bind to the 3' stem-loop structure of the West Nile virus genome RNA. J. Virol. 1995, 69, 5650–5658. [Google Scholar]
- Witwer, K.W.; Sisk, J.M.; Gama, L.; Clements, J.E. MicroRNA regulation of IFN-beta protein expression: rapid and sensitive modulation of the innate immune response. J. Immunol. 2010, 184, 2369–2376. [Google Scholar] [CrossRef]
- Gantier, M.P.; Sadler, A.J.; Williams, B.R. Fine-tuning of the innate immune response by microRNAs. Immunol Cell Biol. 2007, 85, 458–462. [Google Scholar] [CrossRef]
- Pedersen, I.M.; Cheng, G.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roby, J.A.; Pijlman, G.P.; Wilusz, J.; Khromykh, A.A. Noncoding Subgenomic Flavivirus RNA: Multiple Functions in West Nile Virus Pathogenesis and Modulation of Host Responses. Viruses 2014, 6, 404-427. https://doi.org/10.3390/v6020404
Roby JA, Pijlman GP, Wilusz J, Khromykh AA. Noncoding Subgenomic Flavivirus RNA: Multiple Functions in West Nile Virus Pathogenesis and Modulation of Host Responses. Viruses. 2014; 6(2):404-427. https://doi.org/10.3390/v6020404
Chicago/Turabian StyleRoby, Justin A., Gorben P. Pijlman, Jeffrey Wilusz, and Alexander A. Khromykh. 2014. "Noncoding Subgenomic Flavivirus RNA: Multiple Functions in West Nile Virus Pathogenesis and Modulation of Host Responses" Viruses 6, no. 2: 404-427. https://doi.org/10.3390/v6020404