The Role of Chromatin in Adenoviral Vector Function

{kind=link}
Abstract
:1. Introduction
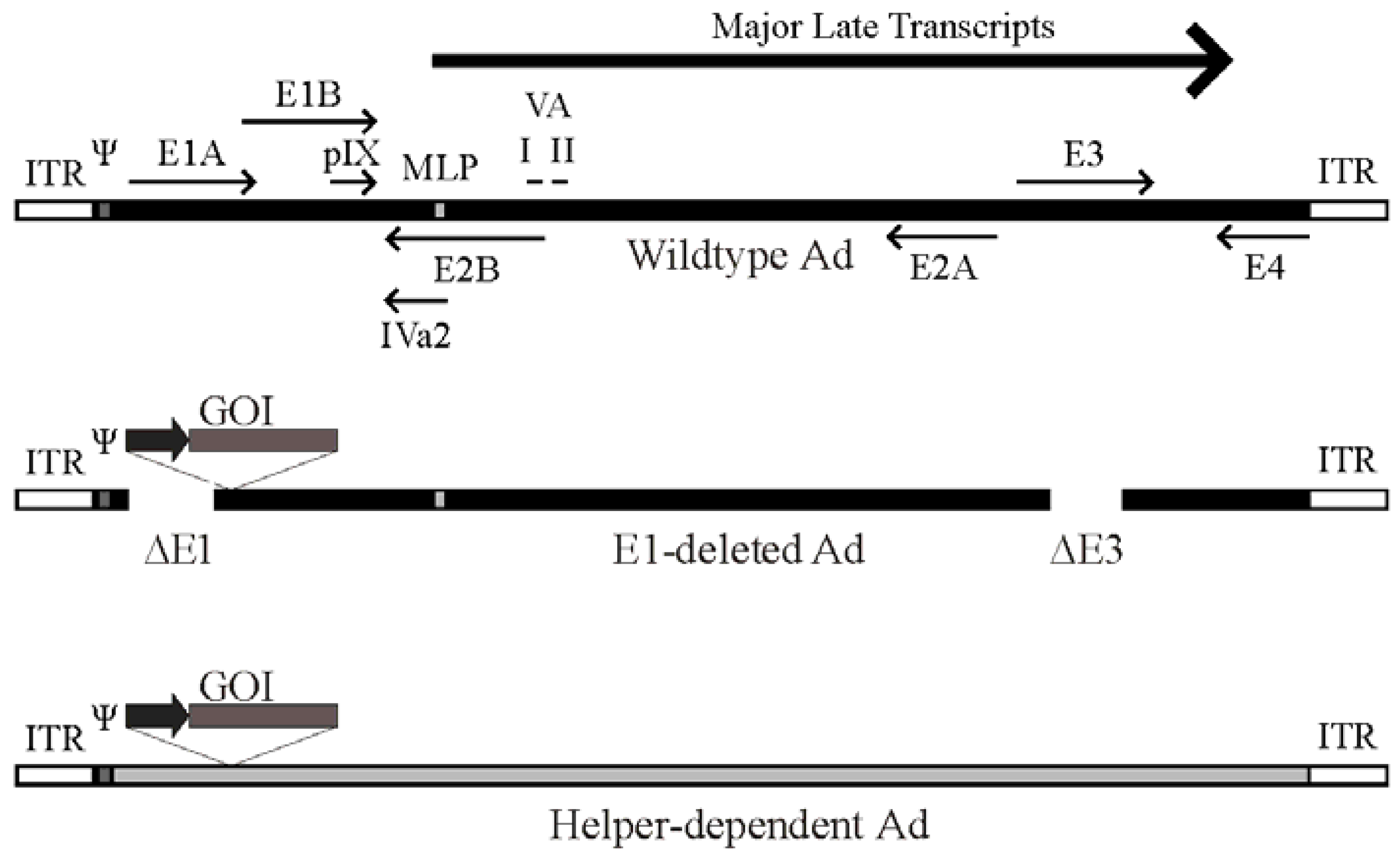
2. Ad Vector Design

3. Ad Virion Structure
4. Ad Infection of a Cell
5. Ad Vector DNA Remains Primarily Episomal within the Infected Cell Nucleus
6. Early Events within the Infected Cell Nucleus
7. Ad Vector DNA Associates with Cellular Histones in the Infected Cell Nucleus
8. Epigenetic Regulation of Ad Vectors
9. Conclusions and Future Perspectives
Acknowledgements
References and Notes
- Hassell, J.A.; Lukanidin, E.; Fey, G.; Sambrook, J. The structure and expression of two defective adenovirus 2/simian virus 40 hybrids. J. Mol. Biol. 1978, 120, 209–247. [Google Scholar] [CrossRef]
- Tjian, R. The binding site on SV40 DNA for a T antigen-related protein. Cell 1978, 13, 165–179. [Google Scholar] [CrossRef]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Giberson, A.N.; Davidson, A.R.; Parks, R.J. Chromatin structure of adenovirus DNA throughout infection. Nucleic Acids Res. 2012, 40, 2369–2376. [Google Scholar] [CrossRef]
- Amalfitano, A.; Parks, R.J. Separating fact from fiction: Assessing the potential of modified adenovirus vectors for use in human gene therapy. Curr. Gene Ther. 2002, 2, 111–133. [Google Scholar] [CrossRef]
- Bett, A.J.; Prevec, L.; Graham, F.L. Packaging capacity and stability of human adenovirus type 5 vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar]
- Nelson, J.E.; Kay, M.A. Persistence of recombinant adenovirus in vivo is not dependent on vector DNA replication. J. Virol. 1997, 71, 8902–8907. [Google Scholar]
- Ahi, Y.S.; Bangari, D.S.; Mittal, S.K. Adenoviral vector immunity: Its implications and circumvention strategies. Curr. Gene Ther. 2011, 11, 307–320. [Google Scholar] [CrossRef]
- Thaci, B.; Ulasov, I.V.; Wainwright, D.A.; Lesniak, M.S. The challenge for gene therapy: Innate immune response to adenoviruses. Oncotarget 2011, 2, 113–121. [Google Scholar]
- Yang, Y.; Nunes, F.A.; Berencsi, K.; Furth, E.E.; Gonczol, E.; Wilson, J.M. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc. Natl. Acad. Sci. USA 1994, 91, 4407–4411. [Google Scholar]
- Berk, A.J. Adenoviridae: The viruses and their replication. In Fields Virology, 5th; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2355–2394. [Google Scholar]
- Thomas, G.P.; Mathews, M.B. DNA replication and the early to late transition in adenovirus infection. Cell 1980, 22, 523–533. [Google Scholar] [CrossRef]
- Parks, R.J.; Chen, L.; Anton, M.; Sankar, U.; Rudnicki, M.A.; Graham, F.L. A helper-dependent adenovirus vector system: Removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. USA 1996, 93, 13565–13570. [Google Scholar]
- Hardy, S.; Kitamura, M.; Harris-Stansil, T.; Dai, Y.; Phipps, M.L. Construction of adenovirus vectors through Cre-lox recombination. J. Virol. 1997, 71, 1842–1849. [Google Scholar]
