HCV and Oxidative Stress in the Liver

 ,
,  , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress in Patients with Chronic Hepatitis C
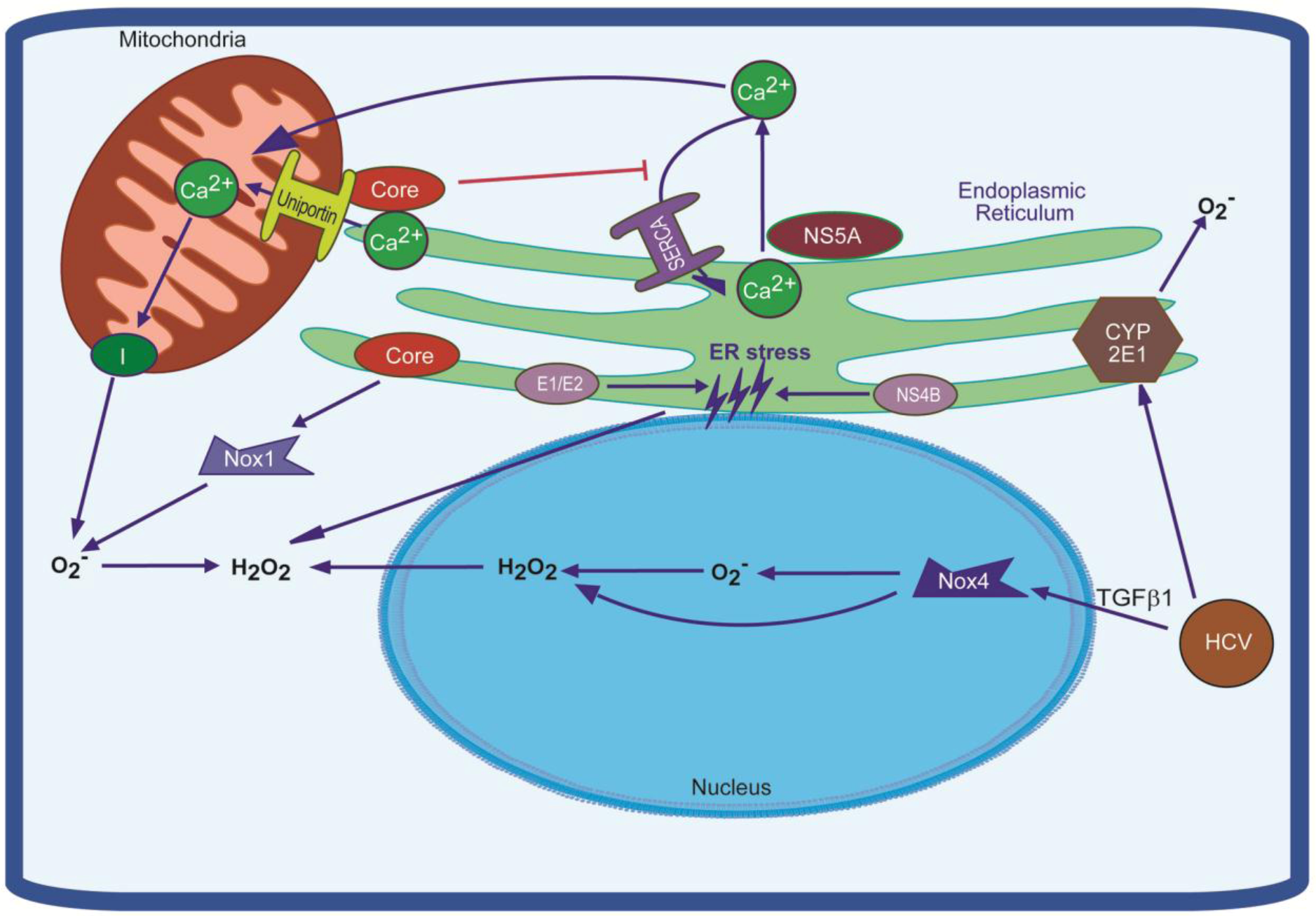
3. Sources of Reactive Oxygen Species in HCV-Infected Cell and their Regulation by HCV
4. Interplay between HCV and the Antioxidant Defense Nrf2/ARE Pathway
5. Effect of Oxidative Stress on HCV Propagation: Facts and Assumptions
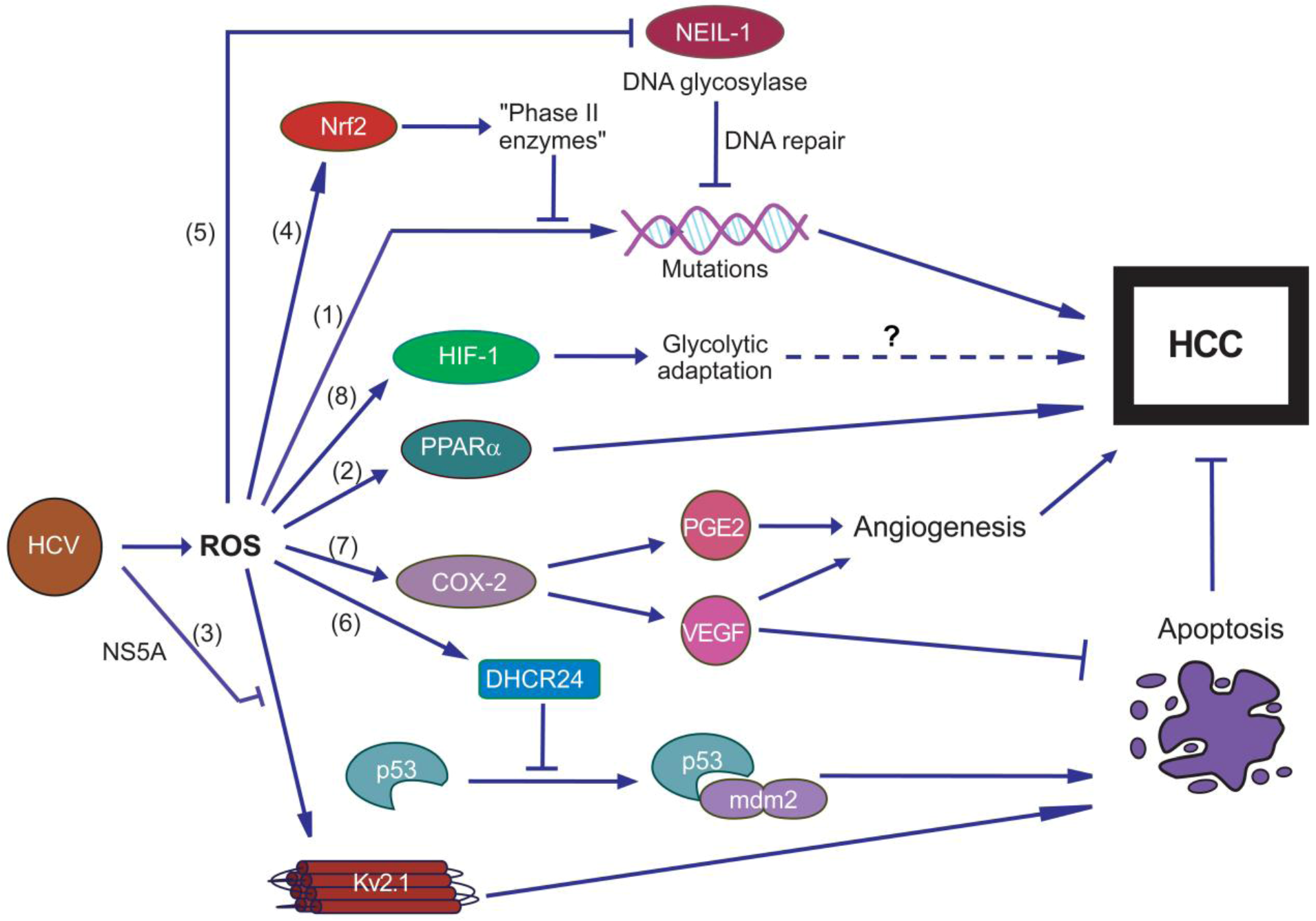
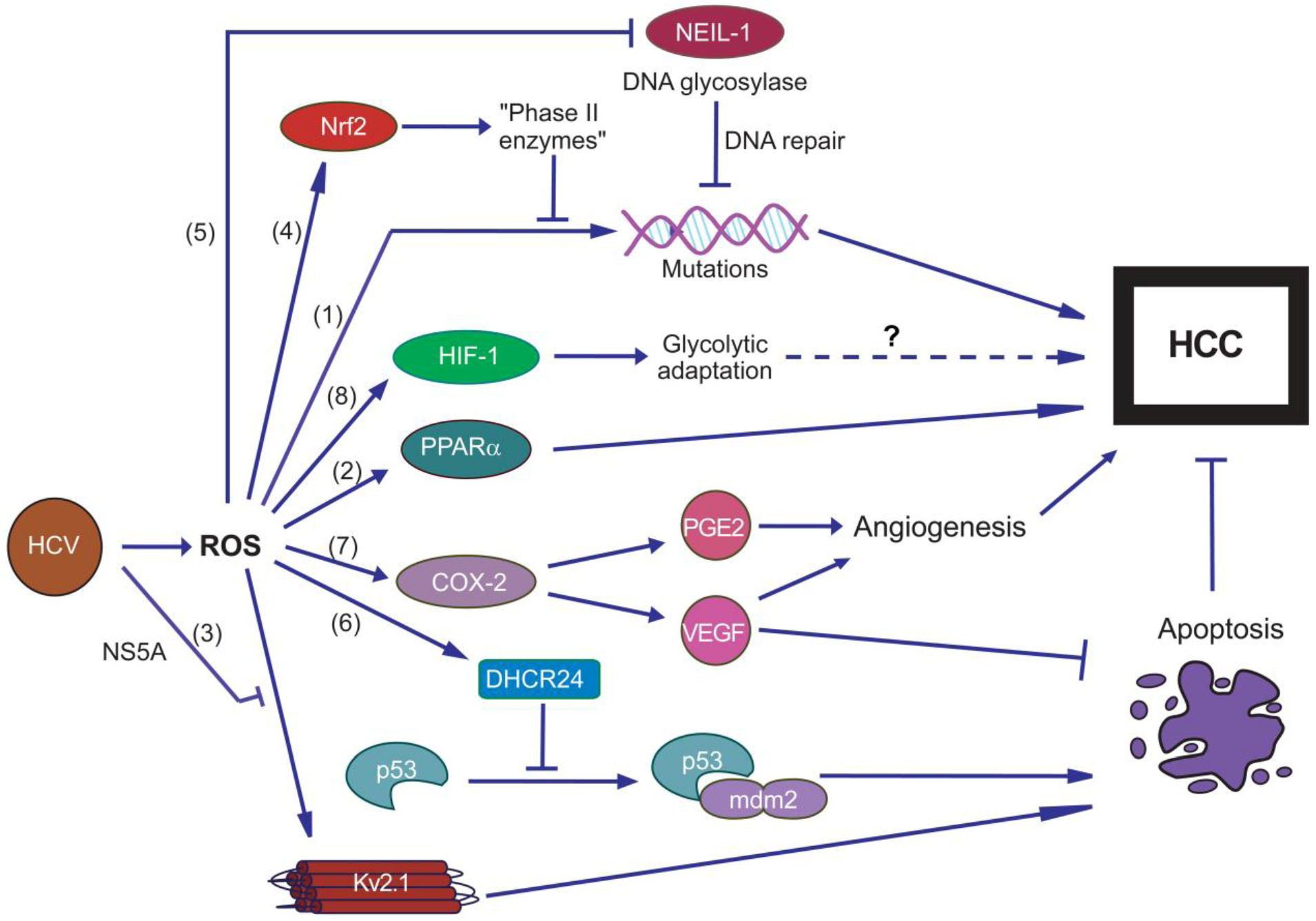
6. Oxidative Stress and HCV-Associated Diseases
6.1. Hepatocellular Carcinoma
6.2. Liver Fibrosis
6.3. Insulin Resistance and Steatosis
7. Oxidative Stress and Iron Overload: “The Chicken or the Egg” Dilemma
8. Future Directions
Acknowledgments
Conflict of Interest
References and Notes
- Hepatitis C. Available online: http://www.who.int/mediacentre/factsheets/fs164/en/ (accessed on 01 December 2012).
- Rosen, H.R.; Gretch, D.R. Hepatitis C virus: Current understanding and prospects for future therapies. Mol. Med. Today 1999, 5, 393–399. [Google Scholar]
- Lemon, S.M.; Walker, C.M.; Alter, M.J.; Yi, M.-K. Hepatitis C virus. In Fields Virology, 5th; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2007; Vol. 1, pp. 1253–1304. [Google Scholar]
- Hartridge-Lambert, S.K.; Stein, E.M.; Markowitz, A.J.; Portlock, C.S. Hepatitis C and non-Hodgkin lymphoma: the clinical perspective. Hepatology 2012, 55, 634–641. [Google Scholar] [CrossRef]
- Nocente, R.; Ceccanti, M.; Bertazzoni, G.; Cammarota, G.; Silveri, N.G.; Gasbarrini, G. HCV infection and extrahepatic manifestations. Hepatogastroenterology 2003, 50, 1149–1154. [Google Scholar]
- Jin, D.Y. Molecular pathogenesis of hepatitis C virus-associated hepatocellular carcinoma. Front Biosci. 2007, 12, 222–233. [Google Scholar]
- Riggio, O.; Montagnese, F.; Fiore, P.; Folino, S.; Giambartolomei, S.; Gandin, C.; Merli, M.; Quinti, I.; Violante, N.; Caroli, S.; et al. Iron overload in patients with chronic viral hepatitis: How common is it? Am. J. Gastroenterol. 1997, 92, 1298–1301. [Google Scholar]
- Adinolfi, L.E.; Restivo, L.; Zampino, R.; Lonardo, A.; Loria, P. Metabolic alterations and chronic hepatitis C: Treatment strategies. Expert Opin. Pharmacother. 2011, 12, 2215–2234. [Google Scholar] [CrossRef]
- Arrese, M.; Riquelme, A.; Soza, A. Insulin resistance, hepatic steatosis and hepatitis C: A complex relationship with relevant clinical implications. Ann. Hepatol. 2010, 9, 112–118. [Google Scholar]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal 2007, 9, 49–89. [Google Scholar]
- Go, Y.M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef]
