Hepatitis C Virus, Cholesterol and Lipoproteins — Impact for the Viral Life Cycle and Pathogenesis of Liver Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lipids, Apolipoproteins, and HCV Pathogenesis
3. Treatments Targeting Lipid Metabolism
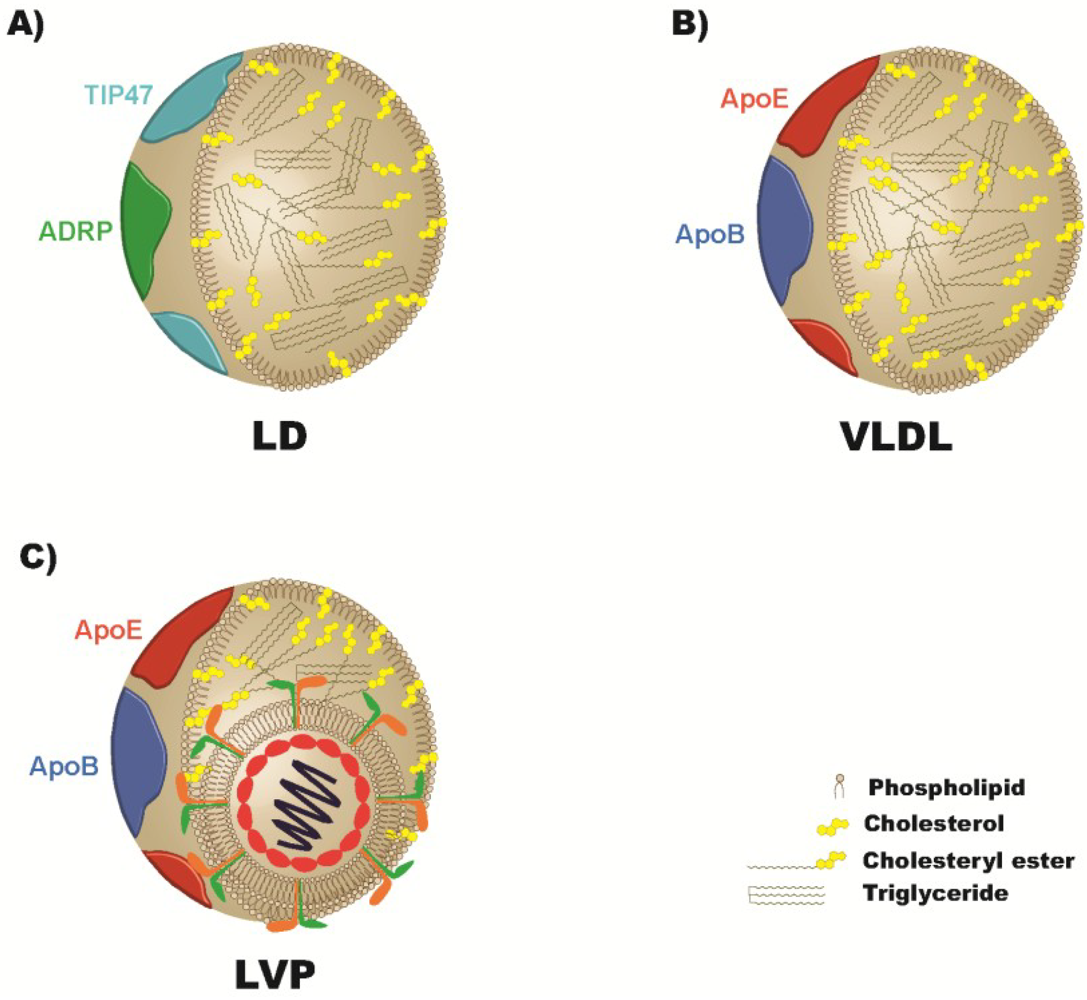
4. Apolipoproteins and the Viral Particle
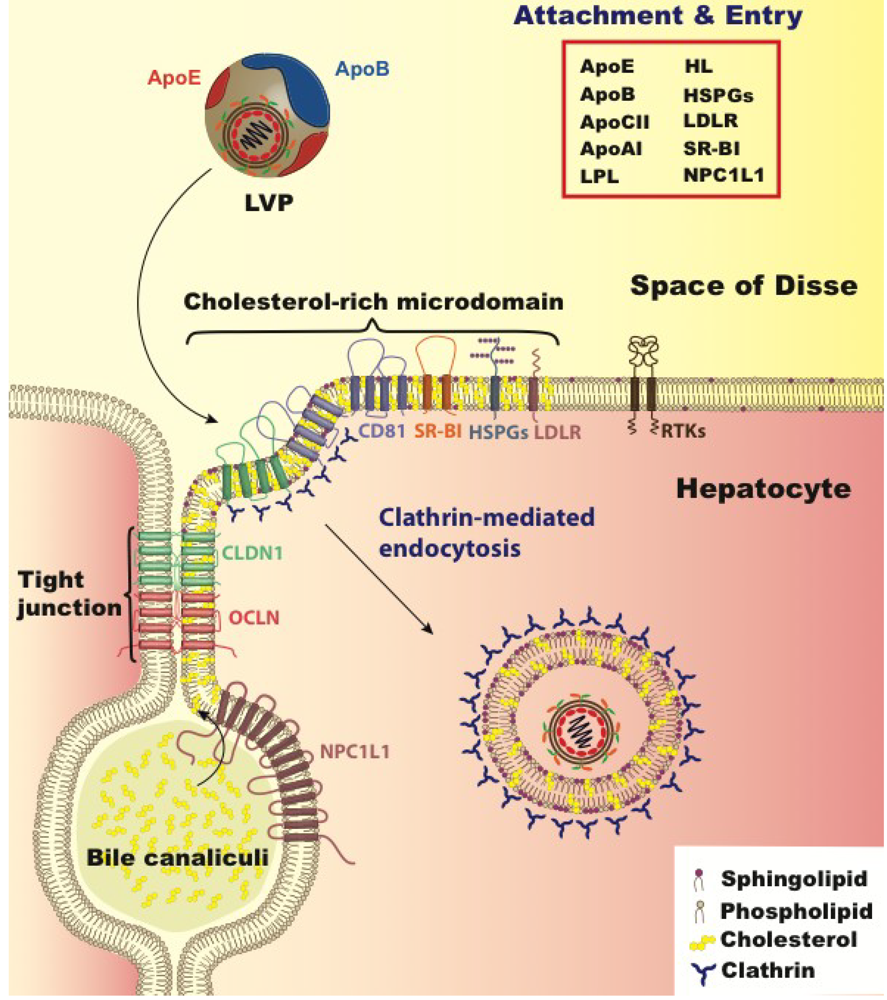
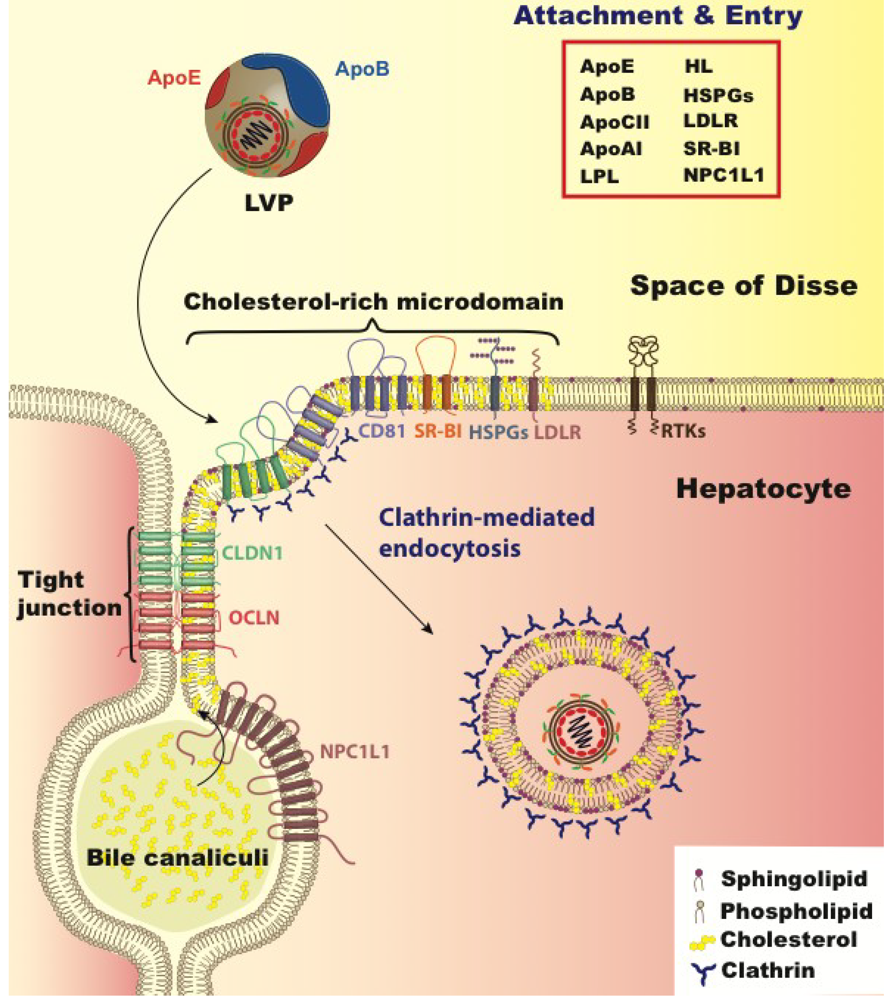
5. Lipoprotein and Cholesterol’s Role in Viral Entry
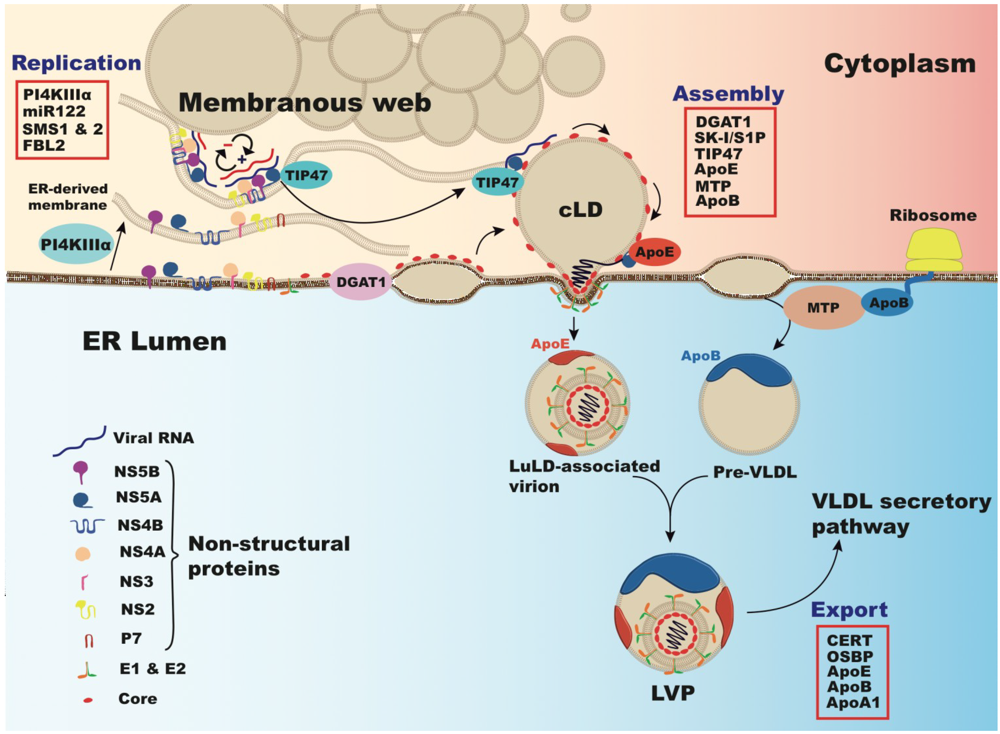
6. Cholesterol and Viral Replication
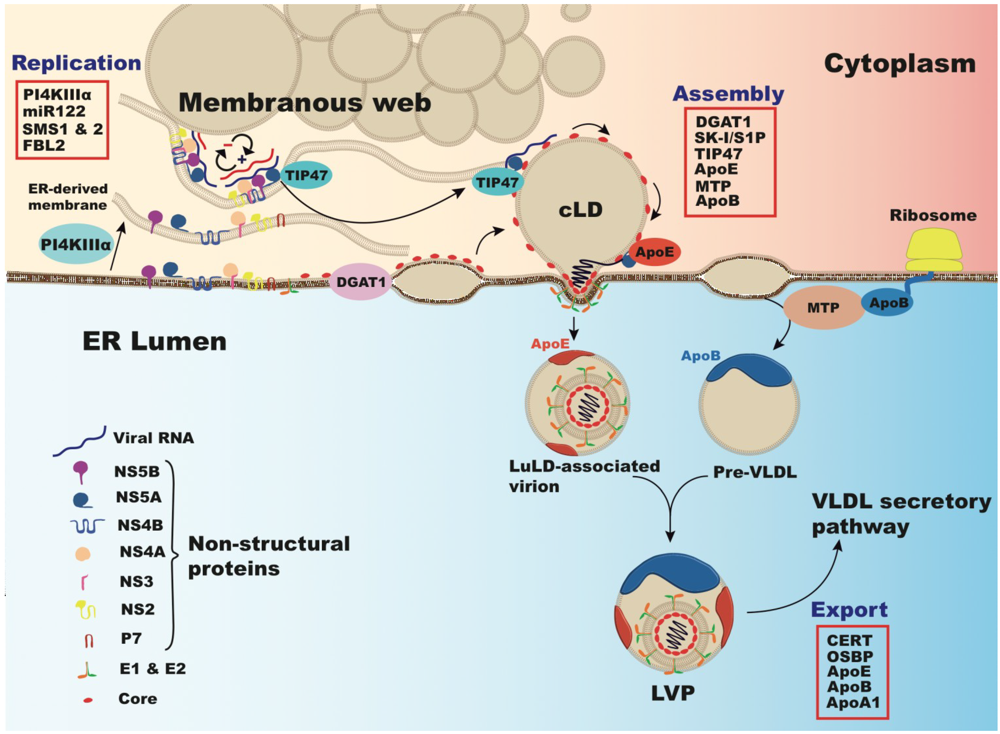
7. Lipoproteins, Lipids and Cholesterol in Virus Production and Secretion
Acknowledgements
Conflict of Interest
References and Notes
- Aghemo, A.; De Francesco, R. New horizons in Hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013. [Google Scholar] [CrossRef]
- Kanwal, F.; Hoang, T.; Kramer, J.R.; Asch, S.M.; Goetz, M.B.; Zeringue, A.; Richardson, P.; El-Serag, H.B. Increasing prevalence of HCC and cirrhosis in patients with chronic hepatitis C virus infection. Gastroenterology 2011, 140, 1182–1188. [Google Scholar] [CrossRef]
- Hezode, C.; Forestier, N.; Dusheiko, G.; Ferenci, P.; Pol, S.; Goeser, T.; Bronowicki, J.P.; Bourliere, M.; Gharakhanian, S.; Bengtsson, L.; et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N. Engl. J. Med. 2009, 360, 1839–1850. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Everson, G.T.; Gordon, S.C.; Jacobson, I.M.; Sulkowski, M.; Kauffman, R.; McNair, L.; Alam, J.; Muir, A.J. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N. Engl. J. Med. 2009, 360, 1827–1838. [Google Scholar] [CrossRef]
- Poordad, F.; McCone, J., Jr.; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; et al. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011, 364, 1195–1206. [Google Scholar] [CrossRef]
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011, 364, 1207–1217. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Manns, M.P.; Muir, A.J.; Terrault, N.A.; Jacobson, I.M.; Afdhal, N.H.; Heathcote, E.J.; Zeuzem, S.; Reesink, H.W.; Garg, J.; et al. Telaprevir for previously treated chronic HCV infection. N. Engl. J. Med. 2010, 362, 1292–1303. [Google Scholar] [CrossRef]
- Dienes, H.P.; Popper, H.; Arnold, W.; Lobeck, H. Histologic observations in human hepatitis non-A, non-B. Hepatology 1982, 2, 562–571. [Google Scholar]
- Thomssen, R.; Bonk, S.; Propfe, C.; Heermann, K.H.; Kochel, H.G.; Uy, A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. 1992, 181, 293–300. [Google Scholar] [CrossRef]
- Thomssen, R.; Bonk, S.; Thiele, A. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. 1993, 182, 329–334. [Google Scholar]
- Diaz, O.; Delers, F.; Maynard, M.; Demignot, S.; Zoulim, F.; Chambaz, J.; Trepo, C.; Lotteau, V.; Andre, P. Preferential association of Hepatitis C virus with apolipoprotein B48-containing lipoproteins. J. Gen. Virol. 2006, 87, 2983–2991. [Google Scholar] [CrossRef]
- Felmlee, D.J.; Sheridan, D.A.; Bridge, S.H.; Nielsen, S.U.; Milne, R.W.; Packard, C.J.; Caslake, M.J.; McLauchlan, J.; Toms, G.L.; Neely, R.D.; et al. Intravascular transfer contributes to postprandial increase in numbers of very-low-density hepatitis C virus particles. Gastroenterology 2010, 139, 1774–1783. [Google Scholar] [CrossRef]
- Scholtes, C.; Ramiere, C.; Rainteau, D.; Perrin-Cocon, L.; Wolf, C.; Humbert, L.; Carreras, M.; Guironnet-Paquet, A.; Zoulim, F.; Bartenschlager, R.; et al. High plasma level of nucleocapsid-free envelope glycoprotein-positive lipoproteins in hepatitis C patients. Hepatology 2012, 56, 39–48. [Google Scholar] [CrossRef]
- Grove, J.; Nielsen, S.; Zhong, J.; Bassendine, M.F.; Drummer, H.E.; Balfe, P.; McKeating, J.A. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J. Virol. 2008, 82, 12020–12029. [Google Scholar]
- Prentoe, J.; Jensen, T.B.; Meuleman, P.; Serre, S.B.; Scheel, T.K.; Leroux-Roels, G.; Gottwein, J.M.; Bukh, J. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J. Virol. 2011, 85, 2224–2234. [Google Scholar]
- Fafi-Kremer, S.; Fofana, I.; Soulier, E.; Carolla, P.; Meuleman, P.; Leroux-Roels, G.; Patel, A.H.; Cosset, F.L.; Pessaux, P.; Doffoel, M.; et al. Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J. Exp. Med. 2010, 207, 2019–2031. [Google Scholar] [CrossRef] [Green Version]
- Fofana, I.; Fafi-Kremer, S.; Carolla, P.; Fauvelle, C.; Zahid, M.N.; Turek, M.; Heydmann, L.; Cury, K.; Hayer, J.; Combet, C.; et al. Mutations that alter use of hepatitis C virus cell entry factors mediate escape from neutralizing antibodies. Gastroenterology 2012, 143, 223–233. [Google Scholar] [CrossRef]
- Bickel, P.E.; Tansey, J.T.; Welte, M.A. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta 2009, 1791, 419–440. [Google Scholar] [CrossRef]
- Brasaemle, D.L. Thematic review series: Adipocyte biology. The perilipin family of structural lipid droplet proteins: Stabilization of lipid droplets and control of lipolysis. J. Lipid. Res. 2007, 48, 2547–2559. [Google Scholar]
- Dallinga-Thie, G.M.; Franssen, R.; Mooij, H.L.; Visser, M.E.; Hassing, H.C.; Peelman, F.; Kastelein, J.J.; Peterfy, M.; Nieuwdorp, M. The metabolism of triglyceride-rich lipoproteins revisited: New players, new insight. Atherosclerosis 2010, 211, 1–8. [Google Scholar]
- Williams, K.J. Molecular processes that handle—and mishandle—dietary lipids. J. Clin. Invest. 2008, 118, 3247–3259. [Google Scholar] [CrossRef]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef]
- Nielsen, S.U.; Bassendine, M.F.; Burt, A.D.; Martin, C.; Pumeechockchai, W.; Toms, G.L. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 2006, 80, 2418–2428. [Google Scholar] [CrossRef]
- Rubbia-Brandt, L.; Quadri, R.; Abid, K.; Giostra, E.; Male, P.J.; Mentha, G.; Spahr, L.; Zarski, J.P.; Borisch, B.; Hadengue, A.; et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J. Hepatol. 2000, 33, 106–115. [Google Scholar] [CrossRef]
- Serfaty, L.; Andreani, T.; Giral, P.; Carbonell, N.; Chazouilleres, O.; Poupon, R. Hepatitis C virus induced hypobetalipoproteinemia: A possible mechanism for steatosis in chronic hepatitis C. J. Hepatol. 2001, 34, 428–434. [Google Scholar]
