Genomic Sequencing and Biological Characteristics of a Novel Escherichia Coli Bacteriophage 9g, a Putative Representative of a New Siphoviridae Genus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage and Bacterial Strains and Their Cultivation
2.2. Bacteriophage Isolation
2.3. Transmission Electron Microscopy (TEM)
2.4. Adsorption Curve Determination and Single Burst Experiment
2.5. Generation and Analysis of Phage-Host Pseudolysogenic Associations (PA)
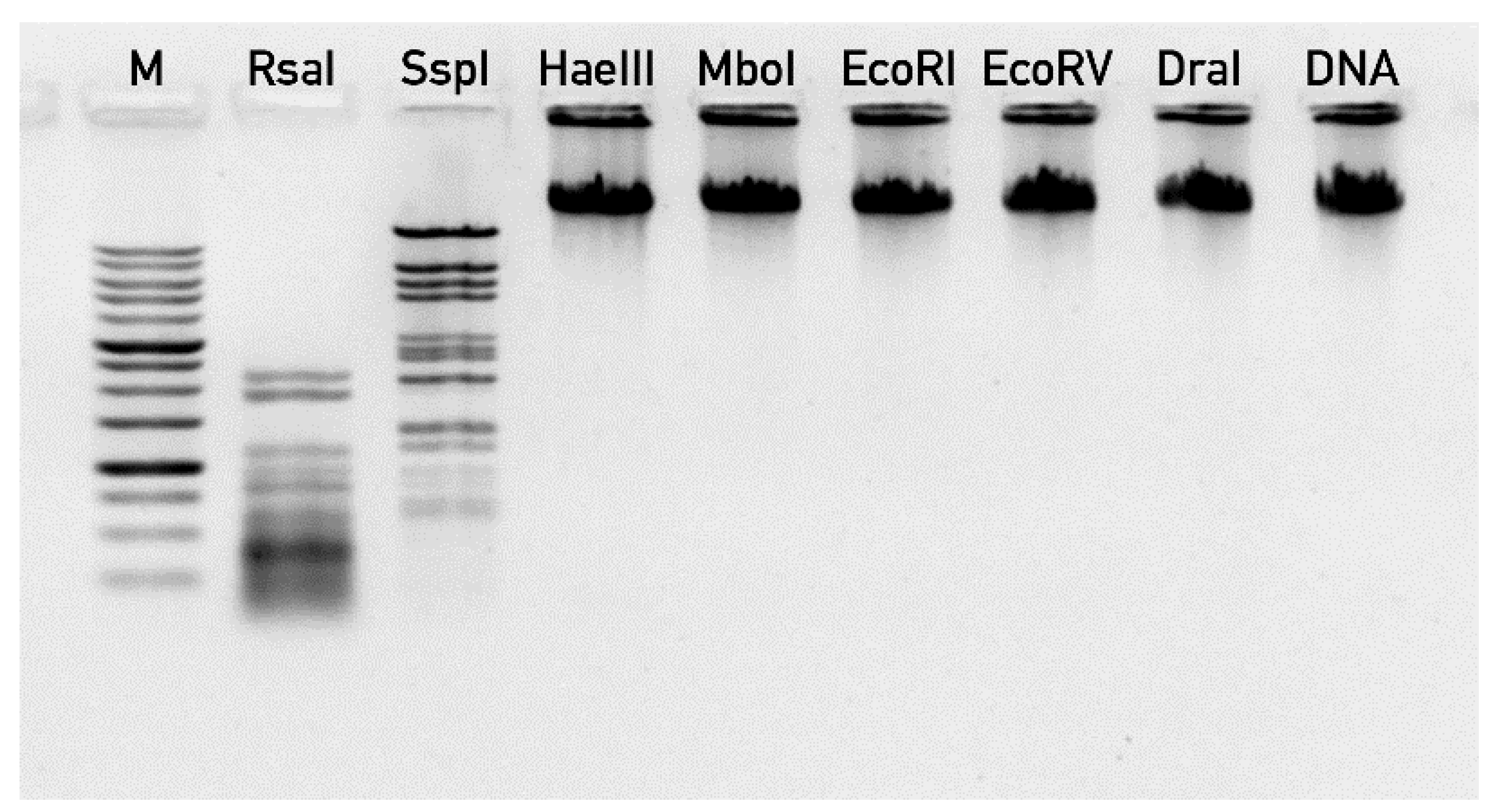
