Avian Influenza: Mixed Infections and Missing Viruses

Abstract
:1. Introduction
2. Results
2.1. AI Matrix Real-Time PCR of VTM and ALF Samples

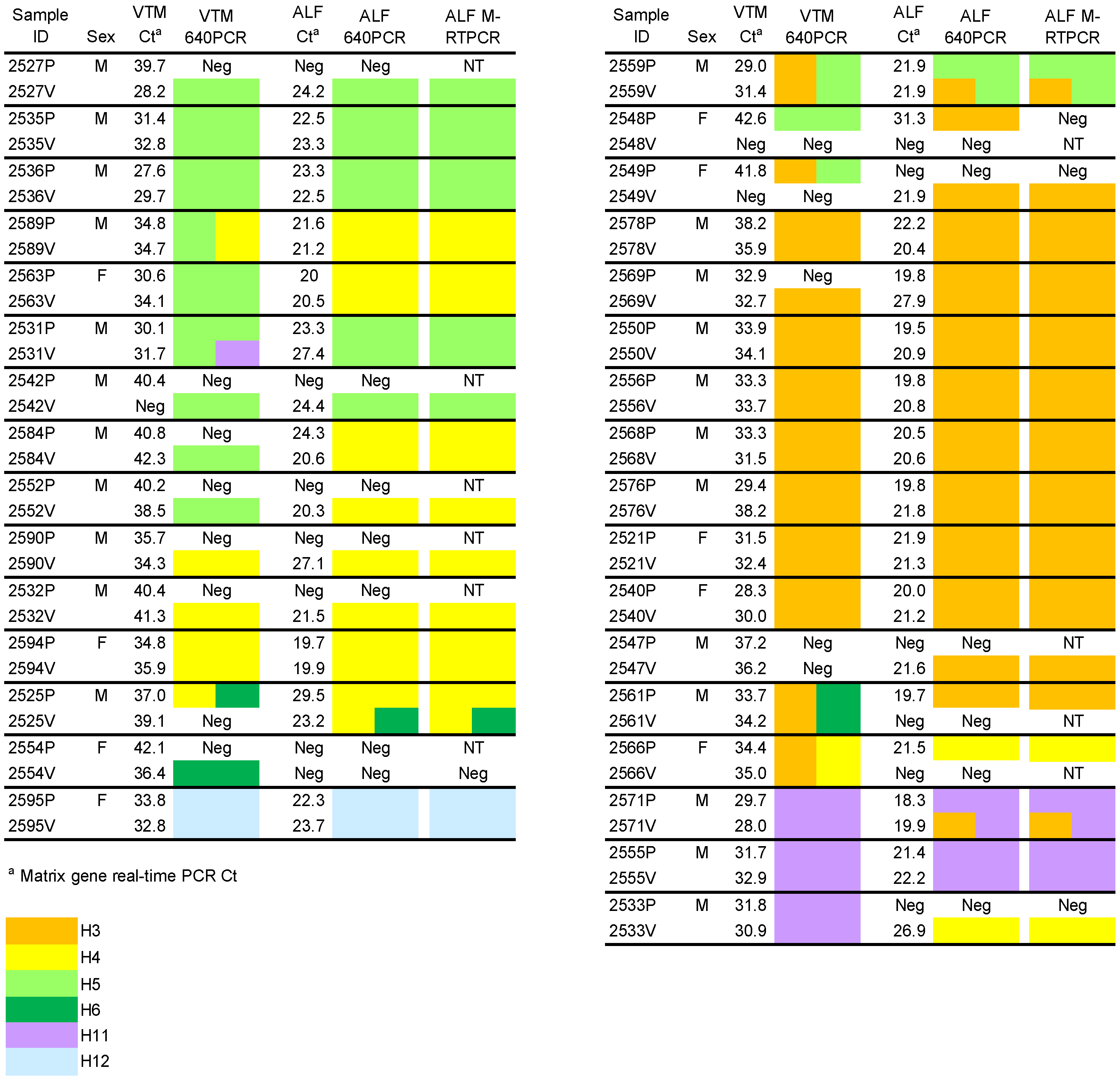{kind=link}
{kind=link}
| Sample Unit | Pre-inoculation (VTM) | Post-inoculation (ALF) | ||||
|---|---|---|---|---|---|---|
| Matrix Ct < 45 | Matrix Ct < 35 | HA positive (640PCR) | Matrix Ct < 45 | 640PCR HA positive | M-RTPCR HA positive | |
| P swab | 56% (44/79) | 25% (20/79) | 29% (23/79) | 29% (23/79) | 29% (23/79) | 28% (22/79) |
| V swab | 47% (37/79) | 24% (19/79) | 35% (28/79) | 35% (28/79) | 35% (28/79) | 35% (28/79) |
| P and V swabs | 51% (81/158) | 25% (39/158) | 32% (51/158) | 32% (51/158) | 32% (51/158) | 32% (51/158) |
| Birds | 58% (46/79) | 28% (22/79) | 39% (31/79) | 39% (31/79) | 39% (31/79) | 38% (30/79) |
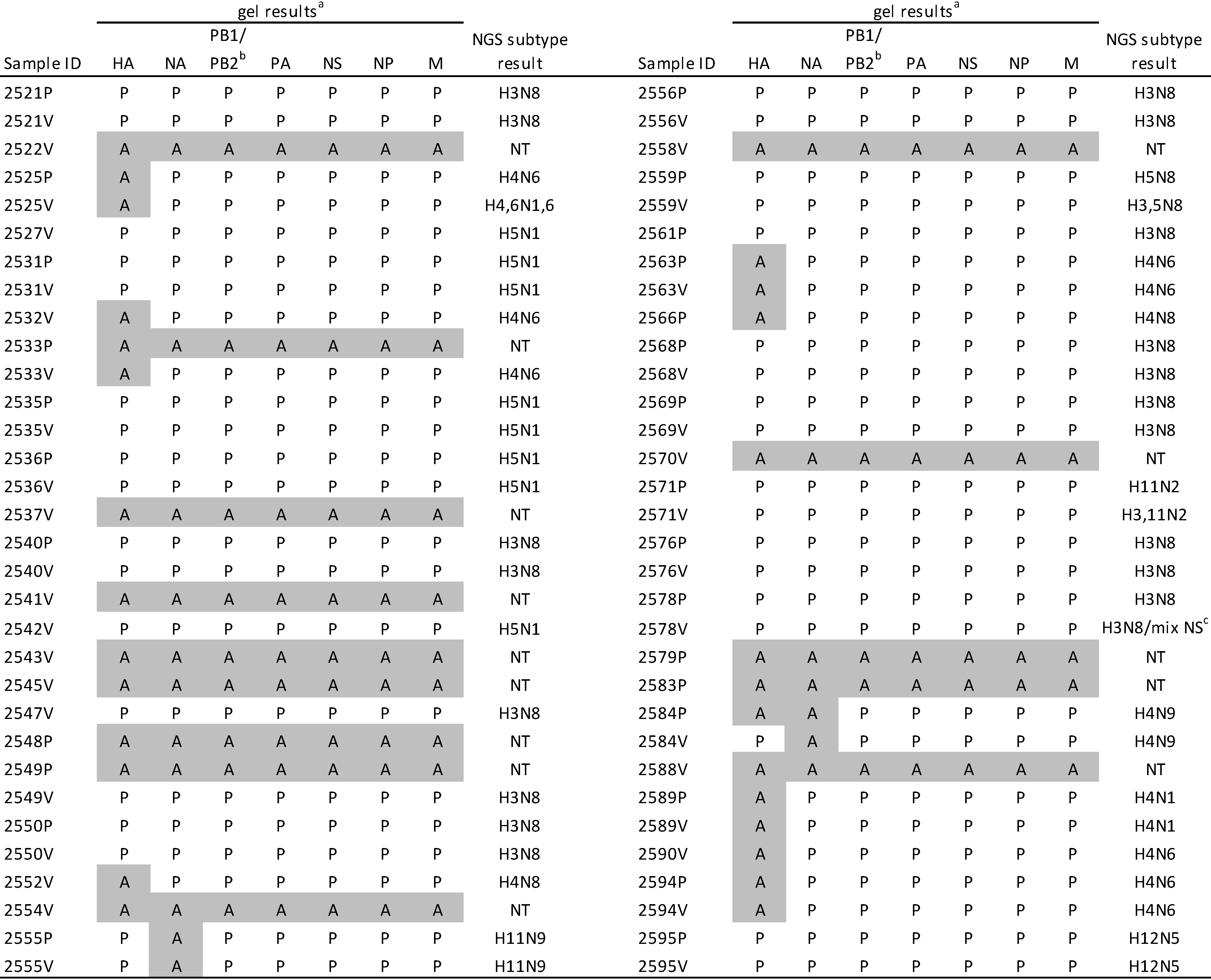
2.2. HA Subtyping
2.3. M-RTPCR Virus Segment Amplification
2.4. Sequence Analysis of Duplicate Samples from Birds
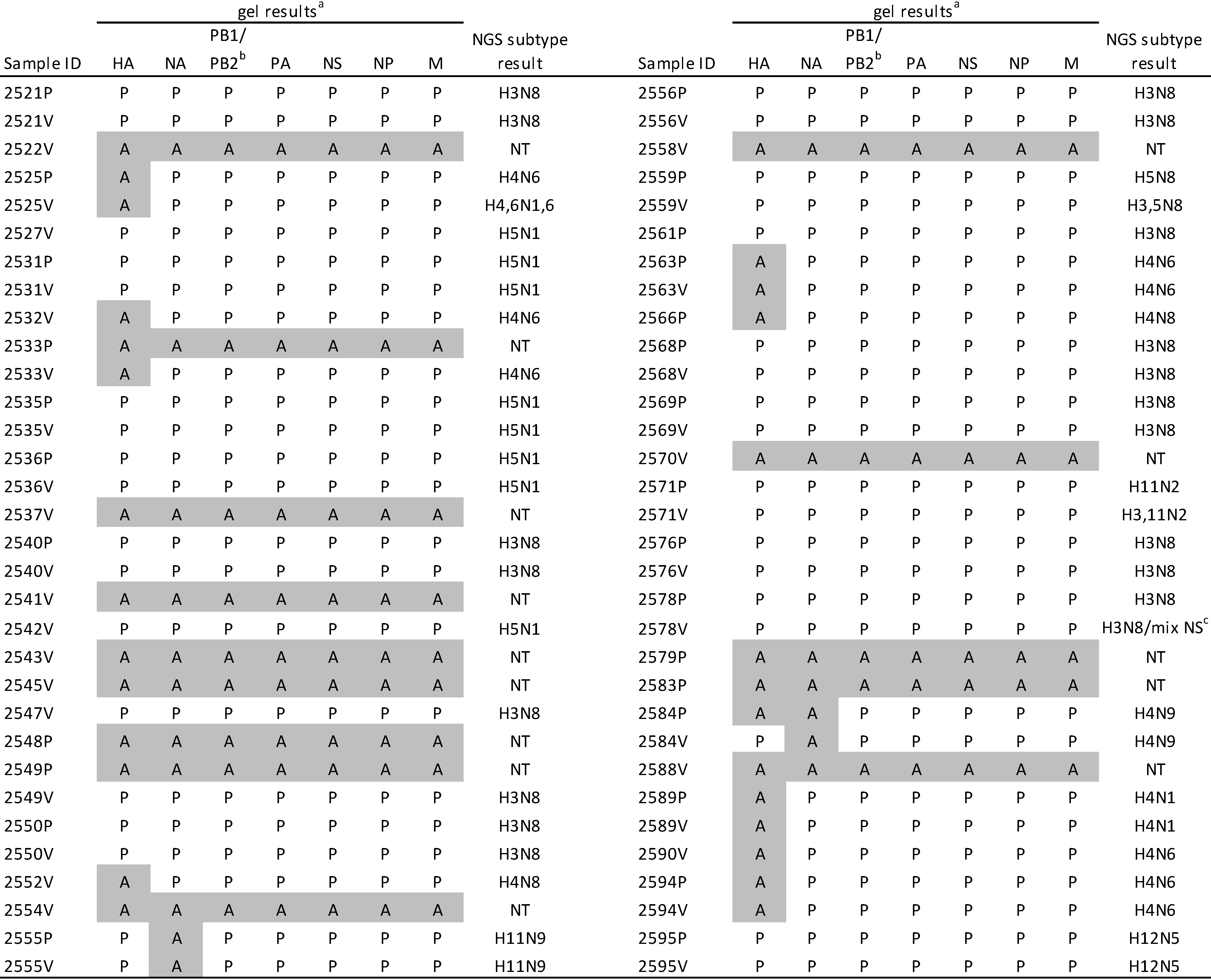
| Bird ID | NGS Subtype | PB2a % identity | PB1 % identity | PA % identity | HA % identity | NP % identity | NA % identity | M % identity | NS % identity |
|---|---|---|---|---|---|---|---|---|---|
| 2521P | H3N8 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| 2521V | H3N8 | ||||||||
| 2525P | H4N6 | 99.96 | 100 | 100 | 100 (H4) | 100 (P,Va)b | 100 | 100 | 100 |
| 2525V | H4,6N1,6 | 92.22 (P,Vb) | |||||||
| 2531P | H5N1 | 98.14 | 100 | 99.77 | 99.94 | 91.76 | 100 | 98.8 | 93.43 |
| 2531V | H5N1 | ||||||||
| 2535P | H5N1 | 100 | 100 | 99.91 | 100 | 100 | 100 | 100 | 100 |
| 2535V | H5N1 | ||||||||
| 2536P | H5N1 | 100 | 99.91 | 100 | 99.94 | 99.93 | 100 | 100 | 100 |
| 2536V | H5N1 | ||||||||