- Smirnova, O.A.; Ivanov, A.V.; Ivanova, O.N.; Valuev-Ellison, V.T.; Kochetkov, S.N. Cell Defense Systems against Oxidative Stress and Endoplasmic Reticulum Stress: Mechanisms of Regulation and the Effect of Hepatitis C Virus. Mol. Biol. (Mosk) 2011, 45, 110–122. [Google Scholar]
- Aleksunes, L.M.; Manautou, J.E. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol. Pathol. 2007, 35, 459–473. [Google Scholar] [CrossRef]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; Van Breusegem, F. ROS signaling: the new wave? Trends Plant. Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef]
- Desikan, R.; Hancock, J.; Neill, S. Reactive Oxygen Species as Signalling Molecules. In Antioxidants and Reactive Oxygen Species in Plants; Blackwell Publishing Ltd: Oxford, UK, 2007; pp. 169–196. [Google Scholar]
- Choi, J. Oxidative stress, endogenous antioxidants, alcohol, and hepatitis C: Pathogenic interactions and therapeutic considerations. Free Radic. Biol. Med. 2012, 52, 1135–1150. [Google Scholar] [CrossRef]
- Valgimigli, M.; Valgimigli, L.; Trere, D.; Gaiani, S.; Pedulli, G.F.; Gramantieri, L.; Bolondi, L. Oxidative stress EPR measurement in human liver by radical-probe technique. Correlation with etiology, histology and cell proliferation. Free Radic. Res. 2002, 36, 939–948. [Google Scholar]
- Valgimigli, L.; Valgimigli, M.; Gaiani, S.; Pedulli, G.F.; Bolondi, L. Measurement of oxidative stress in human liver by EPR spin-probe technique. Free Radic. Res. 2000, 33, 167–178. [Google Scholar] [CrossRef]
- Bhargava, A.; Raghuram, G.V.; Pathak, N.; Varshney, S.; Jatawa, S.K.; Jain, D.; Mishra, P.K. Occult hepatitis C virus elicits mitochondrial oxidative stress in lymphocytes and triggers PI3-kinase-mediated DNA damage response. Free Radic. Biol. Med. 2011, 51, 1806–1814. [Google Scholar] [CrossRef]
- Capone, F.; Guerriero, E.; Sorice, A.; Maio, P.; Colonna, G.; Castello, G.; Costantini, S. Characterization of metalloproteinases, oxidative status and inflammation levels in the different stages of fibrosis in HCV patients. Clin. Biochem. 2012, 45, 525–529. [Google Scholar] [CrossRef]
- Serejo, F.; Emerit, I.; Filipe, P.M.; Fernandes, A.C.; Costa, M.A.; Freitas, J.P.; de Moura, M.C. Oxidative stress in chronic hepatitis C: The effect of interferon therapy and correlation with pathological features. Can. J. Gastroenterol. 2003, 17, 644–650. [Google Scholar]
- Emerit, I.; Serejo, F.; Filipe, P.; Alaoui Youssefi, A.; Fernandes, A.; Costa, A.; Freitas, J.; Ramalho, F.; Baptista, A.; Carneiro de Moura, M. Clastogenic factors as biomarkers of oxidative stress in chronic hepatitis C. Digestion 2000, 62, 200–207. [Google Scholar] [CrossRef]
- Yuan, L.; Kaplowitz, N. Glutathione in liver diseases and hepatotoxicity. Mol. Aspects Med. 2009, 30, 29–41. [Google Scholar] [CrossRef]