2.6. Phage DNA Extraction and Sequencing
3. Results
3.1. Bacteriophage 9g Isolation and Its Host Range
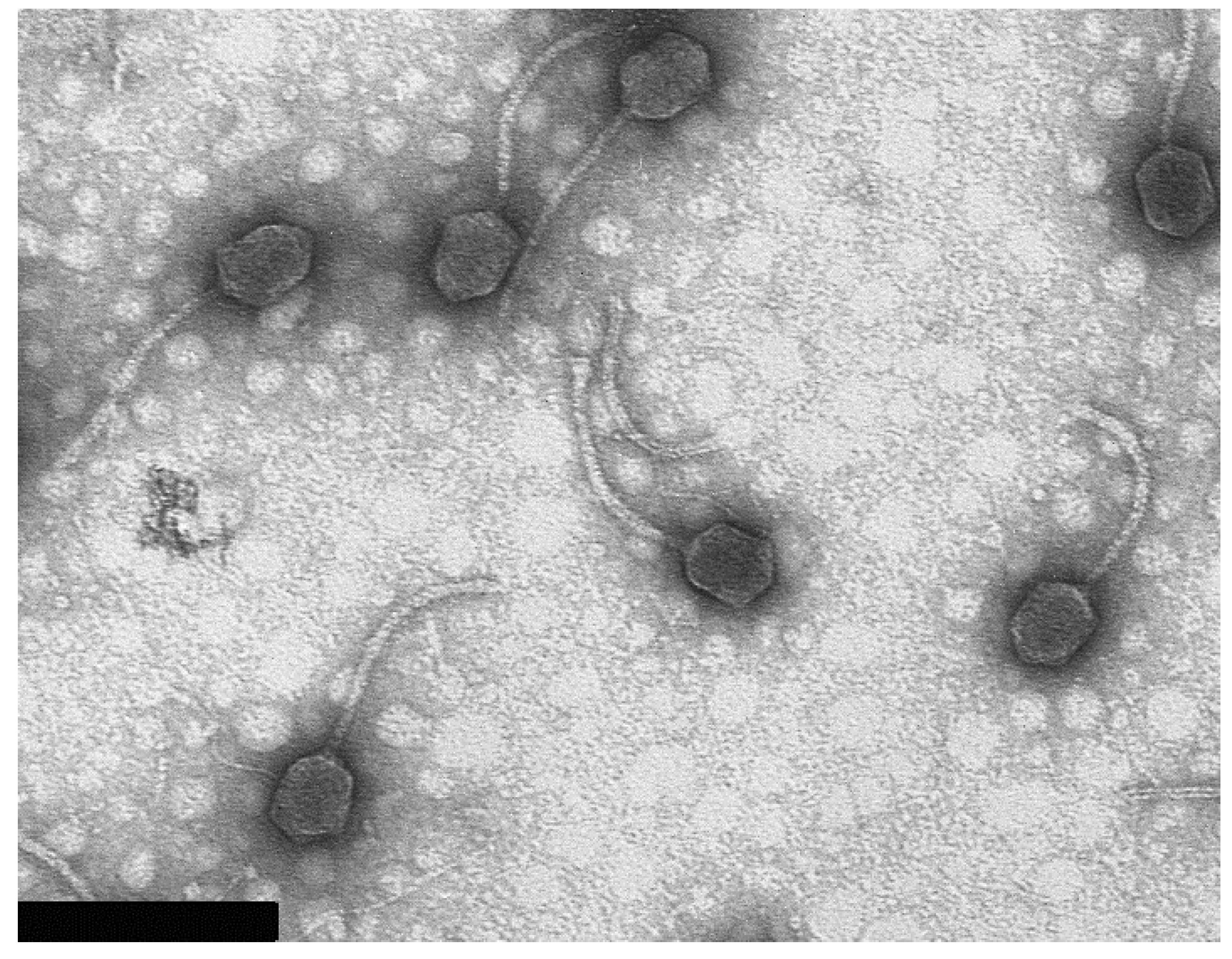
3.2. Virion Morphology
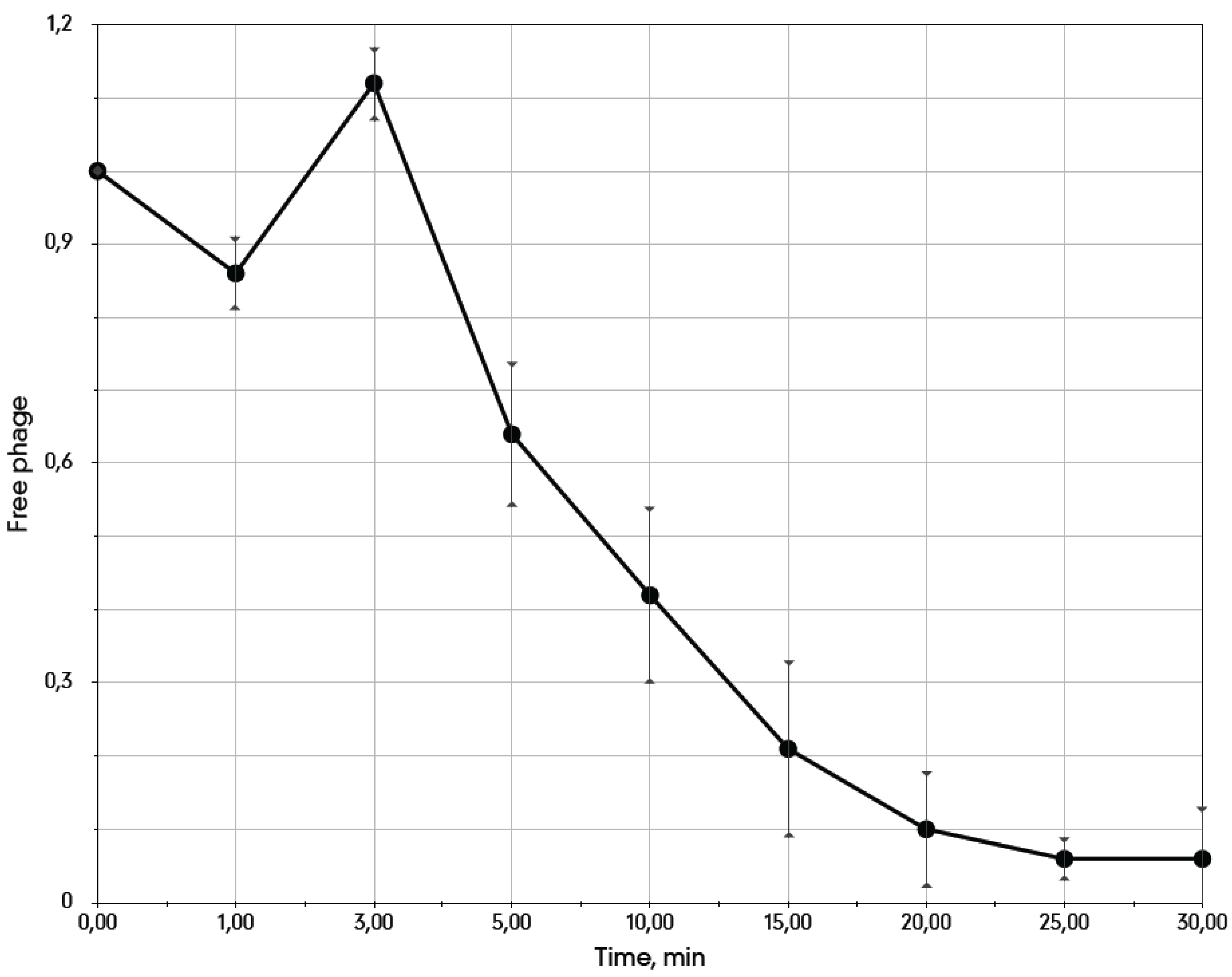
3.3. Life Cycle Parameters