- Umana, P.; Gerdes, C.A.; Stone, D.; Davis, J.R.; Ward, D.; Castro, M.G.; Lowenstein, P.R. Efficient FLPe recombinase enables scalable production of helper- dependent adenoviral vectors with negligible helper-virus contamination. Nat. Biotechnol. 2001, 19, 582–585. [Google Scholar]
- Ng, P.; Beauchamp, C.; Evelegh, C.; Parks, R.; Graham, F.L. Development of a FLP/frt system for generating helper-dependent adenoviral vectors. Mol. Ther. 2001, 3, 809–815. [Google Scholar] [CrossRef]
- Palmer, D.; Ng, P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 2003, 8, 846–852. [Google Scholar] [CrossRef]
- Schiedner, G.; Morral, N.; Parks, R.J.; Wu, Y.; Koopmans, S.C.; Langston, C.; Graham, F.L.; Beaudet, A.L.; Kochanek, S. Genomic DNA transfer with a high-capacity adenovirus vector results in improved in vivo gene expression and decreased toxicity. Nat. Genet. 1998, 18, 180–183. [Google Scholar]
- Kim, I.H.; Jozkowicz, A.; Piedra, P.A.; Oka, K.; Chan, L. Lifetime correction of genetic deficiency in mice with a single injection of helper-dependent adenoviral vector. Proc. Natl. Acad. Sci. USA 2001, 98, 13282–13287. [Google Scholar]
- Oka, K.; Pastore, L.; Kim, I.H.; Merched, A.; Nomura, S.; Lee, H.J.; Merched-Sauvage, M.; Arden-Riley, C.; Lee, B.; Finegold, M.; et al. Long-term stable correction of low-density lipoprotein receptor- deficient mice with a helper-dependent adenoviral vector expressing the very low-density lipoprotein receptor. Circulation 2001, 103, 1274–1281. [Google Scholar] [CrossRef]
- Toietta, G.; Mane, V.P.; Norona, W.S.; Finegold, M.J.; Ng, P.; McDonagh, A.F.; Beaudet, A.L.; Lee, B. Lifelong elimination of hyperbilirubinemia in the Gunn rat with a single injection of helper-dependent adenoviral vector. Proc. Natl. Acad. Sci. USA 2005, 102, 3930–3935. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Nichols, T.C.; McCorquodale, S.; Merricks, E.; Palmer, D.J.; Beaudet, A.L.; Ng, P. Sustained phenotypic correction of canine hemophilia B after systemic administration of helper-dependent adenoviral vector. Hum. Gene Ther. 2005, 16, 811–820. [Google Scholar] [CrossRef]
- Morral, N.; O'Neal, W.; Rice, K.; Leland, M.; Kaplan, J.; Piedra, P.A.; Zhou, H.; Parks, R.J.; Velji, R.; Aguilar-Cordova, E.; et al. Administration of helper-dependent adenoviral vectors and sequential delivery of different vector serotype for long-term liver-directed gene transfer in baboons. Proc. Natl. Acad. Sci. USA 1999, 96, 12816–12821. [Google Scholar] [CrossRef]
- Morral, N.; Parks, R.J.; Zhou, H.; Langston, C.; Schiedner, G.; Quinones, J.; Graham, F.L.; Kochanek, S.; Beaudet, A.L. High doses of a helper-dependent adenoviral vector yield supraphysiological levels of alpha1-antitrypsin with negligible toxicity. Hum. Gene Ther. 1998, 9, 2709–2716. [Google Scholar] [CrossRef]
- Muruve, D.A.; Cotter, M.J.; Zaiss, A.K.; White, L.R.; Liu, Q.; Chan, T.; Clark, S.A.; Ross, P.J.; Meulenbroek, R.A.; Maelandsmo, G.M.; et al. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J. Virol. 2004, 78, 5966–5972. [Google Scholar] [CrossRef]
