KSHV miRNAs Decrease Expression of Lytic Genes in Latently Infected PEL and Endothelial Cells by Targeting Host Transcription Factors

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Antagomir Derepression Assays and Quantitative Reverse Transcription-PCR (RT-qPCR) Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence 5'-3' | Reference |
|---|---|---|
| MYB 3'UTR | FWD- GATGGAGGAGCAGATGACATC REV- AGGTAAAATAAGGGCAC | |
| Ets-1 3'UTR | FWD- CGTGTTGGTTGGACTCTGAA REV- TCTCCAGCAAAATGATGTGC | |
| C/EBPα 3'UTR | FWD-CTTGTGCCTTGGAAATGCAAACTCACC REV- AAGAAGAGAACCAAGCCGTCCTTC | |
| MYB | FWD- TCAGGAAACTTCTTCTGCTCACA REV- AGGTTCCCAGGTACTGCT | |
| Ets-1 | FWD- AAGGGAGATCGAAGGAGGAA REV- TCTGCTATAGGAACTGCAGGAG | |
| C/EBPα | FWD- TGTATACCCCTGGTGGGAGA REV- TCATAACTCCGGTCCCTCTG | |
| RTA | FWD- CACAAAAATGGCGCAAGATGA REV- TGGTAGAGTTGGGCCTTCAGTT | [35] |
| ORF59 | FWD- TTAGAAGTGGAAGGTGTGCC REV- TCCTGGAGTCCGGTATAGAATC | [36] |
| ORF19 | FWD- GGCGAAAAAGTCAGCGGTGGT REV- CGGCGCGTCTTCCCTAAAGA | [37] |
| LANA N-Terminus | FWD- GCGCCCTTAACGAGAGGAAGTT REV- TTCCTTCGCGGTTGTAGATG | |
| β-actin | FWD- CATGTACGTTGCTATCCAGGC REV- CTCCTTAATGTCACGCACGAT | Primerbank ID 4501885a1 |
2.3. Virus Isolation and Quantitation
2.4. Recombineering of miRNA Deletion Mutants in KSHV BAC16
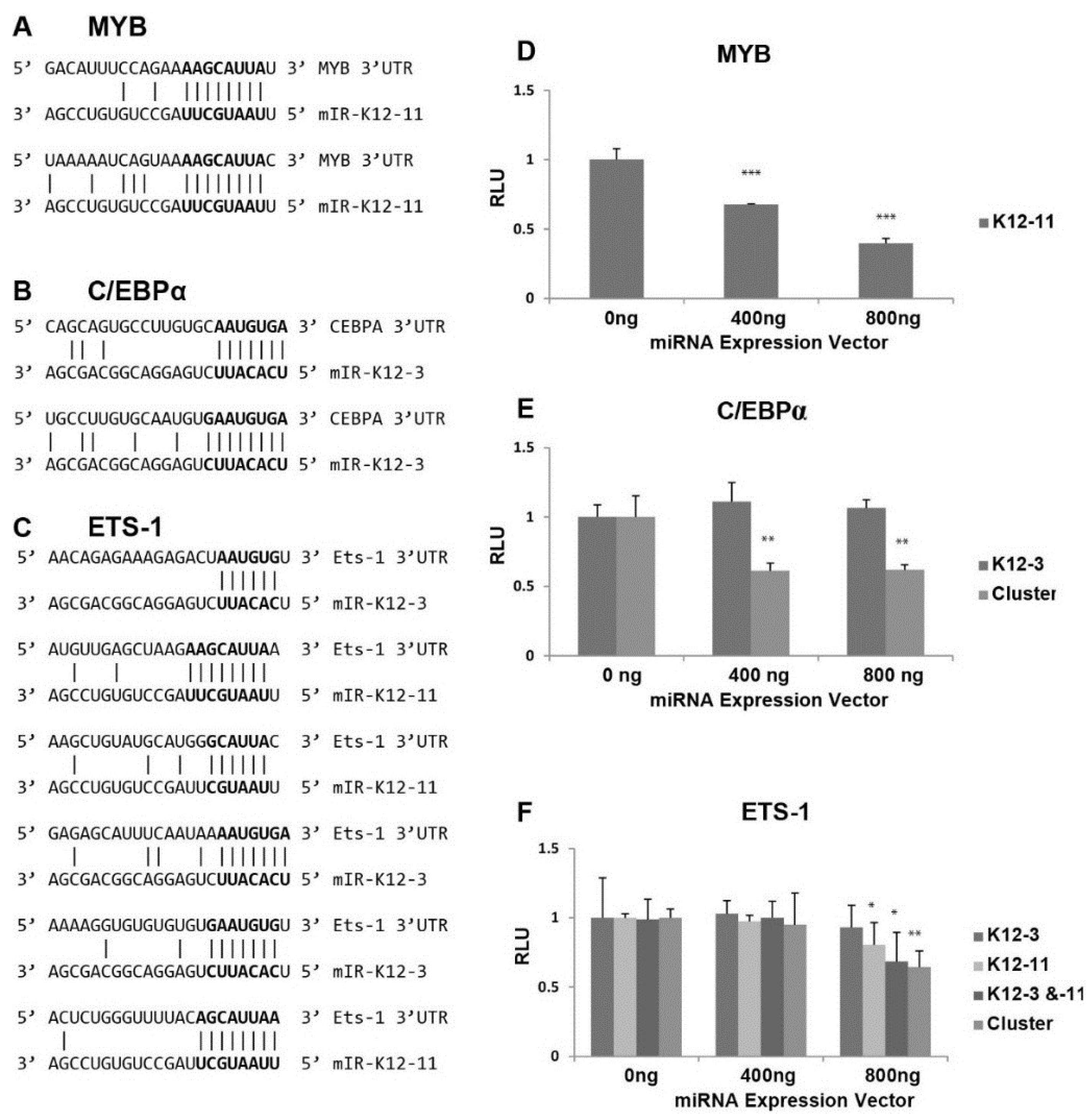
2.5. Reporter Construction and Luciferase Assays
3. Results
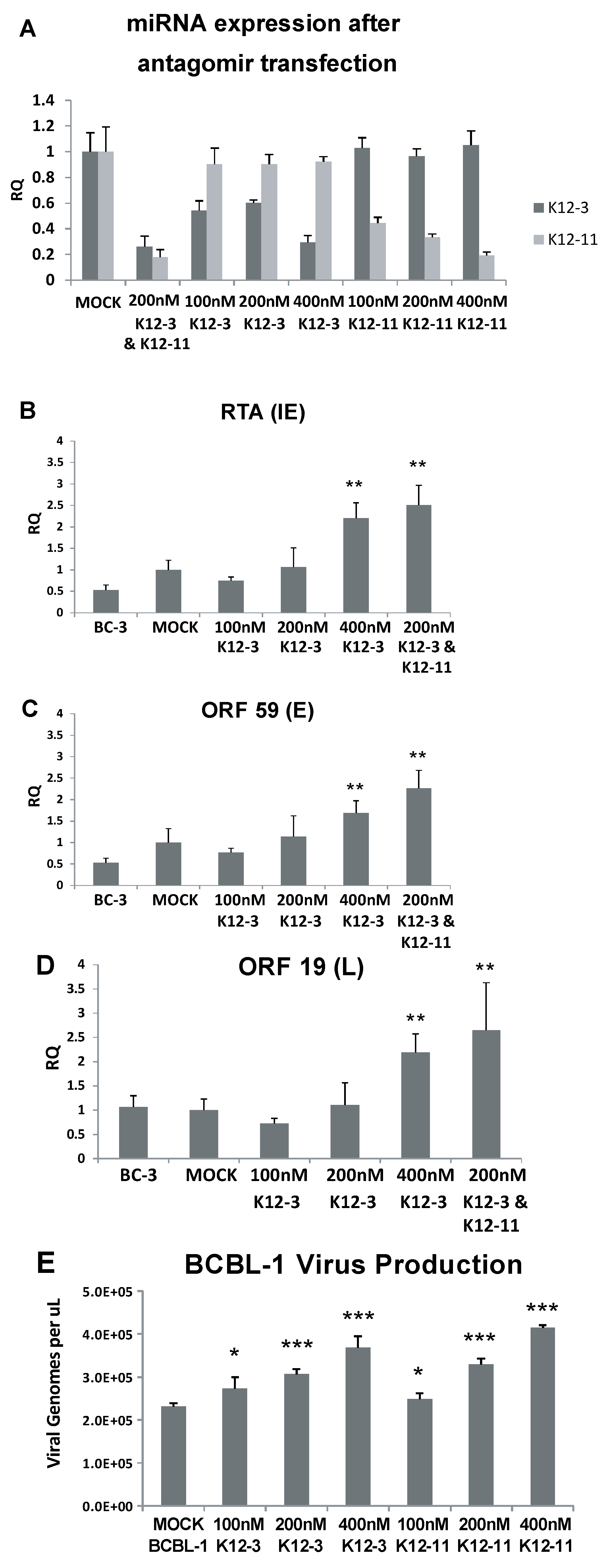
3.1. Screening for Effects of KSHV miRNA Knockdown on Reactivation in BC-3-G Cells
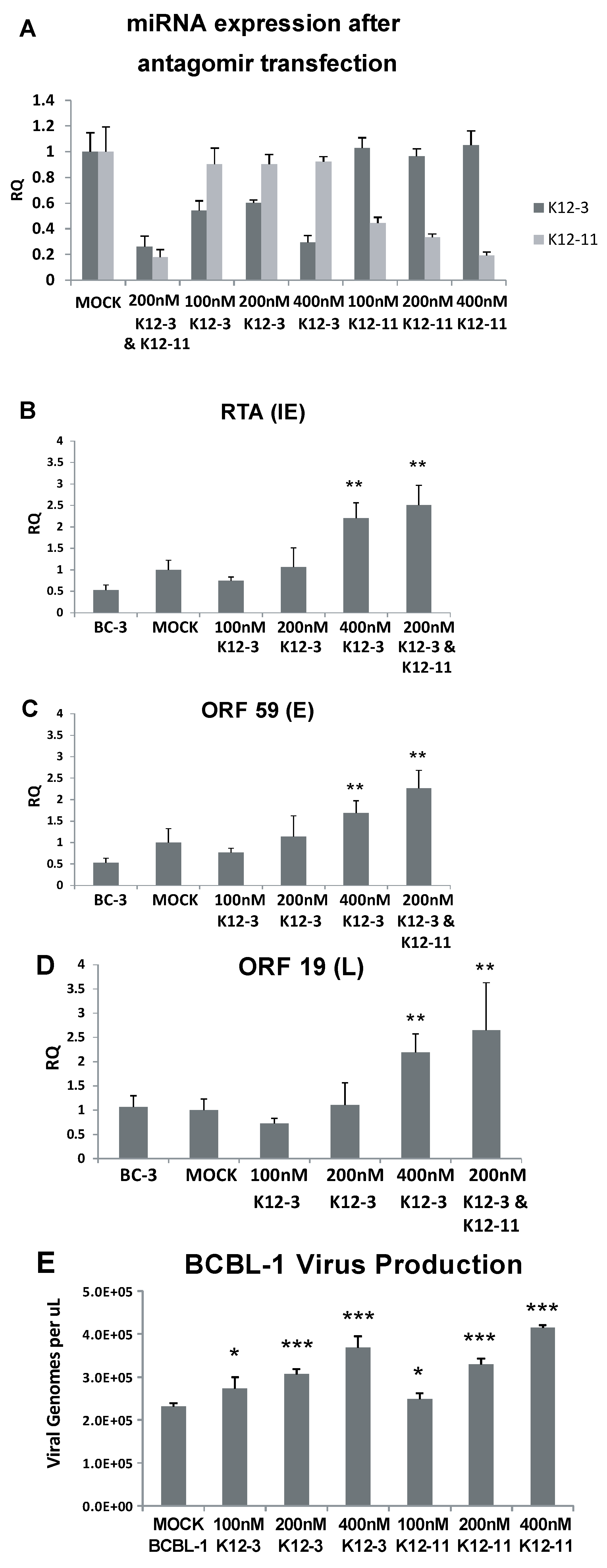
3.2. Lytic Gene Expression and Virus Production Increase upon miR-K12-3 and miR-K12-11 Knockdown in PEL Cells
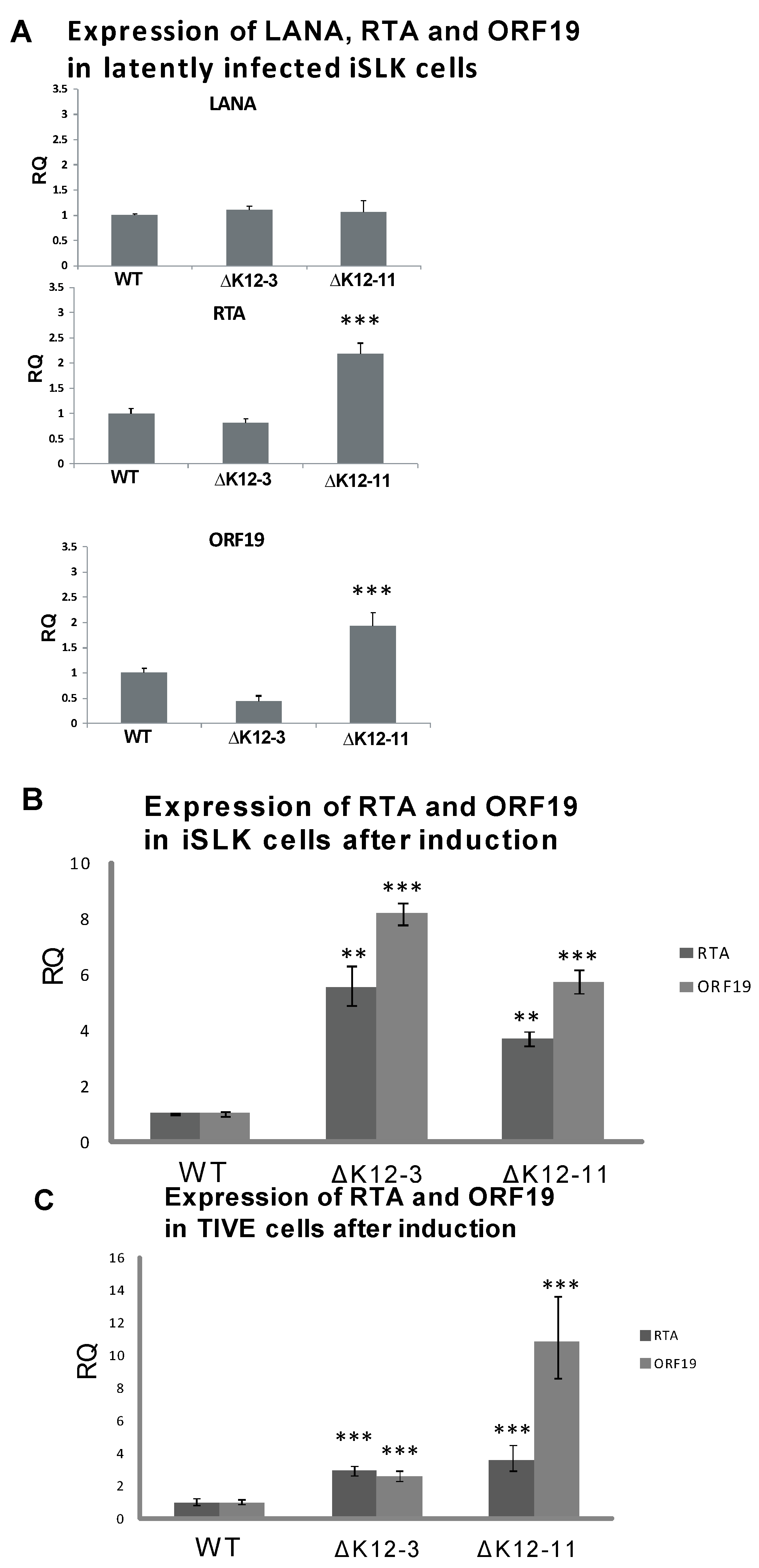
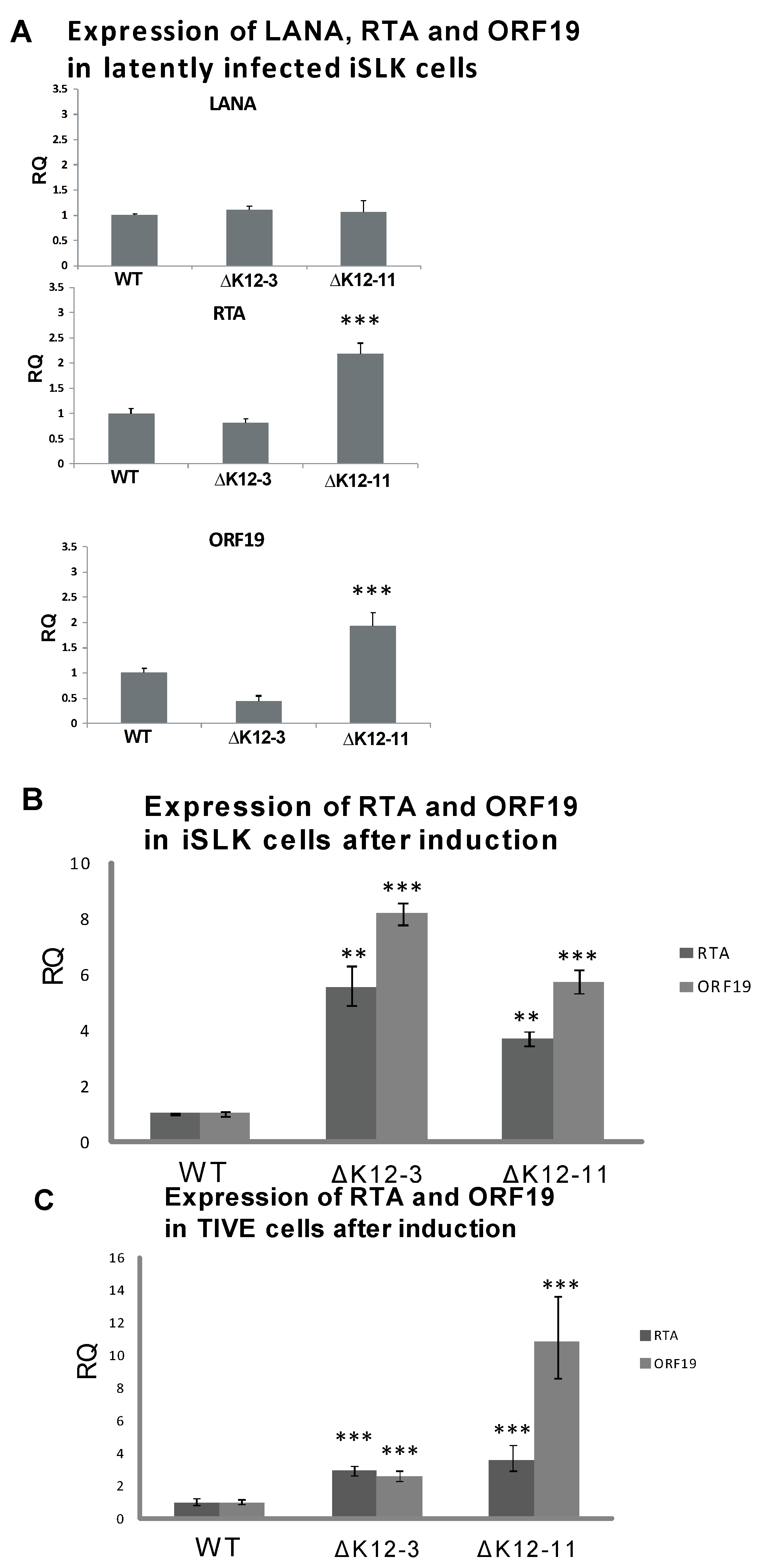
3.3. Generation of KSHV miRNA Deletion Mutants and Latently Infected iSLK Cells