| 2540P | H3N8 | 98.92 | 99.31 | 99.82 | 99.94 | 99.93 | 100 | 100 | 100 |
| 2540V | H3N8 | ||||||||
| 2550P | H3N8 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| 2550V | H3N8 | ||||||||
| 2555P | H11N9 | 100 | 100 | 100 | 99.94 | 100 | 100 | 100 | 99.88 |
| 2555V | H11N9 | ||||||||
| 2556P | H3N8 | 100 | 99.91 | 100 | 100 | 100 | 100 | 100 | 100 |
| 2556V | H3N8 | ||||||||
| 2559P | H5N8 | 99.05 | 96.79 | 96.51 | 99.94 (H5) | 91.63 (P,Va) | 100 | 97.6 | 93.53 |
| 2559V | H3,5N8 | 99.93 (P,Vb) | |||||||
| 2563P | H4N6 | 100 | 99.91 | 100 | 99.94 | 91.9 | 99.72 | 98 | 100 |
| 2563V | H4N6 | ||||||||
| 2568P | H3N8 | 100 | 99.96 | 100 | 100 | 100 | 100 | 100 | 100 |
| 2568V | H3N8 | ||||||||
| 2569P | H3N8 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| 2569V | H3N8 | ||||||||
| 2571P | H11N2 | 100 | 100 | 100 | 99.88 (H11) | 100 | 100 | 100 | 100 |
| 2571V | H3,11N2 | ||||||||
| 2576P | H3N8 | 100 | 100 | 100 | 100 | 100 | 99.93 | 100 | 100 |
| 2576V | H3N8 | ||||||||
| 2578P | H3N8 | 100 | 97 | 99.95 | 100 | 98.69 | 99.93 | 100 | 100 (P,Va) |
| 2578V | H3N8 mix | 93.87 (P,Vb) | |||||||
| 2584P | H4N9 | 99.96 | 100 | 100 | 99.71 | 100 | 100 | 100 | 100 |
| 2584V | H4N9 | ||||||||
| 2589P | H4N1 | 99.91 | 100 | 100 | 99.94 | 100 | 100 | 99.9 | 100 |
| 2589V | H4N1 | ||||||||
| 2594P | H4N6 | 100 | 100 | 100 | 100 | 100 | 100 | 99.9 | 100 |
| 2594V | H4N6 | ||||||||
| 2595P | H12N5 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| 2595V | H12N5 |
3. Discussion
4. Conclusions
5. Experimental Section
5.1. Samples
5.2. RNA Isolation and Reverse Transcription
5.3. Influenza A PCR
5.4. Virus Isolation
5.5. HA Subtyping
5.6. Statistical Analyses
Acknowledgments
Conflict of Interest
References and Notes