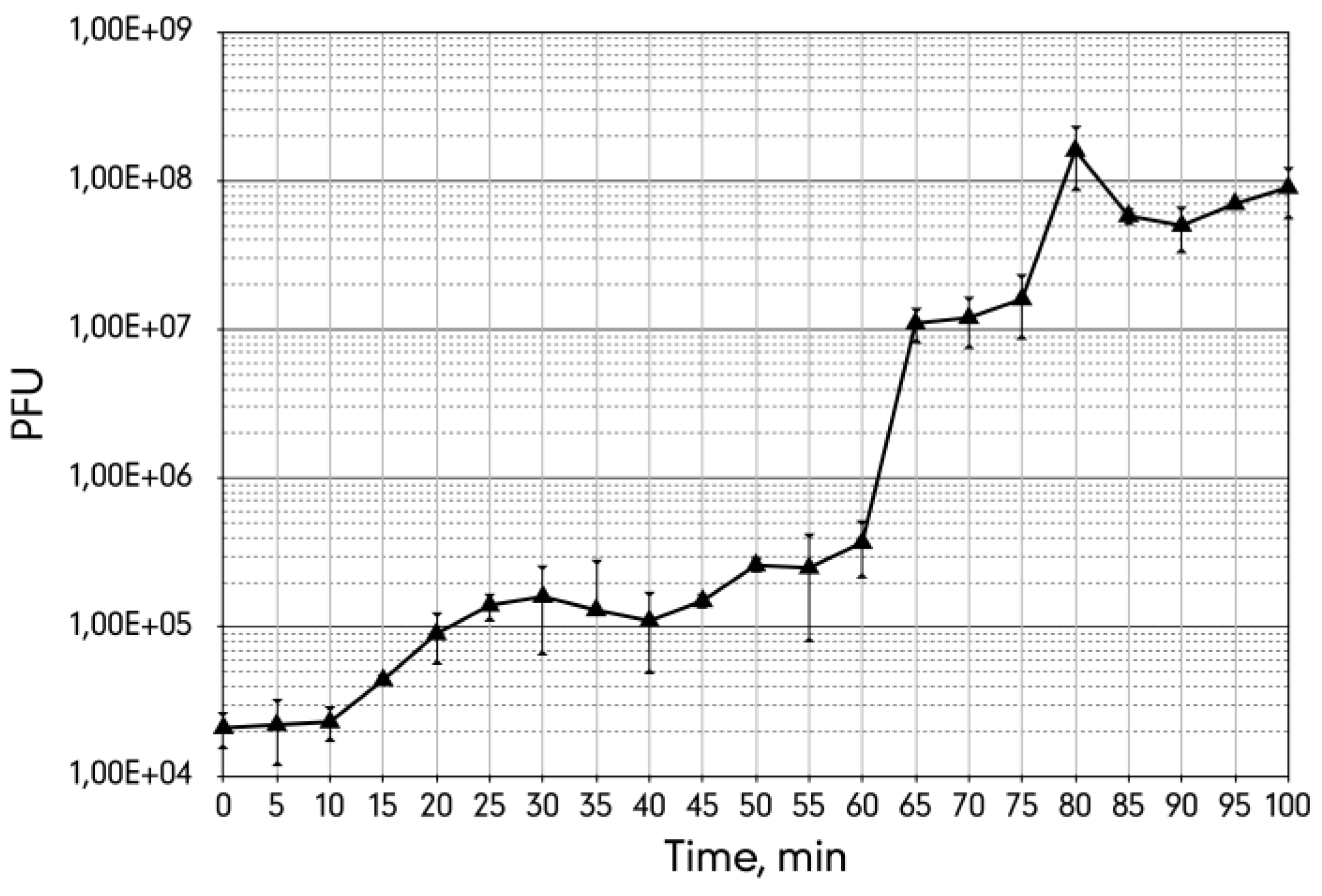
3.4. The Metastable Pseudolysogenic Associations of Phage 9g and Host.

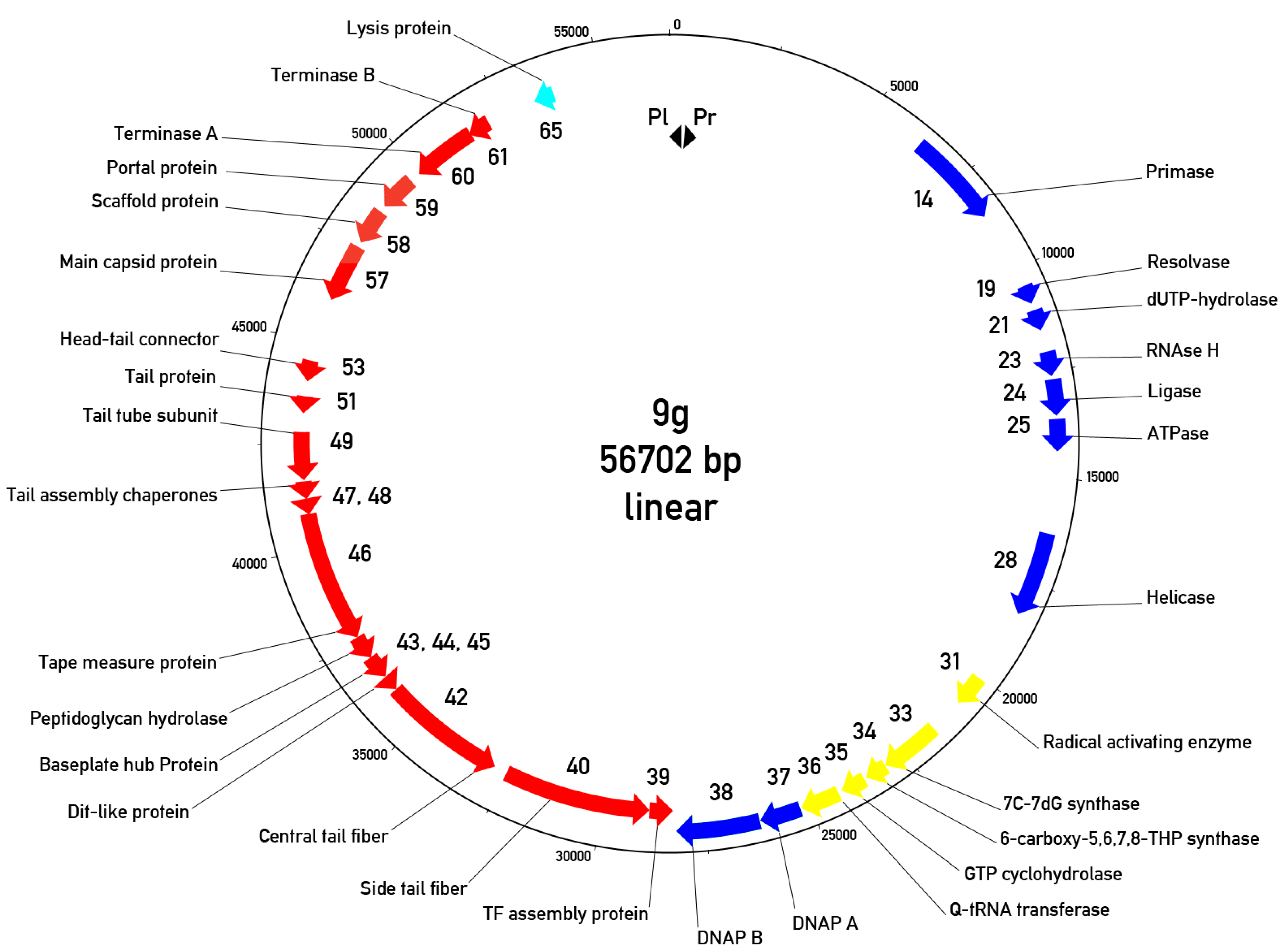
3.5. Overall Genome Organization of Bacteriophage 9g



3.6. The Early or Early/Middle Functions
3.7. The Morphogenetic Proteins and Other Late Functions
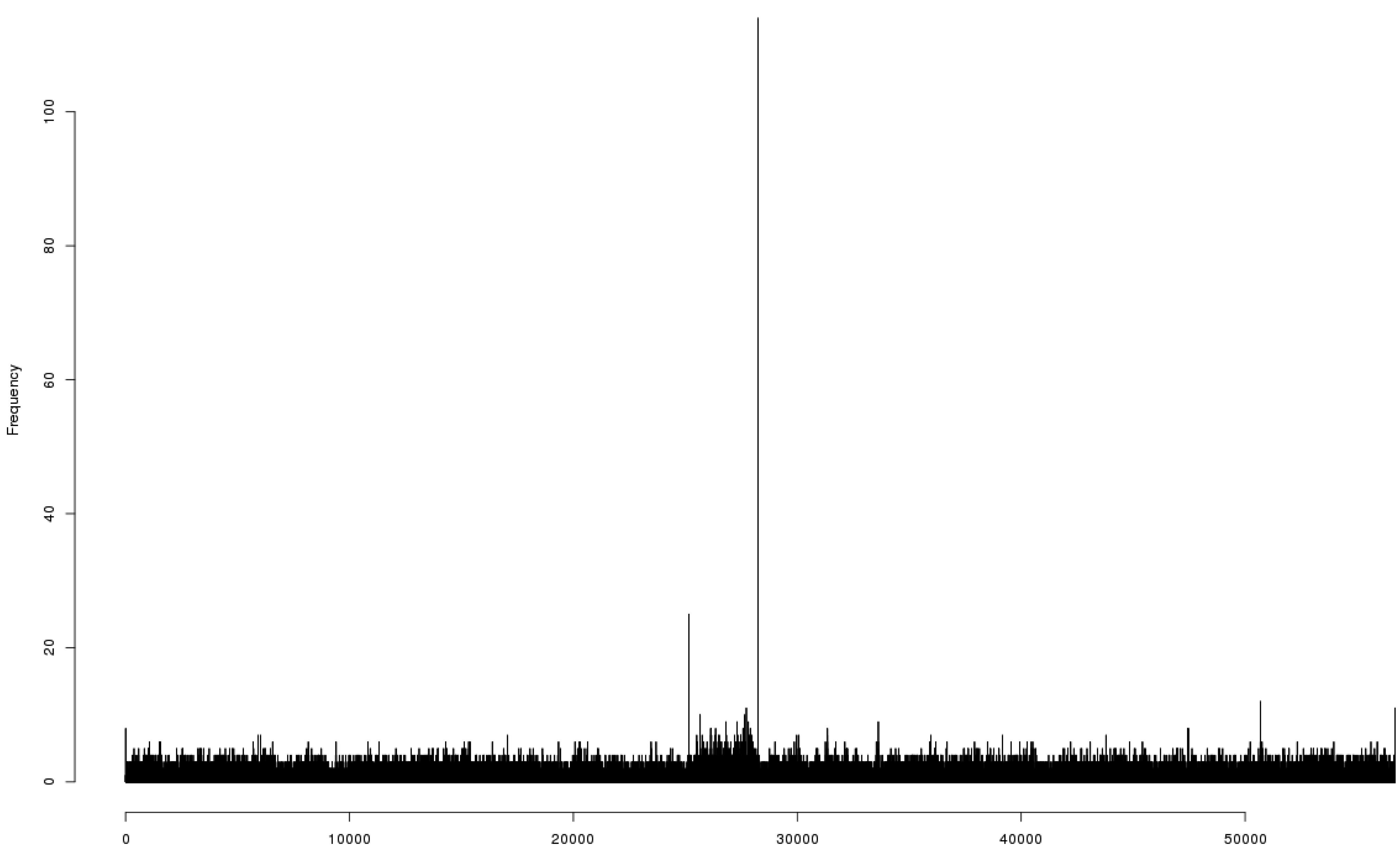
3.8. Relations of the Phage 9g to Other Bacteriophages
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Golomidova, A.; Kulikov, E.; Isaeva, A.; Manykin, A.; Letarov, A. The diversity of coliphages and coliforms in horse feces reveals a complex pattern of ecological interactions. Appl. Environ. Microbiol. 2007, 73, 5975–5981. [Google Scholar] [PubMed]