- Parks, R.J. Improvements in adenoviral vector technology: Overcoming barriers for gene therapy. Clin. Genet. 2000, 58, 1–11. [Google Scholar] [CrossRef]
- Palmer, D.J.; Ng, P. Helper-dependent adenoviral vectors for gene therapy. Hum. Gene Ther. 2005, 16, 1–16. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Ng, P. Progress and prospects: Gene therapy for genetic diseases with helper-dependent adenoviral vectors. Gene Ther. 2008, 15, 553–560. [Google Scholar] [CrossRef]
- Parks, R.J.; Evelegh, C.M.; Graham, F.L. Use of helper-dependent adenoviral vectors of alternative serotypes permits repeat vector administration. Gene Ther. 1999, 6, 1565–1573. [Google Scholar] [CrossRef]
- Smith, A.C.; Poulin, K.L.; Parks, R.J. DNA genome size affects the stability of the adenovirus virion. J. Virol. 2009, 83, 2025–2028. [Google Scholar] [CrossRef]
- Christensen, J.B.; Byrd, S.A.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Imperiale, M.J. Presence of the adenovirus IVa2 protein at a single vertex of the mature virion. J. Virol. 2008, 82, 9086–9093. [Google Scholar] [CrossRef]
- Liu, H.; Jin, L.; Koh, S.B.; Atanasov, I.; Schein, S.; Wu, L.; Zhou, Z.H. Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 2010, 329, 1038–1043. [Google Scholar] [CrossRef]
- Reddy, V.S.; Natchiar, S.K.; Stewart, P.L.; Nemerow, G.R. Crystal structure of human adenovirus at 3.5 A resolution. Science 2010, 329, 1071–1075. [Google Scholar] [CrossRef]
- Russell, W.C. Adenoviruses: Update on structure and function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef]
- Mirza, M.A.; Weber, J. Structure of adenovirus chromatin. Biochim. Biophys. Acta 1982, 696, 76–86. [Google Scholar] [CrossRef]
- Everitt, E.; Sundquist, B.; Pettersson, U.; Philipson, L. Structural proteins of adenoviruses. X. Isolation and topography of low molecular weight antigens from the virion of adenovirus type 2. Virology 1973, 52, 130–147. [Google Scholar] [CrossRef]
- Brown, D.T.; Westphal, M.; Burlingham, B.T.; Winterhoff, U.; Doerfler, W. Structure and composition of the adenovirus type 2 core. J. Virol. 1975, 16, 366–387. [Google Scholar]
- Anderson, C.W.; Young, M.E.; Flint, S.J. Characterization of the adenovirus 2 virion protein, mu. Virology 1989, 172, 506–512. [Google Scholar] [CrossRef]
- Perez-Berna, A.J.; Marabini, R.; Scheres, S.H.; Menendez-Conejero, R.; Dmitriev, I.P.; Curiel, D.T.; Mangel, W.F.; Flint, S.J.; San Martin, C. Structure and uncoating of immature adenovirus. J. Mol. Biol. 2009, 392, 547–557. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nagata, K.; Ui, M.; Hanaoka, F. Template activating factor I, a novel host factor required to stimulate the adenovirus core DNA replication. J. Biol. Chem. 1993, 268, 10582–10587. [Google Scholar]
- Okuwaki, M.; Nagata, K. Template activating factor-I remodels the chromatin structure and stimulates transcription from the chromatin template. J. Biol. Chem. 1998, 273, 34511–34518. [Google Scholar] [CrossRef]