- Fujino, T.; Nakamuta, M.; Yada, R.; Aoyagi, Y.; Yasutake, K.; Kohjima, M.; Fukuizumi, K.; Yoshimoto, T.; Harada, N.; Yada, M.; et al. Expression profile of lipid metabolism-associated genes in hepatitis C virus-infected human liver. Hepatol. Res. 2010, 40, 923–929. [Google Scholar] [CrossRef]
- Nakamuta, M.; Yada, R.; Fujino, T.; Yada, M.; Higuchi, N.; Tanaka, M.; Miyazaki, M.; Kohjima, M.; Kato, M.; Yoshimoto, T.; et al. Changes in the expression of cholesterol metabolism-associated genes in HCV-infected liver: A novel target for therapy? Int. J. Mol. Med. 2009, 24, 825–828. [Google Scholar]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef]
- Chang, M.L.; Yeh, C.T.; Chen, J.C.; Huang, C.C.; Lin, S.M.; Sheen, I.S.; Tai, D.I.; Chu, C.M.; Lin, W.P.; Chang, M.Y.; et al. Altered expression patterns of lipid metabolism genes in an animal model of HCV core-related, nonobese, modest hepatic steatosis. BMC Genomics 2008, 9, e109. [Google Scholar] [CrossRef]
- Lerat, H.; Kammoun, H.L.; Hainault, I.; Merour, E.; Higgs, M.R.; Callens, C.; Lemon, S.M.; Foufelle, F.; Pawlotsky, J.M. Hepatitis C virus proteins induce lipogenesis and defective triglyceride secretion in transgenic mice. J. Biol. Chem. 2009, 284, 33466–33474. [Google Scholar] [CrossRef] [Green Version]
- Oem, J.K.; Jackel-Cram, C.; Li, Y.P.; Zhou, Y.; Zhong, J.; Shimano, H.; Babiuk, L.A.; Liu, Q. Activation of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by hepatitis C virus non-structural protein 2. J. Gen. Virol. 2008, 89, 1225–1230. [Google Scholar] [CrossRef]
- Park, C.Y.; Jun, H.J.; Wakita, T.; Cheong, J.H.; Hwang, S.B. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 2009, 284, 9237–9246. [Google Scholar] [CrossRef]
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130. [Google Scholar] [CrossRef]
- McPherson, S.; Jonsson, J.R.; Barrie, H.D.; O'Rourke, P.; Clouston, A.D.; Powell, E.E. Investigation of the role of SREBP-1c in the pathogenesis of HCV-related steatosis. J. Hepatol. 2008, 49, 1046–1054. [Google Scholar] [CrossRef]
- Lambert, J.E.; Bain, V.G.; Ryan, E.A.; Thomson, A.B.; Clandinin, M.T. Elevated lipogenesis and diminished cholesterol synthesis in patients with hepatitis C viral infection compared to healthy humans. Hepatology 2012, 57, 1697–1704. [Google Scholar]
- Sharma, P.; Balan, V.; Hernandez, J.; Rosati, M.; Williams, J.; Rodriguez-Luna, H.; Schwartz, J.; Harrison, E.; Anderson, M.; Byrne, T.; et al. Hepatic steatosis in hepatitis C virus genotype 3 infection: Does it correlate with body mass index, fibrosis, and HCV risk factors? Dig. Dis. Sci. 2004, 49, 25–29. [Google Scholar] [CrossRef]
- Thomopoulos, K.C.; Arvaniti, V.; Tsamantas, A.C.; Dimitropoulou, D.; Gogos, C.A.; Siagris, D.; Theocharis, G.J.; Labropoulou-Karatza, C. Prevalence of liver steatosis in patients with chronic hepatitis B: A study of associated factors and of relationship with fibrosis. Eur. J. Gastroenterol. Hepatol. 2006, 18, 233–237. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Gambardella, M.; Andreana, A.; Tripodi, M.F.; Utili, R.; Ruggiero, G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 2001, 33, 1358–1364. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Utili, R.; Andreana, A.; Tripodi, M.F.; Marracino, M.; Gambardella, M.; Giordano, M.; Ruggiero, G. Serum HCV RNA levels correlate with histological liver damage and concur with steatosis in progression of chronic hepatitis C. Dig. Dis. Sci. 2001, 46, 1677–1683. [Google Scholar] [CrossRef]
- Hofer, H.; Bankl, H.C.; Wrba, F.; Steindl-Munda, P.; Peck-Radosavljevic, M.; Osterreicher, C.; Mueller, C.; Gangl, A.; Ferenci, P. Hepatocellular fat accumulation and low serum cholesterol in patients infected with HCV-3a. Am. J. Gastroenterol. 2002, 97, 2880–2885. [Google Scholar] [CrossRef]
- Marzouk, D.; Sass, J.; Bakr, I.; El Hosseiny, M.; Abdel-Hamid, M.; Rekacewicz, C.; Chaturvedi, N.; Mohamed, M.K.; Fontanet, A. Metabolic and cardiovascular risk profiles and hepatitis C virus infection in rural Egypt. Gut 2007, 56, 1105–1110. [Google Scholar]
- Clark, P.J.; Thompson, A.J.; Vock, D.M.; Kratz, L.E.; Tolun, A.A.; Muir, A.J.; McHutchison, J.G.; Subramanian, M.; Millington, D.M.; Kelley, R.I.; et al. Hepatitis C virus selectively perturbs the distal cholesterol synthesis pathway in a genotype-specific manner. Hepatology 2012, 56, 49–56. [Google Scholar]
- Domitrovich, A.M.; Felmlee, D.J.; Siddiqui, A. Hepatitis C virus nonstructural proteins inhibit apolipoprotein B100 secretion. J. Biol. Chem. 2005, 280, 39802–39808. [Google Scholar] [CrossRef]
- Mirandola, S.; Realdon, S.; Iqbal, J.; Gerotto, M.; Dal Pero, F.; Bortoletto, G.; Marcolongo, M.; Vario, A.; Datz, C.; Hussain, M.M.; et al. Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis. Gastroenterology 2006, 130, 1661–1669. [Google Scholar] [CrossRef]
- Mancone, C.; Montaldo, C.; Santangelo, L.; Di Giacomo, C.; Costa, V.; Amicone, L.; Ippolito, G.; Pucillo, L.P.; Alonzi, T.; Tripodi, M. Ferritin heavy chain is the host factor responsible for HCV-induced inhibition of apoB-100 production and is required for efficient viral infection. J. Proteome Res. 2012, 11, 2786–2797. [Google Scholar]
- Enjoji, M.; Kohjima, M.; Kotoh, K.; Nakamuta, M. Metabolic disorders and steatosis in patients with chronic hepatitis C: Metabolic strategies for antiviral treatments. Int. J. Hepatol. 2012, 2012, e264017. [Google Scholar]
- Dai, C.Y.; Chuang, W.L.; Ho, C.K.; Hsieh, M.Y.; Huang, J.F.; Lee, L.P.; Hou, N.J.; Lin, Z.Y.; Chen, S.C.; Hsieh, M.Y.; et al. Associations between hepatitis C viremia and low serum triglyceride and cholesterol levels: A community-based study. J. Hepatol. 2008, 49, 9–16. [Google Scholar] [CrossRef]
- Nishimura, M.; Yamamoto, H.; Yoshida, T.; Seimiya, M.; Sawabe, Y.; Matsushita, K.; Umemura, H.; Sogawa, K.; Takizawa, H.; Yokosuka, O.; et al. Decreases in the serum VLDL-TG/non-VLDL-TG ratio from early stages of chronic hepatitis C: Alterations in TG-rich lipoprotein levels. PLoS One 2011, 6, e17309. [Google Scholar]
- Merz, A.; Long, G.; Hiet, M.S.; Brugger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar] [CrossRef]
- Akazawa, D.; Morikawa, K.; Omi, N.; Takahashi, H.; Nakamura, N.; Mochizuki, H.; Date, T.; Ishii, K.; Suzuki, T.; Wakita, T. Production and characterization of HCV particles from serum-free culture. Vaccine 2011, 29, 4821–4828. [Google Scholar] [CrossRef]