3.4. In Silico Seed Sequence Prediction for miR-K12-3 and miR-K12-11 Identified MYB, C/EBPα, and Ets-1 as Potential Targets
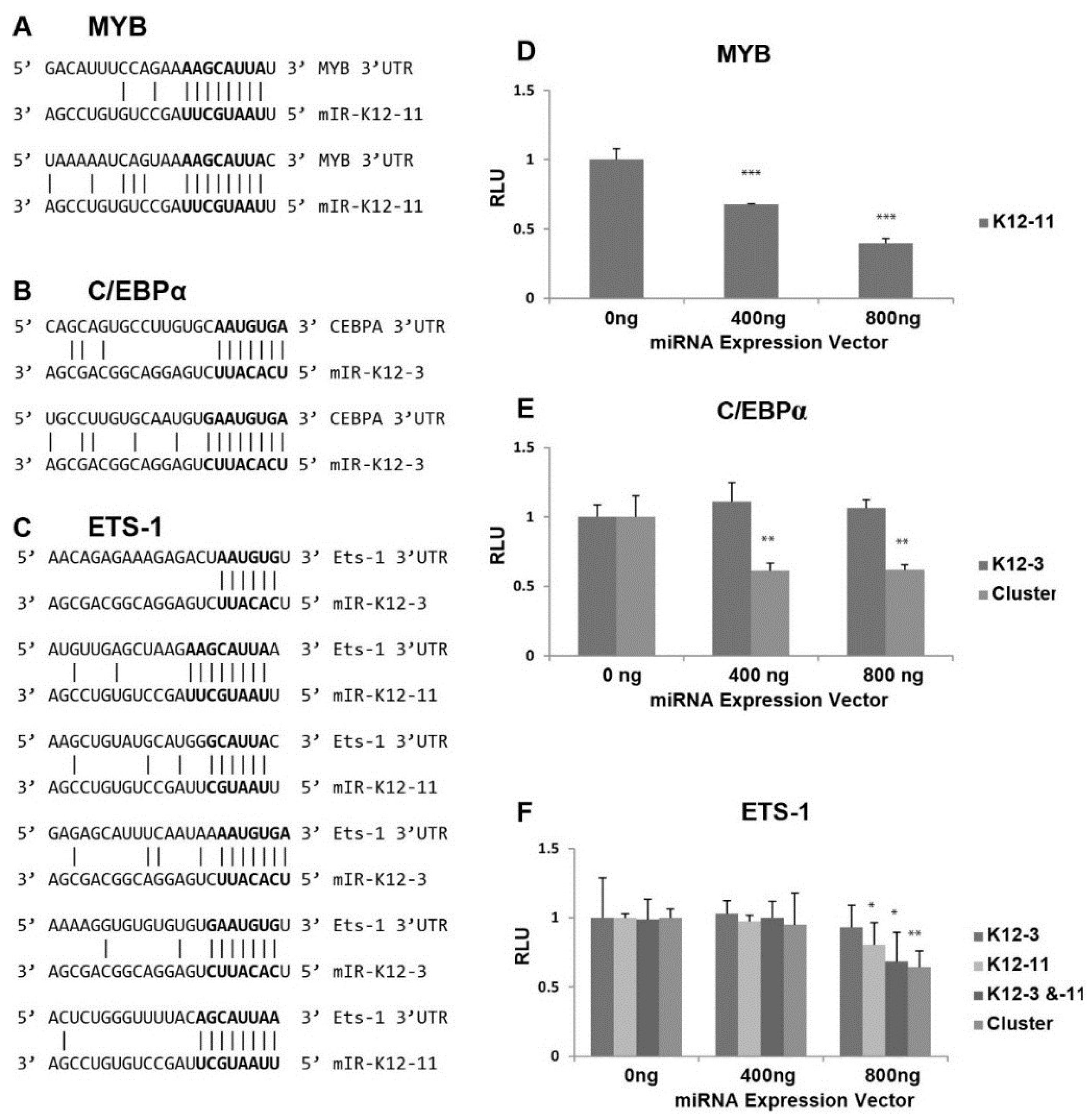
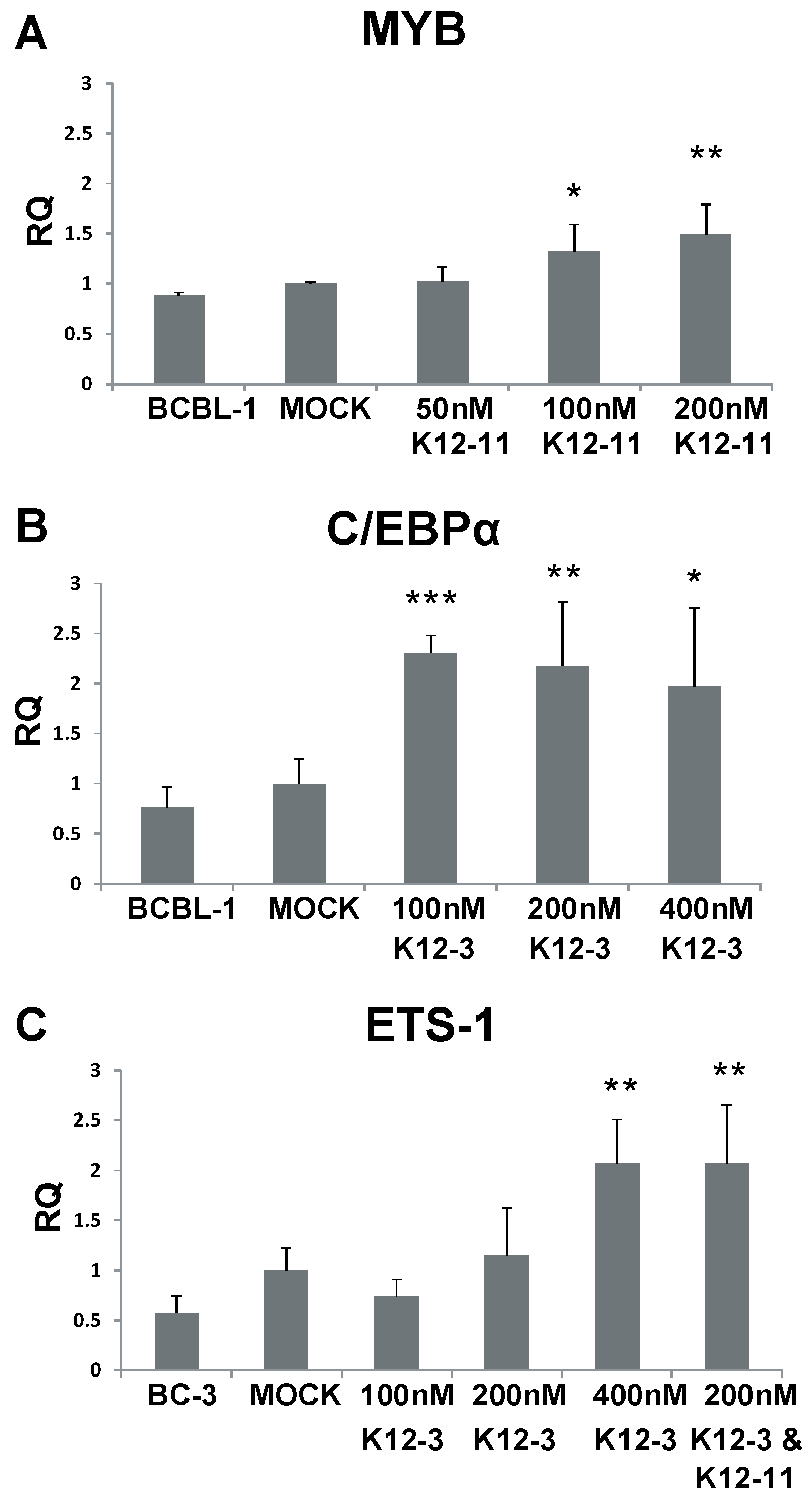
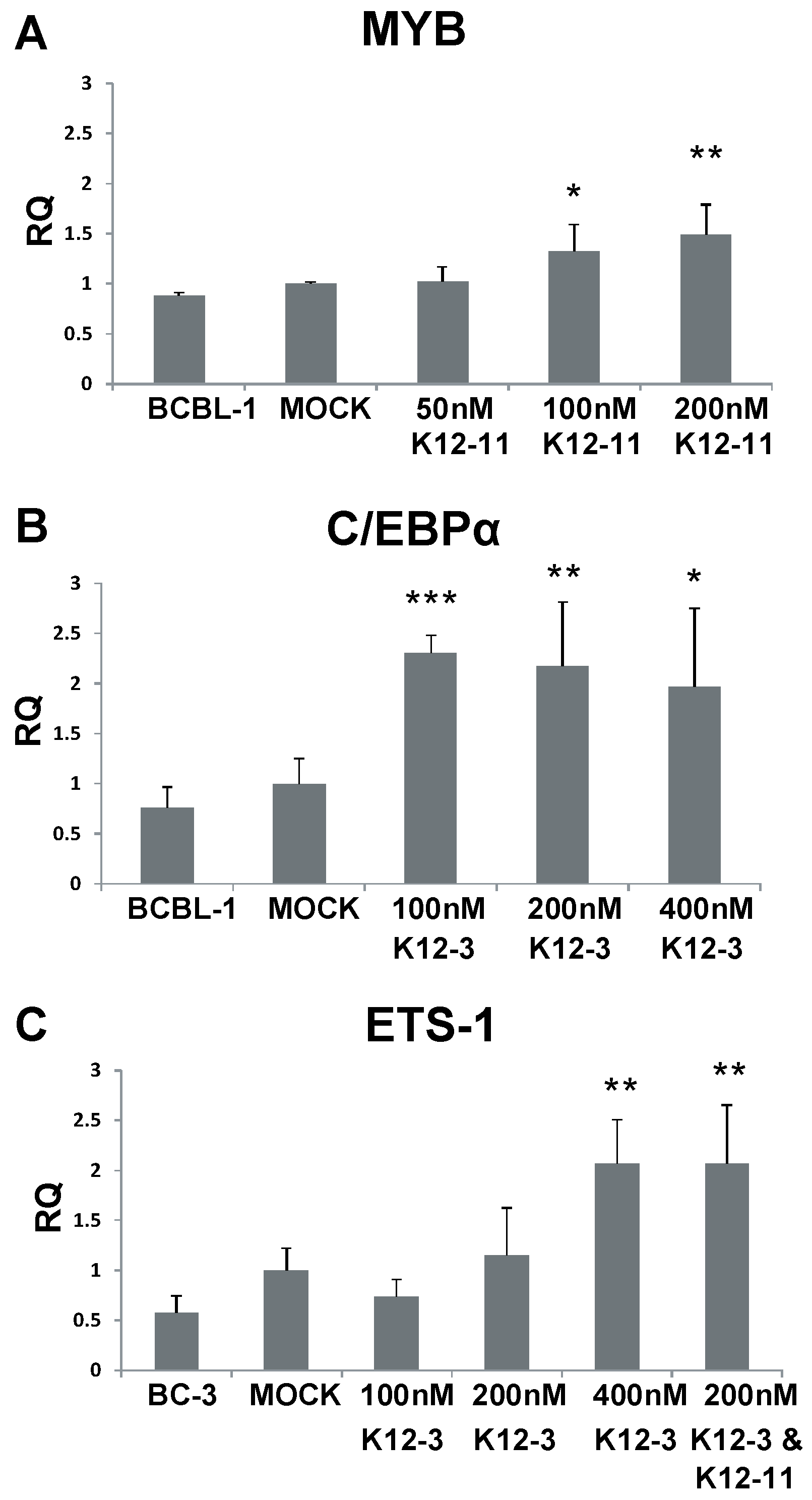
3.5. MYB, C/EBPα and Ets-1 Expression Is Increased upon miRNA Knockdown in PEL Cells
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma (see comments). Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Dittmer, D.; Lagunoff, M.; Renne, R.; Staskus, K.; Haase, A.; Ganem, D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1998, 72, 8309–8315. [Google Scholar] [PubMed]
- Sarid, R.; Flore, O.; Bohenzky, R.A.; Chang, Y.; Moore, P.S. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 1998, 72, 1005–1012. [Google Scholar] [PubMed]
- Talbot, S.J.; Weiss, R.A.; Kellam, P.; Boshoff, C. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 1999, 257, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Rivas, C.; Thlick, A.E.; Parravicini, C.; Moore, P.S.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 2001, 75, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Chandriani, S.; Ganem, D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2010, 84, 5565–5573. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Renne, R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 9301–9305. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Sullivan, C.S.; Ganem, D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 2006, 12, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.J.; Vahrson, W.; Dittmer, D.P. Gene alteration and precursor and mature microRNA transcription changes contribute to the miRNA signature of primary effusion lymphoma. Blood 2008, 111, 2347–2353. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.J.; Chugh, P.; Wang, L.; Netto, E.M.; Luz, E.; Harrington, W.J.; Dezube, B.J.; Damania, B.; Dittmer, D.P. Pre-micro RNA signatures delineate stages of endothelial cell transformation in Kaposi sarcoma. PLoS Pathog. 2009, 5, e1000389. [Google Scholar] [CrossRef] [PubMed]