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenström, J.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Global patterns of in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef]
- Dugan, V.G.; Chen, R.; Spiro, D.J.; Sengamalay, N.; Zaborsky, J.; Ghedin, E.; Nolting, J.; Swayne, D.E.; Runstadler, J.A.; Happ, G.M.; et al. The evolutionary genetics and emergence of avian influenza viruses in wild birds. PLoS Pathog. 2008, 4, e1000076. [Google Scholar] [CrossRef]
- Deliberto, T.J.; Swafford, S.R.; Nolte, D.L.; Pedersen, K.; Lutman, M.W.; Schmit, B.B.; Baroch, J.A.; Kohler, D.J.; Franklin, A. Surveillance for highly pathogenic avian influenza in wild birds in the USA. Integr. Zool. 2009, 4, 426–439. [Google Scholar] [CrossRef]
- Suarez, D.L.; Das, A.; Ellis, E. Review of rapid molecular diagnostic tools for avian influenza virus. Avian Dis. 2007, 51, 201–208. [Google Scholar] [CrossRef]
- Lira, J.; Moresco, K.A.; Stallknecht, D.E.; Swayne, D.E.; Fisher, D.S. Single and combination diagnostic test efficiency and cost analysis for detection and isolation of avian influenza virus from wild bird cloacal swabs. Avian Dis. 2010, 54, 606–612. [Google Scholar] [CrossRef]
- Munster, V.J.; Baas, C.; Lexmond, P.; Bestebroer, T.M.; Guldemeester, J.; Beyer, W.E.; de Wit, E.; Schutten, M.; Rimmelzwaan, G.F.; Osterhaus, A.D.; et al. Practical considerations for high-throughput influenza A virus surveillance studies of wild birds by use of molecular diagnostic tests. J. Clin. Microbiol. 2009, 47, 666–673. [Google Scholar] [CrossRef]
- Brown, J.D.; Poulson, R.; Carter, D.L.; Lebarbenchon, C.; Stallknecht, D.E. Infectivity of avian influenza virus-positive field samples for mallards: What do our diagnostic results mean? J. Wildlife Dis. 2013, 49, 180–185. [Google Scholar] [CrossRef]