- Cun, W.; Jiang, J.; Luo, G. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J. Virol. 2010, 84, 11532–11541. [Google Scholar] [CrossRef]
- Da Costa, D.; Turek, M.; Felmlee, D.J.; Girardi, E.; Pfeffer, S.; Long, G.; Bartenschlager, R.; Zeisel, M.B.; Baumert, T.F. Reconstitution of the entire hepatitis C virus life cycle in nonhepatic cells. J. Virol. 2012, 86, 11919–11925. [Google Scholar] [CrossRef]
- Hishiki, T.; Shimizu, Y.; Tobita, R.; Sugiyama, K.; Ogawa, K.; Funami, K.; Ohsaki, Y.; Fujimoto, T.; Takaku, H.; Wakita, T.; et al. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J. Virol. 2010, 84, 12048–12057. [Google Scholar] [CrossRef]
- Long, G.; Hiet, M.S.; Windisch, M.P.; Lee, J.Y.; Lohmann, V.; Bartenschlager, R. Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology 2011, 141, 1057–1066. [Google Scholar] [CrossRef]
- Price, D.A.; Bassendine, M.F.; Norris, S.M.; Golding, C.; Toms, G.L.; Schmid, M.L.; Morris, C.M.; Burt, A.D.; Donaldson, P.T. Apolipoprotein epsilon3 allele is associated with persistent hepatitis C virus infection. Gut 2006, 55, 715–718. [Google Scholar] [CrossRef]
- Kuhlmann, I.; Minihane, A.M.; Huebbe, P.; Nebel, A.; Rimbach, G. Apolipoprotein E genotype and hepatitis C, HIV and herpes simplex disease risk: A literature review. Lipids Health Dis. 2010, 9, e8. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Itzhaki, R.F.; Faragher, E.B.; James, M.W.; Ryder, S.D.; Irving, W.L. Apolipoprotein E-epsilon 4 protects against severe liver disease caused by hepatitis C virus. Hepatology 2002, 36, 456–463. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.; Han, Q.; Li, Y.; Chen, J. Association of genetic polymorphism of low-density lipoprotein receptor with chronic viral hepatitis C infection in Han Chinese. J. Med. Virol. 2006, 78, 1289–1295. [Google Scholar] [CrossRef]
- Napolitano, M.; Giuliani, A.; Alonzi, T.; Mancone, C.; D'Offizi, G.; Tripodi, M.; Bravo, E. Very low density lipoprotein and low density lipoprotein isolated from patients with hepatitis C infection induce altered cellular lipid metabolism. J. Med. Virol. 2007, 79, 254–258. [Google Scholar] [CrossRef]
- Mancone, C.; Steindler, C.; Santangelo, L.; Simonte, G.; Vlassi, C.; Longo, M.A.; D'Offizi, G.; Di Giacomo, C.; Pucillo, L.P.; Amicone, L.; et al. Hepatitis C virus production requires apolipoprotein A-I and affects its association with nascent low-density lipoproteins. Gut 2011, 60, 378–386. [Google Scholar] [CrossRef]
- Kim, E.; Li, K.; Lieu, C.; Tong, S.; Kawai, S.; Fukutomi, T.; Zhou, Y.; Wands, J.; Li, J. Expression of apolipoprotein C-IV is regulated by Ku antigen/peroxisome proliferator-activated receptor gamma complex and correlates with liver steatosis. J. Hepatol. 2008, 49, 787–798. [Google Scholar] [CrossRef]
- Rowell, J.; Thompson, A.J.; Guyton, J.R.; Lao, X.Q.; McHutchison, J.G.; McCarthy, J.J.; Patel, K. Serum apolipoprotein C-III is independently associated with chronic hepatitis C infection and advanced fibrosis. Hepatol. Int. 2012, 6, 475–481. [Google Scholar] [CrossRef]
- Poynard, T.; Ratziu, V.; McHutchison, J.; Manns, M.; Goodman, Z.; Zeuzem, S.; Younossi, Z.; Albrecht, J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology 2003, 38, 75–85. [Google Scholar]
- Westin, J.; Lagging, M.; Dhillon, A.P.; Norkrans, G.; Romero, A.I.; Pawlotsky, J.M.; Zeuzem, S.; Schalm, S.W.; Verheij-Hart, E.; Negro, F.; et al. Impact of hepatic steatosis on viral kinetics and treatment outcome during antiviral treatment of chronic HCV infection. J. Viral. Hepat. 2007, 14, 29–35. [Google Scholar]
- Zeuzem, S.; Hultcrantz, R.; Bourliere, M.; Goeser, T.; Marcellin, P.; Sanchez-Tapias, J.; Sarrazin, C.; Harvey, J.; Brass, C.; Albrecht, J. Peginterferon alfa-2b plus ribavirin for treatment of chronic hepatitis C in previously untreated patients infected with HCV genotypes 2 or 3. J. Hepatol. 2004, 40, 993–999. [Google Scholar] [CrossRef]
- Corey, K.E.; Kane, E.; Munroe, C.; Barlow, L.L.; Zheng, H.; Chung, R.T. Hepatitis C virus infection and its clearance alter circulating lipids: Implications for long-term follow-up. Hepatology 2009, 50, 1030–1037. [Google Scholar] [CrossRef]
- Gopal, K.; Johnson, T.C.; Gopal, S.; Walfish, A.; Bang, C.T.; Suwandhi, P.; Pena-Sahdala, H.N.; Clain, D.J.; Bodenheimer, H.C., Jr.; Min, A.D. Correlation between beta-lipoprotein levels and outcome of hepatitis C treatment. Hepatology 2006, 44, 335–340. [Google Scholar] [CrossRef]
- Ramcharran, D.; Wahed, A.S.; Conjeevaram, H.S.; Evans, R.W.; Wang, T.; Belle, S.H.; Yee, L.J. Associations between serum lipids and hepatitis C antiviral treatment efficacy. Hepatology 2010, 52, 854–863. [Google Scholar]
- Adinolfi, L.E.; Restivo, L.; Zampino, R.; Guerrera, B.; Lonardo, A.; Ruggiero, L.; Riello, F.; Loria, P.; Florio, A. Chronic HCV infection is a risk of atherosclerosis. Role of HCV and HCV-related steatosis. Atherosclerosis 2012, 221, 496–502. [Google Scholar] [CrossRef]
- Akuta, N.; Suzuki, F.; Kawamura, Y.; Yatsuji, H.; Sezaki, H.; Suzuki, Y.; Hosaka, T.; Kobayashi, M.; Kobayashi, M.; Arase, Y.; et al. Predictive factors of early and sustained responses to peginterferon plus ribavirin combination therapy in Japanese patients infected with hepatitis C virus genotype 1b: Amino acid substitutions in the core region and low-density lipoprotein cholesterol levels. J. Hepatol. 2007, 46, 403–410. [Google Scholar] [CrossRef]
- Sheridan, D.A.; Bridge, S.H.; Felmlee, D.J.; Crossey, M.M.; Thomas, H.C.; Taylor-Robinson, S.D.; Toms, G.L.; Neely, R.D.; Bassendine, M.F. Apolipoprotein-E and hepatitis C lipoviral particles in genotype 1 infection: Evidence for an association with interferon sensitivity. J. Hepatol. 2012, 57, 32–38. [Google Scholar] [CrossRef]
- Bridge, S.H.; Sheridan, D.A.; Felmlee, D.J.; Nielsen, S.U.; Thomas, H.C.; Taylor-Robinson, S.D.; Neely, R.D.; Toms, G.L.; Bassendine, M.F. Insulin resistance and low-density apolipoprotein B-associated lipoviral particles in hepatitis C virus genotype 1 infection. Gut 2011, 60, 680–687. [Google Scholar] [CrossRef]
- Li, J.H.; Lao, X.Q.; Tillmann, H.L.; Rowell, J.; Patel, K.; Thompson, A.; Suchindran, S.; Muir, A.J.; Guyton, J.R.; Gardner, S.D.; et al. Interferon-lambda genotype and low serum low-density lipoprotein cholesterol levels in patients with chronic hepatitis C infection. Hepatology 2010, 51, 1904–1911. [Google Scholar] [CrossRef]