- Kennedy, M.A.; Parks, R.J. Adenovirus virion stability and the viral genome: Size matters. Mol. Ther. 2009, 17, 1664–1666. [Google Scholar] [CrossRef]
- Parks, R.J.; Graham, F.L. A helper-dependent system for adenovirus vector production helps define a lower limit for efficient DNA packaging. J. Virol. 1997, 71, 3293–3298. [Google Scholar]
- Parks, R.J.; Bramson, J.L.; Wan, Y.; Addison, C.L.; Graham, F.L. Effects of stuffer DNA on transgene expression from helper-dependent adenovirus vectors. J. Virol. 1999, 73, 8027–8034. [Google Scholar]
- Ross, P.J.; Kennedy, M.A.; Parks, R.J. Host cell detection of noncoding stuffer DNA contained in helper-dependent adenovirus vectors leads to epigenetic repression of transgene expression. J. Virol. 2009, 83, 8409–8417. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356. [Google Scholar] [CrossRef]
- Smith, T.A.; Idamakanti, N.; Rollence, M.L.; Marshall-Neff, J.; Kim, J.; Mulgrew, K.; Nemerow, G.R.; Kaleko, M.; Stevenson, S.C. Adenovirus serotype 5 fiber shaft influences in vivo gene transfer in mice. Hum. Gene Ther. 2003, 14, 777–787. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.Y.; Lieber, A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.K.; Greig, J.A.; Denby, L.; et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef]
- Kalyuzhniy, O.; Di Paolo, N.C.; Silvestry, M.; Hofherr, S.E.; Barry, M.A.; Stewart, P.L.; Shayakhmetov, D.M. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 5483–5488. [Google Scholar] [CrossRef]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Leopold, P.L.; Ferris, B.; Grinberg, I.; Worgall, S.; Hackett, N.R.; Crystal, R.G. Fluorescent virions: Dynamic tracking of the pathway of adenoviral gene transfer vectors in living cells. Hum. Gene Ther. 1998, 9, 367–378. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef]
- Greber, U.F.; Willetts, M.; Webster, P.; Helenius, A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 1993, 75, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, P.K.; Vayda, M.E.; Flint, S.J. Identification of proteins and protein domains that contact DNA within adenovirus nucleoprotein cores by ultraviolet light crosslinking of oligonucleotides 32P-labelled in vivo. J. Mol. Biol. 1986, 188, 23–37. [Google Scholar] [CrossRef]
- Strunze, S.; Engelke, M.F.; Wang, I.H.; Puntener, D.; Boucke, K.; Schleich, S.; Way, M.; Schoenenberger, P.; Burckhardt, C.J.; Greber, U.F. Kinesin-1-mediated capsid disassembly and disruption of the nuclear pore complex promote virus infection. Cell Host Microbe 2011, 10, 210–223. [Google Scholar] [CrossRef] [Green Version]
- Greber, U.F.; Puntener, D. DNA-tumor virus entry—From plasma membrane to the nucleus. Semin. Cell Dev. Biol. 2009, 20, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, R.; Schmidt, M.; von Kalle, C. Integration of retroviral vectors. Curr. Opin. Immunol. 2012, 24, 592–597. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. New Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Harui, A.; Suzuki, S.; Kochanek, S.; Mitani, K. Frequency and stability of chromosomal integration of adenovirus vectors. J. Virol. 1999, 73, 6141–6146. [Google Scholar]