- Moresco, K.A.; Stallknecht, D.E.; Swayne, D.E. Avian embryos, real-time polymerase chain reaction, and cell culture for detection of avian influenza and Newcastle disease virus in wild bird surveillance samples. J. Vet. Diagn. Invest. 2012, 24, 563–567. [Google Scholar] [CrossRef]
- Stallknecht, D.E.; Luttrell, M.P.; Poulson, R.; Goekjian, V.; Niles, L.; Dey, A.; Krauss, S.; Webster, R.G. Detection of avian influenza viruses from shorebirds: Evaluation of surveillance and testing approaches. J. Wildlife Dis. 2012, 48, 382–393. [Google Scholar]
- Runstadler, J.A.; Happ, G.M.; Slemons, R.D.; Sheng, Z.M.; Gundlach, N.; Petrula, M.; Senne, D.; Nolting, J.; Evers, D.L.; Modrell, A.; et al. Using RRT-PCR analysis and virus isolation to determine the prevalence of avian influenza virus infections in ducks at Minto Flats State Game Refuge, Alaska, during August 2005. Arch. Virol. 2007, 152, 1901–1910. [Google Scholar] [CrossRef]
- Swayne, D.E.; Senne, D.A.; Suarez, D.L. Avian influenza. In A Laboratory Manual for the Isolation and Identification of Avian Pathogens, 5th ed.; Dufour-Zavala, L., Swayne, D.E., Glisson, J.R., Pearson, J.E., Reed, W.M., Jackwood, M.W., Woolcock, P.R., Eds.; American Association of Avian Pathologists: Kennett Square, PA, USA, 2008; pp. 128–134. [Google Scholar]
- Moresco, K.A.; Stallknecht, D.E.; Swayne, D.E. Evaluation and attempted optimization of avian embryos and cell culture methods for efficient isolation and propagation of low pathogenicity avian influenza viruses. Avian Dis. 2010, 54, 622–626. [Google Scholar] [CrossRef]
- Wang, R.; Soll, L.; Dugan, V.; Runstadler, J.; Happ, G.; Slemons, R.D.; Taubenberger, J.K. Examining the hemagglutinin subtype diversity among wild duck-origin influenza A viruses using ethanol-fixed cloacal swabs and a novel RT-PCR method. Virology 2008, 375, 182–189. [Google Scholar] [CrossRef]