- Romero-Gomez, M.; Diago, M.; Andrade, R.J.; Calleja, J.L.; Salmeron, J.; Fernandez-Rodriguez, C.M.; Sola, R.; Garcia-Samaniego, J.; Herrerias, J.M.; De la Mata, M.; et al. Treatment of insulin resistance with metformin in naive genotype 1 chronic hepatitis C patients receiving peginterferon alfa-2a plus ribavirin. Hepatology 2009, 50, 1702–1708. [Google Scholar] [CrossRef]
- Kohjima, M.; Enjoji, M.; Yoshimoto, T.; Yada, R.; Fujino, T.; Aoyagi, Y.; Fukushima, N.; Fukuizumi, K.; Harada, N.; Yada, M.; et al. Add-on therapy of pitavastatin and eicosapentaenoic acid improves outcome of peginterferon plus ribavirin treatment for chronic hepatitis C. J. Med. Virol. 2013, 85, 250–260. [Google Scholar] [CrossRef]
- Chojkier, M.; Elkhayat, H.; Sabry, D.; Donohue, M.; Buck, M. Pioglitazone decreases hepatitis C viral load in overweight, Treatment naive, Genotype 4 infected-patients: A pilot study. PLoS One 2012, 7, e31516. [Google Scholar]
- Harrison, S.A.; Rossaro, L.; Hu, K.Q.; Patel, K.; Tillmann, H.; Dhaliwal, S.; Torres, D.M.; Koury, K.; Goteti, V.S.; Noviello, S.; et al. Serum cholesterol and statin use predict virological response to peginterferon and ribavirin therapy. Hepatology 2010, 52, 864–874. [Google Scholar]
- Atsukawa, M.; Tsubota, A.; Kondo, C.; Itokawa, N.; Narahara, Y.; Nakatsuka, K.; Hashimoto, S.; Fukuda, T.; Matsushita, Y.; Kidokoro, H.; et al. Combination of fluvastatin with pegylated interferon/ribavirin therapy reduces viral relapse in chronic hepatitis C infected with HCV genotype 1b. J. Gastroenterol. Hepatol. 2013, 28, 51–56. [Google Scholar]
- Rao, G.A.; Pandya, P.K. Statin therapy improves sustained virologic response among diabetic patients with chronic hepatitis C. Gastroenterology 2011, 140, 144–152. [Google Scholar] [CrossRef]
- Ye, J.; Wang, C.; Sumpter, R., Jr.; Brown, M.S.; Goldstein, J.L.; Gale, M., Jr. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc. Natl. Acad. Sci. USA 2003, 100, 15865–15870. [Google Scholar]
- Bader, T.; Fazili, J.; Madhoun, M.; Aston, C.; Hughes, D.; Rizvi, S.; Seres, K.; Hasan, M. Fluvastatin inhibits hepatitis C replication in humans. Am. J. Gastroenterol. 2008, 103, 1383–1389. [Google Scholar]
- Forde, K.A.; Law, C.; O'Flynn, R.; Kaplan, D.E. Do statins reduce hepatitis C RNA titers during routine clinical use? World J. Gastroenterol. 2009, 15, 5020–5027. [Google Scholar]
- Milazzo, L.; Meroni, L.; Galazzi, M.; Cesari, M.; Caramma, I.; Marchetti, G.; Galli, M.; Antinori, S. Does fluvastatin favour HCV replication in vivo? A pilot study on HIV-HCV coinfected patients. J. Viral. Hepatitis 2009, 16, 479–484. [Google Scholar] [CrossRef]
- O'Leary, J.G.; Chan, J.L.; McMahon, C.M.; Chung, R.T. Atorvastatin does not exhibit antiviral activity against HCV at conventional doses: A pilot clinical trial. Hepatology 2007, 45, 895–898. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Lawitz, E.J.; Shiffman, M.L.; Muir, A.J.; Galler, G.W.; McCone, J.; Nyberg, L.M.; Lee, W.M.; Ghalib, R.H.; Schiff, E.R.; et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N. Engl. J. Med. 2009, 361, 580–593. [Google Scholar]
- Milazzo, L.; Caramma, I.; Mazzali, C.; Cesari, M.; Olivetti, M.; Galli, M.; Antinori, S. Fluvastatin as an adjuvant to pegylated interferon and ribavirin in HIV/hepatitis C virus genotype 1 co-infected patients: An open-label randomized controlled study. J. Antimicrob. Chemother. 2010, 65, 735–740. [Google Scholar] [CrossRef]
- Sezaki, H.; Suzuki, F.; Akuta, N.; Yatsuji, H.; Hosaka, T.; Kobayashi, M.; Suzuki, Y.; Arase, Y.; Ikeda, K.; Miyakawa, Y.; et al. An open pilot study exploring the efficacy of fluvastatin, pegylated interferon and ribavirin in patients with hepatitis C virus genotype 1b in high viral loads. Intervirology 2009, 52, 43–48. [Google Scholar]
- Raal, F.J.; Santos, R.D.; Blom, D.J.; Marais, A.D.; Charng, M.J.; Cromwell, W.C.; Lachmann, R.H.; Gaudet, D.; Tan, J.L.; Chasan-Taber, S.; et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 375, 998–1006. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar]
- Goldwasser, J.; Cohen, P.Y.; Lin, W.; Kitsberg, D.; Balaguer, P.; Polyak, S.J.; Chung, R.T.; Yarmush, M.L.; Nahmias, Y. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J. Hepatol. 2011, 55, 963–971. [Google Scholar]
- Nahmias, Y.; Goldwasser, J.; Casali, M.; van Poll, D.; Wakita, T.; Chung, R.T.; Yarmush, M.L. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology 2008, 47, 1437–1445. [Google Scholar]
- Ciesek, S.; von Hahn, T.; Colpitts, C.C.; Schang, L.M.; Friesland, M.; Steinmann, J.; Manns, M.P.; Ott, M.; Wedemeyer, H.; Meuleman, P.; et al. The green tea polyphenol, epigallocatechin-3-gallate, Inhibits hepatitis C virus entry. Hepatology 2011, 54, 1947–1955. [Google Scholar]
- Calland, N.; Albecka, A.; Belouzard, S.; Wychowski, C.; Duverlie, G.; Descamps, V.; Hober, D.; Dubuisson, J.; Rouille, Y.; Seron, K. (−)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology 2012, 55, 720–729. [Google Scholar]
- Lin, Y.T.; Wu, Y.H.; Tseng, C.K.; Lin, C.K.; Chen, W.C.; Hsu, Y.C.; Lee, J.C. Green tea phenolic epicatechins inhibit hepatitis C virus replication via cycloxygenase-2 and attenuate virus-induced inflammation. PLoS One 2013, 8, e54466. [Google Scholar]
- Kaito, M.; Watanabe, S.; Tsukiyama-Kohara, K.; Yamaguchi, K.; Kobayashi, Y.; Konishi, M.; Yokoi, M.; Ishida, S.; Suzuki, S.; Kohara, M. Hepatitis C virus particle detected by immunoelectron microscopic study. J. Gen. Virol. 1994, 75, 1755–1760. [Google Scholar]
- Bradley, D.; McCaustland, K.; Krawczynski, K.; Spelbring, J.; Humphrey, C.; Cook, E.H. Hepatitis C virus: Buoyant density of the factor VIII-derived isolate in sucrose. J. Med. Virol. 1991, 34, 206–208. [Google Scholar]
- Bradley, D.W.; McCaustland, K.A.; Cook, E.H.; Schable, C.A.; Ebert, J.W.; Maynard, J.E. Posttransfusion non-A, non-B hepatitis in chimpanzees. Physicochemical evidence that the tubule-forming agent is a small, enveloped virus. Gastroenterology 1985, 88, 773–779. [Google Scholar]
- He, L.F.; Alling, D.; Popkin, T.; Shapiro, M.; Alter, H.J.; Purcell, R.H. Determining the size of non-A, non-B hepatitis virus by filtration. J. Infect. Dis. 1987, 156, 636–640. [Google Scholar] [CrossRef]