- Swietek, K.; Juszczyk, J. Reduced glutathione concentration in erythrocytes of patients with acute and chronic viral hepatitis. J. Viral. Hepat. 1997, 4, 139–141. [Google Scholar]
- Look, M.P.; Gerard, A.; Rao, G.S.; Sudhop, T.; Fischer, H.P.; Sauerbruch, T.; Spengler, U. Interferon/antioxidant combination therapy for chronic hepatitis C—a controlled pilot trial. Antiviral Res. 1999, 43, 113–122. [Google Scholar] [CrossRef]
- Vendemiale, G.; Grattagliano, I.; Portincasa, P.; Serviddio, G.; Palasciamo, G.; Altomare, E. Oxidative stress in symptom-free HCV carriers: relation with ALT flare-up. Eur. J. Clin. Invest. 2001, 31, 54–63. [Google Scholar] [CrossRef]
- Jain, S.K.; Pemberton, P.W.; Smith, A.; McMahon, R.F.; Burrows, P.C.; Aboutwerat, A.; Warnes, T.W. Oxidative stress in chronic hepatitis C: not just a feature of late stage disease. J. Hepatol. 2002, 36, 805–811. [Google Scholar]
- Yadav, D.; Hertan, H.I.; Schweitzer, P.; Norkus, E.P.; Pitchumoni, C.S. Serum and liver micronutrient antioxidants and serum oxidative stress in patients with chronic hepatitis C. Am. J. Gastroenterol. 2002, 97, 2634–2639. [Google Scholar] [CrossRef]
- Salem, T.A.; El-Refaei, M.F.; Badra, G.A. Study of antioxidant enzymes level and phagocytic activity in chronic liver disease patients. Egypt. J. Immunol. 2003, 10, 37–45. [Google Scholar]
- Venturini, D.; Simao, A.N.; Barbosa, D.S.; Lavado, E.L.; Narciso, V.E.; Dichi, I.; Dichi, J.B. Increased oxidative stress, decreased total antioxidant capacity, and iron overload in untreated patients with chronic hepatitis C. Dig. Dis. Sci. 2010, 55, 1120–1127. [Google Scholar] [CrossRef]
- Barbaro, G.; Di Lorenzo, G.; Ribersani, M.; Soldini, M.; Giancaspro, G.; Bellomo, G.; Belloni, G.; Grisorio, B.; Barbarini, G. Serum ferritin and hepatic glutathione concentrations in chronic hepatitis C patients related to the hepatitis C virus genotype. J. Hepatol. 1999, 30, 774–782. [Google Scholar] [CrossRef]
- Bessa, S.S.; Mohamed Ali, E.M.; Abd El-Wahab Ael, S.; Nor El-Din, S.A. Heme oxygenase-1 mRNA expression in egyptian patients with chronic liver disease. Hepat. Mon. 2012, 12, 278–285. [Google Scholar]
- Boya, P.; de la Pena, A.; Beloqui, O.; Larrea, E.; Conchillo, M.; Castelruiz, Y.; Civeira, M.P.; Prieto, J. Antioxidant status and glutathione metabolism in peripheral blood mononuclear cells from patients with chronic hepatitis C. J. Hepatol. 1999, 31, 808–814. [Google Scholar] [CrossRef]
- Levent, G.; Ali, A.; Ahmet, A.; Polat, E.C.; Aytac, C.; Ayse, E.; Ahmet, S. Oxidative stress and antioxidant defense in patients with chronic hepatitis C patients before and after pegylated interferon alfa-2b plus ribavirin therapy. J. Transl. Med. 2006, 4, 25. [Google Scholar]
- Osman, H.G.; Gabr, O.M.; Lotfy, S.; Gabr, S. Serum levels of bcl-2 and cellular oxidative stress in patients with viral hepatitis. Indian J. Med. Microbiol. 2007, 25, 323–329. [Google Scholar] [CrossRef]