- Schild, G.C.; Oxford, J.S.; de Jong, J.C.; Webster, R.G. Evidence for host-cell selection of influenza virus antigenic variants. Nature 1983, 303, 706–709. [Google Scholar] [CrossRef]
- Woolcock, P.R.; McFarland, M.D.; Lai, S.; Chin, R.P. Enhanced recovery of avian influenza virus isolates by a combination of chicken embryo inoculation methods. Avian Dis. 2001, 45, 1030–1035. [Google Scholar] [CrossRef]
- Varich, N.L.; Gitelman, A.K.; Shilov, A.A.; Smirnov, Y.A.; Kaverin, N.V. Deviation from the random distribution pattern of influenza A virus gene segments in reassortants produced under non-selective conditions. Arch. Virol. 2008, 153, 1149–1154. [Google Scholar] [CrossRef]
- Jindal, N.; Chander, Y.; de Abin, M.; Sreevatsan, S.; Stallknecht, D.; Halvorson, D.A.; Goyal, S.M. Amplification of four genes of influenza A viruses using a degenerate primer set in a one step RT-PCR method. J. Virol. Methods 2009, 160, 163–166. [Google Scholar] [CrossRef]
- Dugan, V.G. A robust tool highlights the influence of bird migration on influenza A virus evolution. Mol. Ecol. 2012, 21, 5905–5907. [Google Scholar] [CrossRef]
- Lubeck, M.D.; Palese, P.; Schulman, J.L. Nonrandom association of parental genes in influenza A virus recombinants. Virology 1979, 95, 269–274. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; von Kirchbach, J.C.; Gog, J.R.; Digard, P. Genome packaging in influenza A virus. J. Gen. Virol. 2010, 91, 313–328. [Google Scholar] [CrossRef]