- Kanto, T.; Hayashi, N.; Takehara, T.; Hagiwara, H.; Mita, E.; Naito, M.; Kasahara, A.; Fusamoto, H.; Kamada, T. Density analysis of hepatitis C virus particle population in the circulation of infected hosts: Implications for virus neutralization or persistence. J. Hepatol. 1995, 22, 440–448. [Google Scholar]
- Pumeechockchai, W.; Bevitt, D.; Agarwal, K.; Petropoulou, T.; Langer, B.C.; Belohradsky, B.; Bassendine, M.F.; Toms, G.L. Hepatitis C virus particles of different density in the blood of chronically infected immunocompetent and immunodeficient patients: Implications for virus clearance by antibody. J. Med. Virol. 2002, 68, 335–342. [Google Scholar]
- Nielsen, S.U.; Bassendine, M.F.; Martin, C.; Lowther, D.; Purcell, P.J.; King, B.J.; Neely, D.; Toms, G.L. Characterization of hepatitis C RNA-containing particles from human liver by density and size. J. Gen. Virol. 2008, 89, 2507–2517. [Google Scholar] [CrossRef]
- Hijikata, M.; Shimizu, Y.K.; Kato, H.; Iwamoto, A.; Shih, J.W.; Alter, H.J.; Purcell, R.H.; Yoshikura, H. Equilibrium centrifugation studies of hepatitis C virus: Evidence for circulating immune complexes. J. Virol. 1993, 67, 1953–1958. [Google Scholar]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef]
- Gastaminza, P.; Kapadia, S.B.; Chisari, F.V. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 2006, 80, 11074–11081. [Google Scholar] [CrossRef]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef]
- Meex, S.J.; Andreo, U.; Sparks, J.D.; Fisher, E.A. Huh-7 or HepG2 cells: Which is the better model for studying human apolipoprotein-B100 assembly and secretion? J. Lipid. Res. 2011, 52, 152–158. [Google Scholar]
- Akazawa, D.; Date, T.; Morikawa, K.; Murayama, A.; Omi, N.; Takahashi, H.; Nakamura, N.; Ishii, K.; Suzuki, T.; Mizokami, M.; et al. Characterization of infectious hepatitis C virus from liver-derived cell lines. Biochem. Biophys. Res. Commun. 2008, 377, 747–751. [Google Scholar] [CrossRef]
- Gastaminza, P.; Dryden, K.A.; Boyd, B.; Wood, M.R.; Law, M.; Yeager, M.; Chisari, F.V. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J. Virol. 2010, 84, 10999–11009. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Meuleman, P.; Ploss, A.; Vanwolleghem, T.; Syder, A.J.; McKeating, J.A.; Lanford, R.E.; Feinstone, S.M.; Major, M.E.; Leroux-Roels, G.; et al. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. USA 2006, 103, 3805–3809. [Google Scholar] [CrossRef]
- Podevin, P.; Carpentier, A.; Pene, V.; Aoudjehane, L.; Carriere, M.; Zaidi, S.; Hernandez, C.; Calle, V.; Meritet, J.F.; Scatton, O.; et al. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology 2010, 139, 1355–1364. [Google Scholar] [CrossRef]
- Jammart, B.; Michelet, M.; Pecheur, E.I.; Parent, R.; Bartosch, B.; Zoulim, F.; Durantel, D. VLDL-producing and HCV-replicating HepG2 cells secrete no more LVP than VLDL-deficient Huh7.5 cells. J. Virol. 2013, 87, 5065–5080. [Google Scholar] [CrossRef]
- Aizaki, H.; Morikawa, K.; Fukasawa, M.; Hara, H.; Inoue, Y.; Tani, H.; Saito, K.; Nishijima, M.; Hanada, K.; Matsuura, Y.; et al. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J. Virol. 2008, 82, 5715–5724. [Google Scholar]
- Icard, V.; Diaz, O.; Scholtes, C.; Perrin-Cocon, L.; Ramiere, C.; Bartenschlager, R.; Penin, F.; Lotteau, V.; Andre, P. Secretion of hepatitis C virus envelope glycoproteins depends on assembly of apolipoprotein B positive lipoproteins. PLoS One 2009, 4, e4233. [Google Scholar]
- Meunier, J.C.; Russell, R.S.; Engle, R.E.; Faulk, K.N.; Purcell, R.H.; Emerson, S.U. Apolipoprotein c1 association with hepatitis C virus. J. Virol. 2008, 82, 9647–9656. [Google Scholar]
- Benga, W.J.; Krieger, S.E.; Dimitrova, M.; Zeisel, M.B.; Parnot, M.; Lupberger, J.; Hildt, E.; Luo, G.; McLauchlan, J.; Baumert, T.F.; et al. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 2010, 51, 43–53. [Google Scholar] [CrossRef]
- Owen, D.M.; Huang, H.; Ye, J.; Gale, M., Jr. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 2009, 394, 99–108. [Google Scholar] [CrossRef]
- Tomiyasu, K.; Walsh, B.W.; Ikewaki, K.; Judge, H.; Sacks, F.M. Differential metabolism of human VLDL according to content of ApoE and ApoC-III. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1494–1500. [Google Scholar] [CrossRef]
- Dreux, M.; Boson, B.; Ricard-Blum, S.; Molle, J.; Lavillette, D.; Bartosch, B.; Pecheur, E.I.; Cosset, F.L. The exchangeable apolipoprotein ApoC-I promotes membrane fusion of hepatitis C virus. J. Biol. Chem. 2007, 282, 32357–32369. [Google Scholar] [CrossRef]
- Mazumdar, B.; Banerjee, A.; Meyer, K.; Ray, R. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 2011, 54, 1149–1156. [Google Scholar] [CrossRef]
- Koutsoudakis, G.; Dragun, J.; Perez-Del-Pulgar, S.; Coto-Llerena, M.; Mensa, L.; Crespo, G.; Gonzalez, P.; Navasa, M.; Forns, X. Interplay between basic residues of hepatitis C virus glycoprotein E2 with viral receptors, neutralizing antibodies and lipoproteins. PLoS One 2012, 7, e52651. [Google Scholar]
- Zhong, J.; Gastaminza, P.; Chung, J.; Stamataki, Z.; Isogawa, M.; Cheng, G.; McKeating, J.A.; Chisari, F.V. Persistent hepatitis C virus infection in vitro: Coevolution of virus and host. J. Virol. 2006, 80, 11082–11093. [Google Scholar] [CrossRef]
- Sabahi, A.; Marsh, K.A.; Dahari, H.; Corcoran, P.; Lamora, J.M.; Yu, X.; Garry, R.F.; Uprichard, S.L. The rate of hepatitis C virus infection initiation in vitro is directly related to particle density. Virology 2010, 407, 110–119. [Google Scholar] [CrossRef]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.X. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar]
- Albecka, A.; Belouzard, S.; Op de Beeck, A.; Descamps, V.; Goueslain, L.; Bertrand-Michel, J.; Terce, F.; Duverlie, G.; Rouille, Y.; Dubuisson, J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 2012, 55, 998–1007. [Google Scholar] [CrossRef]
- Bishop, J.R.; Passos-Bueno, M.R.; Fong, L.; Stanford, K.I.; Gonzales, J.C.; Yeh, E.; Young, S.G.; Bensadoun, A.; Witztum, J.L.; Esko, J.D.; et al. Deletion of the basement membrane heparan sulfate proteoglycan type XVIII collagen causes hypertriglyceridemia in mice and humans. PLoS One 2010, 5, e13919. [Google Scholar]