- Kulikov, E.E.; Isaeva, A.S.; Rotkina, A.S.; Manykin, A.A.; Letarov, A.V. Diversity and dynamics of bacteriophages in horse feces. Mikrobiologiia 2007, 76, 271–278. [Google Scholar] [PubMed]
- Letarov, A.; Kulikov, E. The bacteriophages in human- and animal body-associated microbial communities. J. Appl. Microbiol. 2009, 107, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, E.; Kropinski, A.M.; Golomidova, A.; Lingohr, E.; Govorun, V.; Serebryakova, M.; Prokhorov, N.; Letarova, M.; Manykin, A.; Strotskaya, A.; et al. Isolation and characterization of a novel indigenous intestinal N4-related coliphage vB_EcoP_G7C. Virology 2012, 426, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Waller, A.S.; Yamada, T.; Kristensen, D.M.; Kultima, J.R.; Sunagawa, S.; Koonin, E.V.; Bork, P. Classification and quantification of bacteriophage taxa in human gut metagenomes. The ISME J. 2014, 8, 1551–1552. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Letarov, A. Bacteriophages as a part of the human microbiome. In Bacteriophages in Health and Disease; Hyman, P., Abedon, S.T., Eds.; CABI Press: Wallingford, Oxfordshire, UK, 2012; Volume 24, pp. 6–20. [Google Scholar]
- Isaeva, A.S.; Kulikov, E.E.; Tarasyan, K.K.; Letarov, A.V. A Novel High–Resolving Method for Genomic PCR–Fingerprinting of Enterobacteria. Acta Naturae 2010, 2, 82–88. [Google Scholar] [PubMed]
- Virus Taxonomy: 2013 Release. Available online: http://www.ictvonline.org/virusTaxonomy.asp (accessed on 16 December 2014).