- Ramakrishnan, M.A.; Tu, Z.J.; Singh, S.; Chockalingam, A.K.; Gramer, M.R.; Wang, P.; Goyal, S.M.; Yang, M.; Halvorson, D.A.; Sreevatsan, S. The feasibility of using hih resolution genome sequencing of influenza A viruses to detect mixed infections and quasispecies. PLoS One 2009, 4, e7105. [Google Scholar] [CrossRef]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Mertens, E.; Dugan, V.G.; Stockwell, T.B.; Lindsay, L.L.; Plancarte, M.; Boyce, W.M. Evaluation of phenotypic markers in full genome sequences of avian influenza isolates from California. Comp. Immunol. Microbiol. Infect. Dis. 2013, in press. [Google Scholar]
- Saira, K.; Lin, X.; Depasse, J.V.; Halpin, R.; Twaddle, A.; Stockwell, T.; Angus, B.; Cozzi-Lepri, A.; Delfino, M.; Dugan, V.; et al. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J. Virol. 2013, 87, 8064–8074. [Google Scholar] [CrossRef]
- Hill, N.J.; Takekawa, J.Y.; Ackerman, J.T.; Hobson, K.A.; Herring, G.; Cardona, C.J.; Runstadler, J.A.; Boyce, W.M. Migration strategy affects avian influenza dynamics in mallards (Anas platyrhynchos). Mol. Ecol. 2012, 21, 5986–5999. [Google Scholar] [CrossRef]
- Krauss, S.; Walker, D.; Webster, R.G. Influenza virus isolation. Method. Mol. Biol. 2012, 865, 11–24. [Google Scholar] [CrossRef]
- Spackman, E.; Senne, D.A.; Myers, T.J.; Bulaga, L.L.; Garber, L.P.; Perdue, M.L.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J. Clin. Microbiol. 2002, 40, 3256–3260. [Google Scholar] [CrossRef]
- Khalenkov, A.; Laver, W.G.; Webster, R.G. Detection and isolation of H5N1 influenza virus from large volumes of natural water. J. Virol. Methods 2008, 149, 180–183. [Google Scholar] [CrossRef]
- Phipps, L.P.; Essen, S.C.; Brown, I.H. Genetic subtyping of influenza A viruses using RT-PCR with a single set of primers based on conserved sequences within the HA2 coding region. J. Virol. Methods 2004, 122, 119–122. [Google Scholar] [CrossRef]