- Zheng, C.; Murdoch, S.J.; Brunzell, J.D.; Sacks, F.M. Lipoprotein lipase bound to apolipoprotein B lipoproteins accelerates clearance of postprandial lipoproteins in humans. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 891–896. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Felmlee, D.J.; Baumert, T.F. Hepatitis C virus entry. Curr. Top. Microbiol. Immunol. 2013, 369, 87–112. [Google Scholar]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes Hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef]
- Fraser, R.; Dobbs, B.R.; Rogers, G.W. Lipoproteins and the liver sieve: The role of the fenestrated sinusoidal endothelium in lipoprotein metabolism, Atherosclerosis, and cirrhosis. Hepatology 1995, 21, 863–874. [Google Scholar]
- Ishibashi, S.; Perrey, S.; Chen, Z.; Osuga, J.; Shimada, M.; Ohashi, K.; Harada, K.; Yazaki, Y.; Yamada, N. Role of the low density lipoprotein (LDL) receptor pathway in the metabolism of chylomicron remnants. A quantitative study in knockout mice lacking the LDL receptor, apolipoprotein E, or both. J. Biol. Chem. 1996, 271, 22422–22427. [Google Scholar]
- Rohlmann, A.; Gotthardt, M.; Hammer, R.E.; Herz, J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J. Clin. Invest. 1998, 101, 689–695. [Google Scholar] [CrossRef]
- Wilsie, L.C.; Orlando, R.A. The low density lipoprotein receptor-related protein complexes with cell surface heparan sulfate proteoglycans to regulate proteoglycan-mediated lipoprotein catabolism. J. Biol. Chem. 2003, 278, 15758–15764. [Google Scholar] [CrossRef]
- Chen, K.; Liu, M.L.; Schaffer, L.; Li, M.; Boden, G.; Wu, X.; Williams, K.J. Type 2 diabetes in mice induces hepatic overexpression of sulfatase 2, A novel factor that suppresses uptake of remnant lipoproteins. Hepatology 2010, 52, 1957–1967. [Google Scholar] [CrossRef]
- Hassing, H.C.; Mooij, H.; Guo, S.; Monia, B.P.; Chen, K.; Kulik, W.; Dallinga-Thie, G.M.; Nieuwdorp, M.; Stroes, E.S.; Williams, K.J. Inhibition of hepatic sulfatase-2 in vivo: A novel strategy to correct diabetic dyslipidemia. Hepatology 2012, 55, 1746–1753. [Google Scholar] [CrossRef]
- MacArthur, J.M.; Bishop, J.R.; Stanford, K.I.; Wang, L.; Bensadoun, A.; Witztum, J.L.; Esko, J.D. Liver heparan sulfate proteoglycans mediate clearance of triglyceride-rich lipoproteins independently of LDL receptor family members. J. Clin. Invest. 2007, 117, 153–164. [Google Scholar] [CrossRef]
- Stanford, K.I.; Bishop, J.R.; Foley, E.M.; Gonzales, J.C.; Niesman, I.R.; Witztum, J.L.; Esko, J.D. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J. Clin. Invest. 2009, 119, 3236–3245. [Google Scholar]
- Trigatti, B.L.; Krieger, M.; Rigotti, A. Influence of the HDL receptor SR-BI on lipoprotein metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1732–1738. [Google Scholar] [CrossRef]
- Morrone, D.; Weintraub, W.S.; Toth, P.P.; Hanson, M.E.; Lowe, R.S.; Lin, J.; Shah, A.K.; Tershakovec, A.M. Lipid-altering efficacy of ezetimibe plus statin and statin monotherapy and identification of factors associated with treatment response: A pooled analysis of over 21,000 subjects from 27 clinical trials. Atherosclerosis 2012, 223, 251–261. [Google Scholar] [CrossRef]
- Thomssen, R.; Bonk, S. Virolytic action of lipoprotein lipase on hepatitis C virus in human sera. Med. Microbiol. Immunol. 2002, 191, 17–24. [Google Scholar] [CrossRef]
- Andreo, U.; Maillard, P.; Kalinina, O.; Walic, M.; Meurs, E.; Martinot, M.; Marcellin, P.; Budkowska, A. Lipoprotein lipase mediates hepatitis C virus (HCV) cell entry and inhibits HCV infection. Cell Microbiol. 2007, 9, 2445–2456. [Google Scholar] [CrossRef]
- Shimizu, Y.; Hishiki, T.; Sugiyama, K.; Ogawa, K.; Funami, K.; Kato, A.; Ohsaki, Y.; Fujimoto, T.; Takaku, H.; Shimotohno, K. Lipoprotein lipase and hepatic triglyceride lipase reduce the infectivity of hepatitis C virus (HCV) through their catalytic activities on HCV-associated lipoproteins. Virology 2010, 407, 152–159. [Google Scholar] [CrossRef]
- Maillard, P.; Walic, M.; Meuleman, P.; Roohvand, F.; Huby, T.; Le Goff, W.; Leroux-Roels, G.; Pecheur, E.I.; Budkowska, A. Lipoprotein lipase inhibits hepatitis C virus (HCV) infection by blocking virus cell entry. PLoS One 2011, 6, e26637. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.Y.; Lin, C.C.; Lee, J.C.; Wang, S.W.; Cheng, P.N.; Wu, I.C.; Chang, T.T.; Lai, M.D.; Shieh, D.B.; Young, K.C. Very low-density lipoprotein/lipo-viro particles reverse lipoprotein lipase-mediated inhibition of hepatitis C virus infection via apolipoprotein C-III. Gut 2012. [Google Scholar] [CrossRef]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; Van Kuppevelt, T.H.; Depla, E.; et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef]
- Barth, H.; Schnober, E.K.; Zhang, F.; Linhardt, R.J.; Depla, E.; Boson, B.; Cosset, F.L.; Patel, A.H.; Blum, H.E.; Baumert, T.F. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J. Virol. 2006, 80, 10579–10590. [Google Scholar] [CrossRef]
- Jiang, J.; Cun, W.; Wu, X.; Shi, Q.; Tang, H.; Luo, G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 2012, 86, 7256–7267. [Google Scholar] [CrossRef]
- Kapadia, S.B.; Barth, H.; Baumert, T.; McKeating, J.A.; Chisari, F.V. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Viro.l 2007, 81, 374–383. [Google Scholar] [CrossRef]
- Fuki, I.V.; Meyer, M.E.; Williams, K.J. Transmembrane and cytoplasmic domains of syndecan mediate a multi-step endocytic pathway involving detergent-insoluble membrane rafts. Biochem. J. 2000, 351, 607–612. [Google Scholar] [CrossRef]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO. J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef]
- Barth, H.; Cerino, R.; Arcuri, M.; Hoffmann, M.; Schurmann, P.; Adah, M.I.; Gissler, B.; Zhao, X.; Ghisetti, V.; Lavezzo, B.; et al. Scavenger receptor class B type I and hepatitis C virus infection of primary tupaia hepatocytes. J. Virol. 2005, 79, 5774–5785. [Google Scholar] [CrossRef]
- Barth, H.; Schnober, E.K.; Neumann-Haefelin, C.; Thumann, C.; Zeisel, M.B.; Diepolder, H.M.; Hu, Z.; Liang, T.J.; Blum, H.E.; Thimme, R.; et al. Scavenger receptor class B is required for hepatitis C virus uptake and cross-presentation by human dendritic cells. J. Virol. 2008, 82, 3466–3479. [Google Scholar] [CrossRef]