- Larrea, E.; Beloqui, O.; Munoz-Navas, M.A.; Civeira, M.P.; Prieto, J. Superoxide dismutase in patients with chronic hepatitis C virus infection. Free Radic. Biol. Med. 1998, 24, 1235–1241. [Google Scholar] [CrossRef]
- Sumida, Y.; Nakashima, T.; Yoh, T.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Sakamoto, Y.; Okanoue, T.; Kashima, K.; Nakamura, H.; et al. Serum thioredoxin levels as an indicator of oxidative stress in patients with hepatitis C virus infection. J. Hepatol. 2000, 33, 616–622. [Google Scholar] [CrossRef]
- Nakashima, T.; Sumida, Y.; Yoh, T.; Kakisaka, Y.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Kashima, K.; Nakamura, H.; Yodoi, J. Thioredoxin levels in the sera of untreated viral hepatitis patients and those treated with glycyrrhizin or ursodeoxycholic acid. Antioxid. Redox Signal 2000, 2, 687–694. [Google Scholar] [CrossRef]
- Cardin, R.; Saccoccio, G.; Masutti, F.; Bellentani, S.; Farinati, F.; Tiribelli, C. DNA oxidative damage in leukocytes correlates with the severity of HCV-related liver disease: Validation in an open population study. J. Hepatol. 2001, 34, 587–592. [Google Scholar] [CrossRef]
- Zuwala-Jagiello, J.; Pazgan-Simon, M.; Simon, K.; Warwas, M. Advanced oxidation protein products and inflammatory markers in liver cirrhosis: A comparison between alcohol-related and HCV-related cirrhosis. Acta Biochim. Pol. 2011, 58, 59–65. [Google Scholar]
- Kitada, T.; Seki, S.; Iwai, S.; Yamada, T.; Sakaguchi, H.; Wakasa, K. In situ detection of oxidative DNA damage, 8-hydroxydeoxyguanosine, in chronic human liver disease. J. Hepatol. 2001, 35, 613–618. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS One 2011, 6, e24957. [Google Scholar]
- Pal, S.; Polyak, S.J.; Bano, N.; Qiu, W.C.; Carithers, R.L.; Shuhart, M.; Gretch, D.R.; Das, A. Hepatitis C virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme NEIL1. J. Gastroenterol. Hepatol. 2010, 25, 627–634. [Google Scholar] [CrossRef]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 2002, 122, 366–375. [Google Scholar] [CrossRef]
- Garcia-Mediavilla, M.V.; Sanchez-Campos, S.; Gonzalez-Perez, P.; Gomez-Gonzalo, M.; Majano, P.L.; Lopez-Cabrera, M.; Clemente, G.; Garcia-Monzon, C.; Gonzalez-Gallego, J. Differential contribution of hepatitis C virus NS5A and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte-derived cells. J. Hepatol. 2005, 43, 606–613. [Google Scholar] [CrossRef]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar]
- Piccoli, C.; Scrima, R.; Quarato, G.; D'Aprile, A.; Ripoli, M.; Lecce, L.; Boffoli, D.; Moradpour, D.; Capitanio, N. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 2007, 46, 58–65. [Google Scholar] [CrossRef]