- Hillgenberg, M.; Tonnies, H.; Strauss, M. Chromosomal integration pattern of a helper-dependent minimal adenovirus vector with a selectable marker inserted into a 27.4-kilobase genomic stuffer. J. Virol. 2001, 75, 9896–9908. [Google Scholar]
- Stephen, S.L.; Montini, E.; Sivanandam, V.G.; Al-Dhalimy, M.; Kestler, H.A.; Finegold, M.; Grompe, M.; Kochanek, S. Chromosomal integration of adenoviral vector DNA in vivo. J. Virol. 2010, 84, 9987–9994. [Google Scholar] [CrossRef]
- Jager, L.; Ehrhardt, A. Persistence of high-capacity adenoviral vectors as replication-defective monomeric genomes in vitro and in murine liver. Hum. Gene Ther. 2009, 20, 883–896. [Google Scholar] [CrossRef]
- Ehrhardt, A.; Xu, H.; Kay, M.A. Episomal persistence of recombinant adenoviral vector genomes during the cell cycle in vivo. J. Virol. 2003, 77, 7689–7695. [Google Scholar] [CrossRef]
- Rauschhuber, C.; Noske, N.; Ehrhardt, A. New insights into stability of recombinant adenovirus vector genomes in mammalian cells. Eur. J. Cell Biol. 2012, 91, 2–9. [Google Scholar] [CrossRef]
- Trotman, L.C.; Mosberger, N.; Fornerod, M.; Stidwill, R.P.; Greber, U.F. Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat. Cell Biol. 2001, 3, 1092–1100. [Google Scholar] [CrossRef]
- Chatterjee, P.K.; Vayda, M.E.; Flint, S.J. Adenoviral protein VII packages intracellular viral DNA throughout the early phase of infection. EMBO J. 1986, 5, 1633–1644. [Google Scholar]
- Xue, Y.; Johnson, J.S.; Ornelles, D.A.; Lieberman, J.; Engel, D.A. Adenovirus protein VII functions throughout early phase and interacts with cellular proteins SET and pp32. J. Virol. 2005, 79, 2474–2483. [Google Scholar] [CrossRef]
- Chen, J.; Morral, N.; Engel, D.A. Transcription releases protein VII from adenovirus chromatin. Virology 2007, 369, 411–422. [Google Scholar] [CrossRef]
- Karen, K.A.; Hearing, P. Adenovirus core protein VII protects the viral genome from a DNA damage response at early times after infection. J. Virol. 2011, 85, 4135–4142. [Google Scholar] [CrossRef]
- Walkiewicz, M.P.; Morral, N.; Engel, D.A. Accurate single-day titration of adenovirus vectors based on equivalence of protein VII nuclear dots and infectious particles. J. Virol. Methods 2009, 159, 251–258. [Google Scholar] [CrossRef]
- Komatsu, T.; Haruki, H.; Nagata, K. Cellular and viral chromatin proteins are positive factors in the regulation of adenovirus gene expression. Nucleic Acids Res. 2011, 39, 889–901. [Google Scholar] [CrossRef]
- Haruki, H.; Gyurcsik, B.; Okuwaki, M.; Nagata, K. Ternary complex formation between DNA-adenovirus core protein VII and TAF-Ibeta/SET, an acidic molecular chaperone. FEBS Lett. 2003, 555, 521–527. [Google Scholar] [CrossRef]