- Moody, R.; Zhu, Y.; Huang, Y.; Cui, X.; Jones, T.; Bedolla, R.; Lei, X.; Bai, Z.; Gao, S.J. KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog. 2013, 9, e1003857. [Google Scholar] [CrossRef] [PubMed]
- Plaisance-Bonstaff, K.; Renne, R. Viral miRNAs. Methods Mol. Biol. 2011, 721, 43–66. [Google Scholar] [PubMed]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Haecker, I.; Yang, Y.; Gao, S.J.; Renne, R. gamma-Herpesvirus-encoded miRNAs and their roles in viral biology and pathogenesis. Curr. Opin. Virol. 2013, 3, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Vanicek, J.; Robins, H.; Shenk, T.; Levine, A.J. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: Implications for latency. Proc. Natl. Acad. Sci. USA 2008, 105, 5453–5458. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; Tuschl, T. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Pfuhl, T.; Mamiani, A.; Ehses, C.; Roemer, K.; Kremmer, E.; Jaker, C.; Hock, J.; Meister, G.; Grasser, F.A. Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic Acids Res. 2008, 36, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, J.M.; Sullivan, C.S.; Ganem, D. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat. Genet. 2009, 41, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma- associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [PubMed]
- Lu, F.; Stedman, W.; Yousef, M.; Renne, R.; Lieberman, P.M. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 2010, 84, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- b>Lin, X.; Liang, D.; He, Z.; Deng, Q.; Robertson, E.S.; Lan, K. miR-K12–7-5p encoded by Kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS One 2011, 6, e16224. [Google Scholar]