- Ming-Ju, H.; Yih-Shou, H.; Tzy-Yen, C.; Hui-Ling, C. Hepatitis C virus E2 protein induce reactive oxygen species (ROS)-related fibrogenesis in the HSC-T6 hepatic stellate cell line. J. Cell Biochem. 2011, 112, 233–243. [Google Scholar] [CrossRef]
- Li, S.; Ye, L.; Yu, X.; Xu, B.; Li, K.; Zhu, X.; Liu, H.; Wu, X.; Kong, L. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology 2009, 391, 257–264. [Google Scholar] [CrossRef]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. P. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef]
- Ando, M.; Korenaga, M.; Hino, K.; Ikeda, M.; Kato, N.; Nishina, S.; Hidaka, I.; Sakaida, I. Mitochondrial electron transport inhibition in full genomic hepatitis C virus replicon cells is restored by reducing viral replication. Liver Int. 2008, 28, 1158–1166. [Google Scholar] [CrossRef]
- Wang, T.; Campbell, R.V.; Yi, M.K.; Lemon, S.M.; Weinman, S.A. Role of Hepatitis C virus core protein in viral-induced mitochondrial dysfunction. J. Viral Hepat. 2010, 17, 784–793. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Matsuda, M.; Aizaki, H.; Moriya, K.; Miyoshi, H.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Miyamura, T.; Suzuki, T.; et al. Proteomics analysis of mitochondrial proteins reveals overexpression of a mitochondrial protein chaperon, prohibitin, in cells expressing hepatitis C virus core protein. Hepatology 2009, 50, 378–386. [Google Scholar] [CrossRef]
- Bourges, I.; Ramus, C.; Mousson de Camaret, B.; Beugnot, R.; Remacle, C.; Cardol, P.; Hofhaus, G.; Issartel, J.P. Structural organization of mitochondrial human complex I: Role of the ND4 and ND5 mitochondria-encoded subunits and interaction with prohibitin. Biochem. J. 2004, 383, 491–499. [Google Scholar] [CrossRef]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef]
- Presser, L.D.; Haskett, A.; Waris, G. Hepatitis C virus-induced furin and thrombospondin-1 activate TGF-beta1: role of TGF-beta1 in HCV replication. Virology 2011, 412, 284–296. [Google Scholar] [CrossRef]
- Li, Y.; Boehning, D.F.; Qian, T.; Popov, V.L.; Weinman, S.A. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. Faseb J. 2007, 21, 2474–2485. [Google Scholar] [CrossRef]
- Dionisio, N.; Garcia-Mediavilla, M.V.; Sanchez-Campos, S.; Majano, P.L.; Benedicto, I.; Rosado, J.A.; Salido, G.M.; Gonzalez-Gallego, J. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J. Hepatol. 2009, 50, 872–882. [Google Scholar] [CrossRef]
- Robinson, L.C.; Marchant, J.S. Enhanced Ca2+ leak from ER Ca2+ stores induced by hepatitis C NS5A protein. Biochem. Biophys. Res. Commun. 2008, 368, 593–599. [Google Scholar] [CrossRef]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; De Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef]