- Ross, P.J.; Kennedy, M.A.; Christou, C.; Risco Quiroz, M.; Poulin, K.L.; Parks, R.J. Assembly of helper-dependent adenovirus DNA into chromatin promotes efficient gene expression. J. Virol. 2011, 85, 3950–3958. [Google Scholar]
- Komatsu, T.; Nagata, K. Replication-uncoupled histone deposition during adenovirus DNA replication. J. Virol. 2012, 86, 6701–6711. [Google Scholar] [CrossRef]
- Kawase, H.; Okuwaki, M.; Miyaji, M.; Ohba, R.; Handa, H.; Ishimi, Y.; Fujii-Nakata, T.; Kikuchi, A.; Nagata, K. NAP-I is a functional homologue of TAF-I that is required for replication and transcription of the adenovirus genome in a chromatin-like structure. Gene. Cell. 1996, 1, 1045–1056. [Google Scholar]
- Okuwaki, M.; Iwamatsu, A.; Tsujimoto, M.; Nagata, K. Identification of nucleophosmin/B23, an acidic nucleolar protein, as a stimulatory factor for in vitro replication of adenovirus DNA complexed with viral basic core proteins. J. Mol. Biol. 2001, 311, 41–55. [Google Scholar] [CrossRef]
- Gyurcsik, B.; Haruki, H.; Takahashi, T.; Mihara, H.; Nagata, K. Binding modes of the precursor of adenovirus major core protein VII to DNA and template activating factor I: Implication for the mechanism of remodeling of the adenovirus chromatin. Biochemistry 2006, 45, 303–313. [Google Scholar]
- Johnson, J.S.; Osheim, Y.N.; Xue, Y.; Emanuel, M.R.; Lewis, P.W.; Bankovich, A.; Beyer, A.L.; Engel, D.A. Adenovirus protein VII condenses DNA, represses transcription, and associates with transcriptional activator E1A. J. Virol. 2004, 78, 6459–6468. [Google Scholar] [CrossRef]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef]
- Smith, S.; Stillman, B. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 1989, 58, 15–25. [Google Scholar] [CrossRef]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef]
- Schneiderman, J.I.; Orsi, G.A.; Hughes, K.T.; Loppin, B.; Ahmad, K. Nucleosome-depleted chromatin gaps recruit assembly factors for the H3.3 histone variant. Proc. Natl. Acad. Sci. USA 2012, 109, 19721–19726. [Google Scholar]
- Placek, B.J.; Huang, J.; Kent, J.R.; Dorsey, J.; Rice, L.; Fraser, N.W.; Berger, S.L. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J. Virol. 2009, 83, 1416–1421. [Google Scholar] [CrossRef]
- Pastore, L.; Morral, N.; Zhou, H.; Garcia, R.; Parks, R.J.; Kochanek, S.; Graham, F.L.; Lee, B.; Beaudet, A.L. Use of a liver-specific promoter reduces immune response to the transgene in adenoviral vectors. Hum. Gene Ther. 1999, 10, 1773–1781. [Google Scholar] [CrossRef]
- Schiedner, G.; Hertel, S.; Johnston, M.; Biermann, V.; Dries, V.; Kochanek, S. Variables affecting in vivo performance of high-capacity adenovirus vectors. J. Virol. 2002, 76, 1600–1609. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Smith, C.L.; Hager, G.L. Transcriptional regulation of mammalian genes in vivo. A tale of two templates. J. Biol. Chem. 1997, 272, 27493–27496. [Google Scholar] [CrossRef]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. Genet. 2009, 10, 161–172. [Google Scholar] [CrossRef]