- Yu, F.; Harada, J.N.; Brown, H.J.; Deng, H.; Song, M.J.; Wu, T.T.; Kato-Stankiewicz, J.; Nelson, C.G.; Vieira, J.; Tamanoi, F.; et al. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 2007, 3, e44. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, V.; Nicot, C.; Gessain, A.; Valensi, F.; Gabarre, J.; Matta, H.; Chaudhary, P.M.; Mahieux, R. In primary effusion lymphoma cells, MYB transcriptional repression is associated with v-FLIP expression during latent KSHV infection while both v-FLIP and v-GPCR become involved during the lytic cycle. Br. J. Haematol. 2007, 138, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Wu, F.Y.; Yu, Y.; Hayward, G.S. CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 2003, 77, 9590–9612. [Google Scholar] [CrossRef] [PubMed]
- Myoung, J.; Ganem, D. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: Maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 2011, 174, 12–21. [Google Scholar] [CrossRef] [PubMed]
- An, F.Q.; Folarin, H.M.; Compitello, N.; Roth, J.; Gerson, S.L.; McCrae, K.R.; Fakhari, F.D.; Dittmer, D.P.; Renne, R. Long-term-infected telomerase-immortalized endothelial cells: A model for Kaposi’s sarcoma-associated herpesvirus latency in vitro and in vivo. J. Virol. 2006, 80, 4833–4846. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Samols, M.A.; Plaisance, K.B.; Boss, I.W.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J. Virol. 2007, 81, 12836–12845. [Google Scholar] [CrossRef] [PubMed]