- Bergqvist, A.; Sundstrom, S.; Dimberg, L.Y.; Gylfe, E.; Masucci, M.G. The hepatitis C virus core protein modulates T cell responses by inducing spontaneous and altering T-cell receptor-triggered Ca2+ oscillations. J. Biol. Chem. 2003, 278, 18877–18883. [Google Scholar]
- Choi, J.; Forman, H.J.; Ou, J.H.; Lai, M.M.; Seronello, S.; Nandipati, A. Redox modulation of the hepatitis C virus replication complex is calcium dependent. Free Radic. Biol. Med. 2006, 41, 1488–1498. [Google Scholar] [CrossRef]
- Schwer, B.; Ren, S.; Pietschmann, T.; Kartenbeck, J.; Kaehlcke, K.; Bartenschlager, R.; Yen, T.S.; Ott, M. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J. Virol. 2004, 78, 7958–7968. [Google Scholar]
- Suzuki, R.; Sakamoto, S.; Tsutsumi, T.; Rikimaru, A.; Tanaka, K.; Shimoike, T.; Moriishi, K.; Iwasaki, T.; Mizumoto, K.; Matsuura, Y.; et al. Molecular determinants for subcellular localization of hepatitis C virus core protein. J. Virol. 2005, 79, 1271–1281. [Google Scholar]
- Williamson, C.D.; Colberg-Poley, A.M. Access of viral proteins to mitochondria via mitochondria-associated membranes. Rev. Med. Virol. 2009, 19, 147–164. [Google Scholar] [CrossRef]
- McLauchlan, J.; Lemberg, M.K.; Hope, G.; Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. Embo J. 2002, 21, 3980–3988. [Google Scholar] [CrossRef]
- Kang, S.M.; Kim, S.J.; Kim, J.H.; Lee, W.; Kim, G.W.; Lee, K.H.; Choi, K.Y.; Oh, J.W. Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-alpha-mediated apoptosis. Cancer Lett. 2009, 279, 230–237. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar]
- Bureau, C.; Bernad, J.; Chaouche, N.; Orfila, C.; Beraud, M.; Gonindard, C.; Alric, L.; Vinel, J.P.; Pipy, B. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J. Biol. Chem. 2001, 276, 23077–23083. [Google Scholar]
- Thoren, F.; Romero, A.; Lindh, M.; Dahlgren, C.; Hellstrand, K. A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: role of NADPH oxidase-derived oxygen radicals. J. Leukoc. Biol. 2004, 76, 1180–1186. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: A new contributor to HCV-induced oxidative stress. J. Virol. 2009, 83, 12934–12946. [Google Scholar] [CrossRef]
- de Mochel, N.S.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 2005, 10, 1139–1151. [Google Scholar] [CrossRef]
- Weyemi, U.; Dupuy, C. The emerging role of ROS-generating NADPH oxidase NOX4 in DNA-damage responses. Mutat. Res. 2012, 751, 77–81. [Google Scholar] [CrossRef]
- Avadhani, N.G.; Sangar, M.C.; Bansal, S.; Bajpai, P. Bimodal targeting of cytochrome P450s to endoplasmic reticulum and mitochondria: the concept of chimeric signals. FEBS J. 2011, 278, 4218–4229. [Google Scholar] [CrossRef]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef]