- Lomvardas, S.; Thanos, D. Modifying gene expression programs by altering core promoter chromatin architecture. Cell 2002, 110, 261–271. [Google Scholar] [CrossRef]
- Schreiner, S.; Wimmer, P.; Sirma, H.; Everett, R.D.; Blanchette, P.; Groitl, P.; Dobner, T. Proteasome-dependent degradation of Daxx by the viral E1B-55K protein in human adenovirus-infected cells. J. Virol. 2010, 84, 7029–7038. [Google Scholar] [CrossRef]
- Ullman, A.J.; Hearing, P. Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J. Virol. 2008, 82, 7325–7335. [Google Scholar] [CrossRef]
- Schreiner, S.; Burck, C.; Glass, M.; Groitl, P.; Wimmer, P.; Kinkley, S.; Mund, A.; Everett, R.D.; Dobner, T. Control of human adenovirus type 5 gene expression by cellular Daxx/ATRX chromatin-associated complexes. Nucleic Acids Res. 2013, 41, 3532–3550. [Google Scholar] [CrossRef]
- Schreiner, S.; Martinez, R.; Groitl, P.; Rayne, F.; Vaillant, R.; Wimmer, P.; Bossis, G.; Sternsdorf, T.; Marcinowski, L.; Ruzsics, Z.; et al. Transcriptional activation of the adenoviral genome is mediated by capsid protein VI. PLoS Path. 2012, 8, e1002549. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Cerullo, V.; Bertin, T.K.; Cela, R.; Clarke, C.; Guenther, M.; Brunetti-Pierri, N.; Lee, B. MyD88-dependent silencing of transgene expression during the innate and adaptive immune response to helper-dependent adenovirus. Hum. Gene Ther. 2010, 21, 325–336. [Google Scholar] [CrossRef]
- Cerullo, V.; Seiler, M.P.; Mane, V.; Brunetti-Pierri, N.; Clarke, C.; Bertin, T.K.; Rodgers, J.R.; Lee, B. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol. Ther. 2007, 15, 378–385. [Google Scholar] [CrossRef]
- Brestovitsky, A.; Sharf, R.; Mittelman, K.; Kleinberger, T. The adenovirus E4orf4 protein targets PP2A to the ACF chromatin-remodeling factor and induces cell death through regulation of SNF2h-containing complexes. Nucleic Acids Res. 2011, 39, 6414–6427. [Google Scholar] [CrossRef]
- Lavoie, J.N.; Nguyen, M.; Marcellus, R.C.; Branton, P.E.; Shore, G.C. E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J. Cell Biol. 1998, 140, 637–645. [Google Scholar] [CrossRef]
- Marcellus, R.C.; Lavoie, J.N.; Boivin, D.; Shore, G.C.; Ketner, G.; Branton, P.E. The early region 4 orf4 protein of human adenovirus type 5 induces p53- independent cell death by apoptosis. J. Virol. 1998, 72, 7144–7153. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wong, C.M.; McFall, E.R.; Burns, J.K.; Parks, R.J. The Role of Chromatin in Adenoviral Vector Function. Viruses 2013, 5, 1500-1515. https://doi.org/10.3390/v5061500
Wong CM, McFall ER, Burns JK, Parks RJ. The Role of Chromatin in Adenoviral Vector Function. Viruses. 2013; 5(6):1500-1515. https://doi.org/10.3390/v5061500
Chicago/Turabian StyleWong, Carmen M., Emily R. McFall, Joseph K. Burns, and Robin J. Parks. 2013. "The Role of Chromatin in Adenoviral Vector Function" Viruses 5, no. 6: 1500-1515. https://doi.org/10.3390/v5061500