- Bragstad, K.; Jorgensen, P.H.; Handberg, K.J.; Mellergaard, S.; Corbet, S.; Fomsgaard, A. New avian influenza A virus subtype combination H5N7 identified in Danish mallard ducks. Virus Res. 2005, 109, 181–190. [Google Scholar] [CrossRef]
- Basic Local Alignment Search Tool. Available online: http://www.ncbi.nlm.nih.gov/BLAST (accessed on 30 January 2013).
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef]
- Zhou, B.; Wentworth, D.E. Influenza A virus molecular virology techniques. Method. Mol. Biol. 2012, 865, 175–192. [Google Scholar] [CrossRef]
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef]
- R. Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria, 2011. ISBN 3-900051-07-0. Available online: http://www.R-project.org (accessed on 15 January 2011).
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lindsay, L.L.; Kelly, T.R.; Plancarte, M.; Schobel, S.; Lin, X.; Dugan, V.G.; Wentworth, D.E.; Boyce, W.M. Avian Influenza: Mixed Infections and Missing Viruses. Viruses 2013, 5, 1964-1977. https://doi.org/10.3390/v5081964
Lindsay LL, Kelly TR, Plancarte M, Schobel S, Lin X, Dugan VG, Wentworth DE, Boyce WM. Avian Influenza: Mixed Infections and Missing Viruses. Viruses. 2013; 5(8):1964-1977. https://doi.org/10.3390/v5081964
Chicago/Turabian StyleLindsay, LeAnn L., Terra R. Kelly, Magdalena Plancarte, Seth Schobel, Xudong Lin, Vivien G. Dugan, David E. Wentworth, and Walter M. Boyce. 2013. "Avian Influenza: Mixed Infections and Missing Viruses" Viruses 5, no. 8: 1964-1977. https://doi.org/10.3390/v5081964
APA StyleLindsay, L. L., Kelly, T. R., Plancarte, M., Schobel, S., Lin, X., Dugan, V. G., Wentworth, D. E., & Boyce, W. M. (2013). Avian Influenza: Mixed Infections and Missing Viruses. Viruses, 5(8), 1964-1977. https://doi.org/10.3390/v5081964