- Rigamonti, C.; Mottaran, E.; Reale, E.; Rolla, R.; Cipriani, V.; Capelli, F.; Boldorini, R.; Vidali, M.; Sartori, M.; Albano, E. Moderate alcohol consumption increases oxidative stress in patients with chronic hepatitis C. Hepatology 2003, 38, 42–49. [Google Scholar]
- Hutchinson, S.J.; Bird, S.M.; Goldberg, D.J. Influence of alcohol on the progression of hepatitis C virus infection: a meta-analysis. Clin. Gastroenterol Hepatol. 2005, 3, 1150–1159. [Google Scholar] [CrossRef]
- Nakai, K.; Tanaka, H.; Hanada, K.; Ogata, H.; Suzuki, F.; Kumada, H.; Miyajima, A.; Ishida, S.; Sunouchi, M.; Habano, W.; et al. Decreased expression of cytochromes P450 1A2, 2E1, and 3A4 and drug transporters Na+-taurocholate-cotransporting polypeptide, organic cation transporter 1, and organic anion-transporting peptide-C correlates with the progression of liver fibrosis in chronic hepatitis C patients. Drug Metab. Dispos. 2008, 36, 1786–1793. [Google Scholar] [CrossRef]
- Wen, F.; Abdalla, M.Y.; Aloman, C.; Xiang, J.; Ahmad, I.M.; Walewski, J.; McCormick, M.L.; Brown, K.E.; Branch, A.D.; Spitz, D.R.; et al. Increased prooxidant production and enhanced susceptibility to glutathione depletion in HepG2 cells co-expressing HCV core protein and CYP2E1. J. Med. Virol. 2004, 72, 230–240. [Google Scholar] [CrossRef]
- Otani, K.; Korenaga, M.; Beard, M.R.; Li, K.; Qian, T.; Showalter, L.A.; Singh, A.K.; Wang, T.; Weinman, S.A. Hepatitis C virus core protein, cytochrome P450 2E1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology 2005, 128, 96–107. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Merquiol, E.; Uzi, D.; Mueller, T.; Goldenberg, D.; Nahmias, Y.; Xavier, R.J.; Tirosh, B.; Shibolet, O. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One 2011, 6, e24660. [Google Scholar]
- Chan, S.W.; Egan, P.A. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. Faseb J. 2005, 19, 1510–1512. [Google Scholar]
- Zheng, Y.; Gao, B.; Ye, L.; Kong, L.; Jing, W.; Yang, X.; Wu, Z.; Ye, L. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 2005, 43, 529–536. [Google Scholar]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar]
- Burdette, D.; Olivarez, M.; Waris, G. Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J. Gen. Virol. 2010, 91, 681–690. [Google Scholar]
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J. Biol. Chem. 2011, 286, 8941–8951. [Google Scholar]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- Ghaziani, T.; Shan, Y.; Lambrecht, R.W.; Donohue, S.E.; Pietschmann, T.; Bartenschlager, R.; Bonkovsky, H.L. HCV proteins increase expression of heme oxygenase-1 (HO-1) and decrease expression of Bach1 in human hepatoma cells. J. Hepatol. 2006, 45, 5–12. [Google Scholar] [CrossRef]