- Comeau, A.M.; Bertrand, C.; Letarov, A.; Tétart, F.; Krisch, H.M. Modular architecture of the T4 phage superfamily: A conserved core genome and a plastic periphery. Virology 2007, 362, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, Y.; Chen, F.; Jiao, N. Complete genome sequence of a marine roseophage provides evidence into the evolution of gene transfer agents in alphaproteobacteria. Virology J. 2011, 8, e124. [Google Scholar] [CrossRef]
- Lohr, J.E.; Chen, F.; Hill, R.T. Genomic analysis of bacteriophage PhiJL001: Insights into its interaction with a sponge–associated alpha–proteobacterium. Appl Environ Microbiol 2005, 71, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Clokie, M.R.J.; Kropinski, A.M. Bacteriophages: methods and protocols; Humana Press: New York, NY, USA, 2009. [Google Scholar]
- Ackermann, H.W. Basic phage electron microscopy. Methods Mol. Biol. 2009, 501, 113–126. [Google Scholar] [PubMed]
- Ackermann, H.W. Bacteriophage electron microscopy. Adv. Virus Res. 2012, 82, 1–32. [Google Scholar] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular cloning: a laboratory manual, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Besemer, J.; Borodovsky, M. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 1999, 27, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- GeneMark. Available online: http://opal.biology.gatech.edu/GeneMark (accessed on 16 Deccember 2014).
- Van Domselaar, G.H.; Stothard, P.; Shrivastava, S.; Cruz, J.A.; Guo, A.; Dong, X.; Lu, P.; Szafron, D.; Greiner, R.; Wishart, D.S. BASys: a web server for automated bacterial genome annotation. Nucleic Acids Res. 2005, 33, W455–W459. [Google Scholar] [CrossRef] [PubMed]
- BASYS Bacterial Annotation System. Available online: http://www.basys.ca (accessed on 16 Deccember 2014).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- BLAST® Basic Local Alignment Search Tool. Available online: http://www.ncbi.nlm.nih.gov/blast/Blast.cgi (accessed on 16 Deccember 2014).
- HHpred - Homology detection & structure prediction by HMM–HMM comparison. Available online: http://toolkit.tuebingen.mpg.de/hhpred (accessed on 16 Deccember 2014).
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- ARAGORN, tRNA (and tmRNA) detection. Available online: http://130.235.46.10/ARAGORN/ (accessed on 16 Deccember 2014).
- Lowe, T.M.; Eddy, S.R. tRNAscan–SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lowe Lab tRNAscan–SE Search Server. Available online: http://lowelab.ucsc.edu/tRNAscan–SE/ (accessed on 16 Deccember 2014).