- Boss, I.W.; Nadeau, P.E.; Abbott, J.R.; Yang, Y.; Mergia, A.; Renne, R. A KSHV encoded ortholog of miR-155 induces human splenic B-cell expansion in NOD/LtSz-scid IL2R{gamma}null mice. J. Virol. 2011, 85, 9877–9886. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Bai, Z.; Ye, F.; Xie, J.; Kim, C.G.; Huang, Y.; Gao, S.J. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 2010, 12, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Uranishi, H.; Kruhlak, M.; Pilkington, G.R.; Massimelli, M.J.; Bear, J.; Pavlakis, G.N.; Felber, B.K.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesvirus ORF57 interacts with cellular RNA export cofactors RBM15 and OTT3 to promote expression of viral ORF59. J. Virol. 2011, 85, 1528–1540. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.M.; Wilson, A.C. Wide-scale use of Notch signaling factor CSL/RBP-Jkappa in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J. Virol. 2010, 84, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Brulois, K.; Toth, Z.; Wong, L.-Y.; Feng, P.; Gao, S.-J.; Ensser, A.; Jung, J.U. Kaposi’s Sarcoma-Associated Herpesvirus K3 and K5 Ubiquitin E3 Ligases Have Stage-Specific Immune Evasion Roles during Lytic Replication. J. Virol. 2014, 88, 9335–9349. [Google Scholar] [CrossRef] [PubMed]
- Brulois, K.F.; Chang, H.; Lee, A.S.; Ensser, A.; Wong, L.Y.; Toth, Z.; Lee, S.H.; Lee, H.R.; Myoung, J.; Ganem, D.; et al. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 2012, 86, 9708–9720. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Myoung, J.; Ganem, D. Infection of lymphoblastoid cell lines by Kaposi’s sarcoma-associated herpesvirus: Critical role of cell-associated virus. J. Virol. 2011, 85, 9767–9777. [Google Scholar] [CrossRef] [PubMed]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Maldonado, A.M.