- Catanese, M.T.; Ansuini, H.; Graziani, R.; Huby, T.; Moreau, M.; Ball, J.K.; Paonessa, G.; Rice, C.M.; Cortese, R.; Vitelli, A.; et al. Role of scavenger receptor class B type I in hepatitis C virus entry: Kinetics and molecular determinants. J. Virol. 2010, 84, 34–43. [Google Scholar] [CrossRef]
- Dreux, M.; Dao Thi, V.L.; Fresquet, J.; Guerin, M.; Julia, Z.; Verney, G.; Durantel, D.; Zoulim, F.; Lavillette, D.; Cosset, F.L.; et al. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog. 2009, 5, e1000310. [Google Scholar] [CrossRef]
- Dreux, M.; Pietschmann, T.; Granier, C.; Voisset, C.; Ricard-Blum, S.; Mangeot, P.E.; Keck, Z.; Foung, S.; Vu-Dac, N.; Dubuisson, J.; et al. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J. Biol. Chem. 2006, 281, 18285–18295. [Google Scholar] [CrossRef]
- Haberstroh, A.; Schnober, E.K.; Zeisel, M.B.; Carolla, P.; Barth, H.; Blum, H.E.; Cosset, F.L.; Koutsoudakis, G.; Bartenschlager, R.; Union, A.; et al. Neutralizing host responses in hepatitis C virus infection target viral entry at postbinding steps and membrane fusion. Gastroenterology 2008, 135, 1719–1728. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Da Costa, D.; Baumert, T.F. Opening the door for hepatitis C virus infection in genetically humanized mice. Hepatology 2011, 54, 1873–1875. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F.L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C.; et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef]
- Masson, D.; Koseki, M.; Ishibashi, M.; Larson, C.J.; Miller, S.G.; King, B.D.; Tall, A.R. Increased HDL cholesterol and apoA-I in humans and mice treated with a novel SR-BI inhibitor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2054–2060. [Google Scholar] [CrossRef]
- Syder, A.J.; Lee, H.; Zeisel, M.B.; Grove, J.; Soulier, E.; Macdonald, J.; Chow, S.; Chang, J.; Baumert, T.F.; McKeating, J.A.; et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J. Hepatol. 2011, 54, 48–55. [Google Scholar] [CrossRef]
- Out, R.; Hoekstra, M.; de Jager, S.C.; de Vos, P.; van der Westhuyzen, D.R.; Webb, N.R.; Van Eck, M.; Biessen, E.A.; Van Berkel, T.J. Adenovirus-mediated hepatic overexpression of scavenger receptor class B type I accelerates chylomicron metabolism in C57BL/6J mice. J. Lipid Res. 2005, 46, 1172–1181. [Google Scholar] [CrossRef]
- Van Eck, M.; Hoekstra, M.; Out, R.; Bos, I.S.; Kruijt, J.K.; Hildebrand, R.B.; Van Berkel, T.J. Scavenger receptor BI facilitates the metabolism of VLDL lipoproteins in vivo. J. Lipid Res. 2008, 49, 136–146. [Google Scholar] [CrossRef]
- Dao Thi, V.L.; Granier, C.; Zeisel, M.B.; Guerin, M.; Mancip, J.; Granio, O.; Penin, F.; Lavillette, D.; Bartenschlager, R.; Baumert, T.F.; et al. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J. Biol. Chem. 2012, 287, 31242–31257. [Google Scholar]
- Zahid, M.N.; Turek, M.; Xiao, F.; Thi, V.L.; Guerin, M.; Fofana, I.; Bachellier, P.; Thompson, J.; Delang, L.; Neyts, J.; et al. The post-binding activity of scavenger receptor BI mediates initiation of hepatitis C virus infection and viral dissemination. Hepatology 2013, 57, 492–504. [Google Scholar] [CrossRef]
- Sainz, B., Jr.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef]
- Haid, S.; Pietschmann, T.; Pecheur, E.I. Low pH-dependent hepatitis C virus membrane fusion depends on E2 integrity, target lipid composition, and density of virus particles. J. Biol. Chem. 2009, 284, 17657–17667. [Google Scholar]
- Pecheur, E.I.; Diaz, O.; Molle, J.; Icard, V.; Bonnafous, P.; Lambert, O.; Andre, P. Morphological characterization and fusion properties of triglyceride-rich lipoproteins obtained from cells transduced with hepatitis C virus glycoproteins. J. Biol. Chem. 2010, 285, 25802–25811. [Google Scholar]
- Voisset, C.; Lavie, M.; Helle, F.; Op De Beeck, A.; Bilheu, A.; Bertrand-Michel, J.; Terce, F.; Cocquerel, L.; Wychowski, C.; Vu-Dac, N.; et al. Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell Microbiol. 2008, 10, 606–617. [Google Scholar]
- Rocha-Perugini, V.; Lavie, M.; Delgrange, D.; Canton, J.; Pillez, A.; Potel, J.; Lecoeur, C.; Rubinstein, E.; Dubuisson, J.; Wychowski, C.; et al. The association of CD81 with tetraspanin-enriched microdomains is not essential for Hepatitis C virus entry. BMC Microbiol. 2009, 9, 111. [Google Scholar]
- Packard, C.J.; Shepherd, J. Lipoprotein heterogeneity and apolipoprotein B metabolism. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3542–3556. [Google Scholar]
- Yamamoto, M.; Aizaki, H.; Fukasawa, M.; Teraoka, T.; Miyamura, T.; Wakita, T.; Suzuki, T. Structural requirements of virion-associated cholesterol for infectivity, buoyant density and apolipoprotein association of hepatitis C virus. J. Gen. Virol. 2011, 92, 2082–2087. [Google Scholar] [CrossRef]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef]
- Aizaki, H.; Lee, K.J.; Sung, V.M.; Ishiko, H.; Lai, M.M. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 2004, 324, 450–461. [Google Scholar] [CrossRef]
- Wang, C.; Gale, M., Jr.; Keller, B.C.; Huang, H.; Brown, M.S.; Goldstein, J.L.; Ye, J. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol. Cell 2005, 18, 425–434. [Google Scholar] [CrossRef]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2566. [Google Scholar] [CrossRef]
- Scholtes, C.; Diaz, O.; Icard, V.; Kaul, A.; Bartenschlager, R.; Lotteau, V.; Andre, P. Enhancement of genotype 1 hepatitis C virus replication by bile acids through FXR. J. Hepatol. 2008, 48, 192–199. [Google Scholar]
- Chhatwal, P.; Bankwitz, D.; Gentzsch, J.; Frentzen, A.; Schult, P.; Lohmann, V.; Pietschmann, T. Bile acids specifically increase hepatitis C virus RNA-replication. PLoS One 2012, 7, e36029. [Google Scholar]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V., Jr.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar]
- Ploen, D.; Hafirassou, M.L.; Himmelsbach, K.; Sauter, D.; Biniossek, M.L.; Weiss, T.S.; Baumert, T.F.; Schuster, C.; Hildt, E. TIP47 plays a crucial role in the life cycle of hepatitis C virus. J. Hepatol. 2013. [Google Scholar] [CrossRef]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar]
- Shimizu, Y.; Hishiki, T.; Ujino, S.; Sugiyama, K.; Funami, K.; Shimotohno, K. Lipoprotein component associated with hepatitis C virus is essential for virus infectivity. Curr. Opin. Virol. 2011, 1, 19–26. [Google Scholar] [CrossRef]