- Appleyard, R.K. Segregation of New Lysogenic Types during Growth of a Doubly Lysogenic Strain Derived from Escherichia Coli K12. Genetics 1954, 39, 440–452. [Google Scholar] [PubMed]
- Knirel, Y.A.; Prokhorov, N.S.; Shashkov, A.S.; Ovchinnikova, O.G.; Zdorovenko, E.L.; Liu, B.; Kostryukova, E.S.; Larin, A.K.; Golomidova, A.K.; Letarov, A.V. Variations in the O–antigen biosynthesis and O–acetylation associated with the altered phage sensitivity in Escherichia coli 4s. J. Bacteriol. 2015. [Google Scholar] [CrossRef]
- Davidson, A.R.; Cardarelli, L.; Pell, L.G.; Radford, D.R.; Maxwell, K.L. Long noncontractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 2012, 726, 115–142. [Google Scholar] [PubMed]
- Heller, K.; Braun, V. Polymannose O-antigens of Escherichia coli, the binding sites for the reversible adsorption of bacteriophage T5+ via the L–shaped tail fibers. J. Virol. 1982, 41, 222–227. [Google Scholar] [PubMed]
- Reader, J.S.; Metzgar, D.; Schimmel, P.; de Crecy–Lagard, V. Identification of four genes necessary for biosynthesis of the modified nucleoside queuosine. J. Biol. Chem. 2004, 279, 6280–6285. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hendrix, R.W.; Duda, R.L. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol. Cell 2004, 16, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Young, R.; Texas University, Austin, TX, USA. Personal communication, 2014.
- Young, R. Bacteriophage holins: deadly diversity. J. Mol. Microbiol. Biotechnol. 2002, 4, 21–36. [Google Scholar] [PubMed]
- Virology Division, International Union of Microbiological Societies. Virus taxonomy: Classification and nomenclature of viruses. Ninth Report of the International Committee on Taxonomy of Viruses; Academic Press: London; Waltham, MA; San Diego, CA, USA, 2012; p. 1327. [Google Scholar]
- Sabri, M.; Hauser, R.; Ouellette, M.; Liu, J.; Dehbi, M.; Moeck, G.; Garcia, E.; Titz, B.; Uetz, P.; Moineau, S. Genome annotation and intraviral interactome for the Streptococcus pneumoniae virulent phage Dp–1. J Bacteriol. 2011, 193, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Летарова, М.А.; Куликов, Е.Е.; Голомидова, А.К.; Прохоров, Н.С.; Кутузова, Н.М.; Стрелкова, Д.М.; Бакумова, А.Д. Метастабильные ассоциации, формируемые в системе фаг – хозяин, выделенной из фекалий лошади. Вестник Ульяновской государственной сельскохозяйственной академии 2013, 3, 57–61. (In Russian) [Google Scholar]Letarova, M.A.; Kulikov, E.E.; Golomidova, A.K.; Prokhorov, N.S.; Kutuzova, N.M.; Strelkova, D.M.; Bakumova, A.D.; Letarov, A.V. Metastable associations formed in the phage–host system isolated from the horse feces. Vestnik Ulyanovskoi gosudarstvennoi selskokhozyaistvennoi akademii 2013, 3, 57–61. [Google Scholar]
- Abedon, S.T. Disambiguating bacteriophage pseudolysogeny: an historical analysis of lysogeny, pseudolysogeny, and the phage carrier state. In Contemporary trends in bacteriophage research; Adams, H.T., Ed.; Nova Science Publishers: New York, NY, USA, 2009; pp. 285–307. [Google Scholar]
- Stent, G.S. Molecular biology of bacterial viruses; W. H. Freeman: San Francisco, CA, USA, 1963; p. 474. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulikov, E.E.; Golomidova, A.K.; Letarova, M.A.; Kostryukova, E.S.; Zelenin, A.S.; Prokhorov, N.S.; Letarov, A.V. Genomic Sequencing and Biological Characteristics of a Novel Escherichia Coli Bacteriophage 9g, a Putative Representative of a New Siphoviridae Genus. Viruses 2014, 6, 5077-5092. https://doi.org/10.3390/v6125077
Kulikov EE, Golomidova AK, Letarova MA, Kostryukova ES, Zelenin AS, Prokhorov NS, Letarov AV. Genomic Sequencing and Biological Characteristics of a Novel Escherichia Coli Bacteriophage 9g, a Putative Representative of a New Siphoviridae Genus. Viruses. 2014; 6(12):5077-5092. https://doi.org/10.3390/v6125077
Chicago/Turabian StyleKulikov, Eugene E., Alla K. Golomidova, Maria A. Letarova, Elena S. Kostryukova, Alexandr S. Zelenin, Nikolai S. Prokhorov, and Andrey V. Letarov. 2014. "Genomic Sequencing and Biological Characteristics of a Novel Escherichia Coli Bacteriophage 9g, a Putative Representative of a New Siphoviridae Genus" Viruses 6, no. 12: 5077-5092. https://doi.org/10.3390/v6125077