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Corcoran, D.L.; Mukherjee, N.; Skalsky, R.L.; Hafner, M.; Nusbaum, J.D.; Shamulailatpam, P.; Love, C.L.; Dave, S.S.; Tuschl, T.; et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 2011, 10, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Haecker, I.; Gay, L.A.; Yang, Y.; Hu, J.; Morse, A.M.; McIntyre, L.; Renne, R. Ago-HITS-CLIP Expands Understanding of Kaposi’s Sarcoma-associated Herpesvirus miRNA Function in Primary Effusion Lymphomas. PLoS Pathog. 2012, 8, e1002884. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Mukherjee, N.; Sachse, C.; Frenzel, C.; Majoros, W.H.; Chi, J.T.; Braich, R.; Manoharan, M.; Soutschek, J.; Ohler, U.; et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature 2007, 450, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Krueger, B.; Plaisance, K.; Sangani, R.; Lanier, C.; Jain, V.; Hu, J.; Renne, R. A core laboratory for the generation of quality-controlled g-herpesvirus bacmids: Generation of KSHV miRNA mutants. Infect. Agents Cancer 2012, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Krueger, B.; Jain, V.; Plaisance-Bonstaff, K.; Renne, R. Generation of quality-controlled γ-herpesvirus bacmids to create KSHV miRNA mutants. University of Florida: Gainesville, FL, USA, Unpublished work. 2014. [Google Scholar]
- Sturzl, M.; Gaus, D.; Dirks, W.G.; Ganem, D.; Jochmann, R. Kaposi’s sarcoma-derived cell line SLK is not of endothelial origin, but is a contaminant from a known renal carcinoma cell line. Int. J. Cancer 2013, 132, 1954–1958. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Koschmieder, S.; Halmos, B.; Levantini, E.; Tenen, D.G. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J. Clin. Oncol. 2009, 27, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Wasylyk, B.; Hagman, J.; Gutierrez-Hartmann, A. Ets transcription factors: Nuclear effectors of the Ras-MAP-kinase signaling pathway. Trends Biochem. Sci. 1998, 23, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Taddei, A.; Randi, A.M. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim. Biophys. Acta 2007, 1775, 298–312. [Google Scholar] [PubMed]