- Shi, S.T.; Lee, K.J.; Aizaki, H.; Hwang, S.B.; Lai, M.M. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 2003, 77, 4160–4168. [Google Scholar]
- Hirata, Y.; Ikeda, K.; Sudoh, M.; Tokunaga, Y.; Suzuki, A.; Weng, L.; Ohta, M.; Tobita, Y.; Okano, K.; Ozeki, K.; et al. Self-enhancement of hepatitis C virus replication by promotion of specific sphingolipid biosynthesis. PLoS Pathog. 2012, 8, e1002860. [Google Scholar]
- Sakamoto, H.; Okamoto, K.; Aoki, M.; Kato, H.; Katsume, A.; Ohta, A.; Tsukuda, T.; Shimma, N.; Aoki, Y.; Arisawa, M.; et al. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat. Chem. Biol. 2005, 1, 333–337. [Google Scholar]
- Weng, L.; Hirata, Y.; Arai, M.; Kohara, M.; Wakita, T.; Watashi, K.; Shimotohno, K.; He, Y.; Zhong, J.; Toyoda, T. Sphingomyelin activates hepatitis C virus RNA polymerase in a genotype-specific manner. J. Virol. 2010, 84, 11761–11770. [Google Scholar]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Invest. 2012, 122, 2871–2883. [Google Scholar]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Invest. 2012, 122, 2884–2897. [Google Scholar]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar]
- Sarasin-Filipowicz, M.; Krol, J.; Markiewicz, I.; Heim, M.H.; Filipowicz, W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat. Med. 2009, 15, 31–33. [Google Scholar]
- Bulankina, A.V.; Deggerich, A.; Wenzel, D.; Mutenda, K.; Wittmann, J.G.; Rudolph, M.G.; Burger, K.N.; Honing, S. TIP47 functions in the biogenesis of lipid droplets. J. Cell Biol. 2009, 185, 641–655. [Google Scholar]
- Martin, S.; Parton, R.G. Lipid droplets: A unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar]
- Sato, S.; Fukasawa, M.; Yamakawa, Y.; Natsume, T.; Suzuki, T.; Shoji, I.; Aizaki, H.; Miyamura, T.; Nishijima, M. Proteomic profiling of lipid droplet proteins in hepatoma cell lines expressing hepatitis C virus core protein. J. Biochem. 2006, 139, 921–930. [Google Scholar] [CrossRef]
- Boulant, S.; Douglas, M.W.; Moody, L.; Budkowska, A.; Targett-Adams, P.; McLauchlan, J. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 2008, 9, 1268–1282. [Google Scholar] [CrossRef]
- Boulant, S.; Targett-Adams, P.; McLauchlan, J. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 2007, 88, 2204–2213. [Google Scholar]
- Shavinskaya, A.; Boulant, S.; Penin, F.; McLauchlan, J.; Bartenschlager, R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 2007, 282, 37158–37169. [Google Scholar]
- Targett-Adams, P.; Hope, G.; Boulant, S.; McLauchlan, J. Maturation of hepatitis C virus core protein by signal peptide peptidase is required for virus production. J. Biol. Chem. 2008, 283, 16850–16859. [Google Scholar]
- Schuster, C.; Lefevre, M.; Baumert, T.F. Triglyceride synthesis and hepatitis C virus production: Identification of a novel host factor as antiviral target. Hepatology 2011, 53, 1046–1048. [Google Scholar]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Zayas, M.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009, 5, e1000546. [Google Scholar] [CrossRef]
- Anderson, L.J.; Lin, K.; Compton, T.; Wiedmann, B. Inhibition of cyclophilins alters lipid trafficking and blocks hepatitis C virus secretion. Virol. J. 2011, 8, e329. [Google Scholar]
- Olmstead, A.D.; Knecht, W.; Lazarov, I.; Dixit, S.B.; Jean, F. Human subtilase SKI-1/S1P is a master regulator of the HCV Lifecycle and a potential host cell target for developing indirect-acting antiviral agents. PLoS Pathog. 2012, 8, e1002468. [Google Scholar] [CrossRef]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M., Jr.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar]
- Perlemuter, G.; Sabile, A.; Letteron, P.; Vona, G.; Topilco, A.; Chretien, Y.; Koike, K.; Pessayre, D.; Chapman, J.; Barba, G.; et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: A model of viral-related steatosis. FASEB. J. 2002, 16, 185–194. [Google Scholar] [CrossRef]
- Petit, J.M.; Benichou, M.; Duvillard, L.; Jooste, V.; Bour, J.B.; Minello, A.; Verges, B.; Brun, J.M.; Gambert, P.; Hillon, P. Hepatitis C virus-associated hypobetalipoproteinemia is correlated with plasma viral load, steatosis, and liver fibrosis. Am. J. Gastroenterol. 2003, 98, 1150–1154. [Google Scholar]
- Jiang, J.; Luo, G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 2009, 83, 12680–12691. [Google Scholar] [CrossRef]
- Steenbergen, R.H.; Joyce, M.A.; Lund, G.; Lewis, J.; Chen, R.; Barsby, N.; Douglas, D.; Zhu, L.F.; Tyrrell, D.L.; Kneteman, N.M. Lipoprotein profiles in SCID/uPA mice transplanted with human hepatocytes become human-like and correlate with HCV infection success. Am. J. Physiol. Gastrointest. L. 2010, 299, G844–G854. [Google Scholar] [CrossRef]
- Coller, K.E.; Heaton, N.S.; Berger, K.L.; Cooper, J.D.; Saunders, J.L.; Randall, G. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 2012, 8, e1002466. [Google Scholar] [CrossRef]
- Amako, Y.; Sarkeshik, A.; Hotta, H.; Yates, J., 3rd; Siddiqui, A. Role of oxysterol binding protein in hepatitis C virus infection. J. Virol. 2009, 83, 9237–9246. [Google Scholar]
- Amako, Y.; Syed, G.H.; Siddiqui, A. Protein kinase D negatively regulates hepatitis C virus secretion through phosphorylation of oxysterol-binding protein and ceramide transfer protein. J. Biol. Chem. 2011, 286, 11265–11274. [Google Scholar]
- Roe, B.; Kensicki, E.; Mohney, R.; Hall, W.W. Metabolomic profile of hepatitis C virus-infected hepatocytes. PLoS One 2011, 6, e23641. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Felmlee, D.J.; Hafirassou, M.L.; Lefevre, M.; Baumert, T.F.; Schuster, C. Hepatitis C Virus, Cholesterol and Lipoproteins — Impact for the Viral Life Cycle and Pathogenesis of Liver Disease. Viruses 2013, 5, 1292-1324. https://doi.org/10.3390/v5051292
Felmlee DJ, Hafirassou ML, Lefevre M, Baumert TF, Schuster C. Hepatitis C Virus, Cholesterol and Lipoproteins — Impact for the Viral Life Cycle and Pathogenesis of Liver Disease. Viruses. 2013; 5(5):1292-1324. https://doi.org/10.3390/v5051292
Chicago/Turabian StyleFelmlee, Daniel J., Mohamed Lamine Hafirassou, Mathieu Lefevre, Thomas F. Baumert, and Catherine Schuster. 2013. "Hepatitis C Virus, Cholesterol and Lipoproteins — Impact for the Viral Life Cycle and Pathogenesis of Liver Disease" Viruses 5, no. 5: 1292-1324. https://doi.org/10.3390/v5051292