- Gottwein, E.; Cai, X.; Cullen, B.R. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J. Virol. 2006, 80, 5321–5326. [Google Scholar] [CrossRef] [PubMed]
- Marshall, V.; Parks, T.; Bagni, R.; Wang, C.D.; Samols, M.A.; Hu, J.; Wyvil, K.M.; Aleman, K.; Little, R.F.; Yarchoan, R.; et al. Conservation of virally encoded microRNAs in Kaposi sarcoma—Associated herpesvirus in primary effusion lymphoma cell lines and in patients with Kaposi sarcoma or multicentric Castleman disease. J. Infect. Dis. 2007, 195, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Marshall, V.; Barsov, E.; Quinones, O.; Ray, A.; Labo, N.; Trivett, M.; Ott, D.; Renne, R.; Whitby, D. Kaposi’s sarcoma-associated herpesvirus microRNA single-nucleotide polymorphisms identified in clinical samples can affect microRNA processing, level of expression, and silencing activity. J. Virol. 2013, 87, 12237–12248. [Google Scholar] [CrossRef] [PubMed]
- Bellare, P.; Ganem, D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: An evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 2009, 6, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Boss, I.; McIntyre, L.; Renne, R. A systems biology approach identified different regulatory networks targeted by KSHV miR-K12-11 in B cells and endothelial cells. BMC Genomics 2014, 15, 668. [Google Scholar] [CrossRef] [PubMed]
- Reese, T.A.; Wakeman, B.S.; Choi, H.S.; Hufford, M.M.; Huang, S.C.; Zhang, X.; Buck, M.D.; Jezewski, A.; Kambal, A.; Liu, C.Y.; et al. Helminth infection reactivates latent gamma-herpesvirus via cytokine competition at a viral promoter. Science 2014, 345, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Speck, S.H.; Ganem, D. Viral latency and its regulation: Lessons from the gamma-herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef] [PubMed]
- An, F.Q.; Compitello, N.; Horwitz, E.; Sramkoski, M.; Knudsen, E.S.; Renne, R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J. Biol. Chem. 2005, 280, 3862–3874. [Google Scholar] [CrossRef] [PubMed]
- Baccarini, A.; Chauhan, H.; Gardner, T.J.; Jayaprakash, A.D.; Sachidanandam, R.; Brown, B.D. Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr. Biol. 2011, 21, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, S.; Ebert, M.S.; Zheng, G.X.; Tsang, J.S.; Sharp, P.A.; van Oudenaarden, A. MicroRNAs can generate thresholds in target gene expression. Nat. Genet. 2011, 43, 854–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plaisance-Bonstaff, K.; Choi, H.S.; Beals, T.; Krueger, B.J.; Boss, I.W.; Gay, L.A.; Haecker, I.; Hu, J.; Renne, R. KSHV miRNAs Decrease Expression of Lytic Genes in Latently Infected PEL and Endothelial Cells by Targeting Host Transcription Factors. Viruses 2014, 6, 4005-4023. https://doi.org/10.3390/v6104005
Plaisance-Bonstaff K, Choi HS, Beals T, Krueger BJ, Boss IW, Gay LA, Haecker I, Hu J, Renne R. KSHV miRNAs Decrease Expression of Lytic Genes in Latently Infected PEL and Endothelial Cells by Targeting Host Transcription Factors. Viruses. 2014; 6(10):4005-4023. https://doi.org/10.3390/v6104005
Chicago/Turabian StylePlaisance-Bonstaff, Karlie, Hong Seok Choi, Tyler Beals, Brian J. Krueger, Isaac W. Boss, Lauren A. Gay, Irina Haecker, Jianhong Hu, and Rolf Renne. 2014. "KSHV miRNAs Decrease Expression of Lytic Genes in Latently Infected PEL and Endothelial Cells by Targeting Host Transcription Factors" Viruses 6, no. 10: 4005-4023. https://doi.org/10.3390/v6104005