- Hou, W.H.; Rossi, L.; Shan, Y.; Zheng, J.Y.; Lambrecht, R.W.; Bonkovsky, H.L. Iron increases HMOX1 and decreases hepatitis C viral expression in HCV-expressing cells. World J. Gastroenterol. 2009, 15, 4499–4510. [Google Scholar] [CrossRef]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. Embo J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Abdalla, M.Y.; Britigan, B.E.; Wen, F.; Icardi, M.; McCormick, M.L.; LaBrecque, D.R.; Voigt, M.; Brown, K.E.; Schmidt, W.N. Down-regulation of heme oxygenase-1 by hepatitis C virus infection in vivo and by the in vitro expression of hepatitis C core protein. J. Infect. Dis. 2004, 190, 1109–1118. [Google Scholar] [CrossRef]
- Wen, F.; Brown, K.E.; Britigan, B.E.; Schmidt, W.N. Hepatitis C core protein inhibits induction of heme oxygenase-1 and sensitizes hepatocytes to cytotoxicity. Cell Biol. Toxicol. 2008, 24, 175–188. [Google Scholar] [CrossRef]
- Tang, W.; Lazaro, C.A.; Campbell, J.S.; Parks, W.T.; Katze, M.G.; Fausto, N. Responses of nontransformed human hepatocytes to conditional expression of full-length hepatitis C virus open reading frame. Am. J. Pathol. 2007, 171, 1831–1846. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, L.; Thiagalingam, A.; Nelkin, B.D.; Casero, R.A., Jr. The identification of a cis-element and a trans-acting factor involved in the response to polyamines and polyamine analogues in the regulation of the human spermidine/spermine N1-acetyltransferase gene transcription. J. Biol. Chem. 1998, 273, 34623–34630. [Google Scholar]
- Smirnova, O.A.; Isaguliants, M.G.; Hyvonen, M.T.; Keinanen, T.A.; Tunitskaya, V.L.; Vepsalainen, J.; Alhonen, L.; Kochetkov, S.N.; Ivanov, A.V. Chemically induced oxidative stress increases polyamine levels by activating the transcription of ornithine decarboxylase and spermidine/spermine-N1-acetyltransferase in human hepatoma HUH7 cells. Biochimie 2012, 94, 1876–1883. [Google Scholar] [CrossRef]
- Casero, R.A.; Pegg, A.E. Polyamine catabolism and disease. Biochem. J. 2009, 421, 323–338. [Google Scholar] [CrossRef]
- Blackham, S.; Baillie, A.; Al-Hababi, F.; Remlinger, K.; You, S.; Hamatake, R.; McGarvey, M.J. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 2010, 84, 5404–5414. [Google Scholar] [CrossRef]
- Walters, K.A.; Syder, A.J.; Lederer, S.L.; Diamond, D.L.; Paeper, B.; Rice, C.M.; Katze, M.G. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog. 2009, 5, e1000269. [Google Scholar]
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J. Biol. Chem. 2008, 283, 33554–33562. [Google Scholar]
- Biswas, M.; Chan, J.Y. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 2010, 244, 16–20. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, L.; Leung, L.; Yen, T.S.; Lee, C.; Chan, J.Y. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. USA 2005, 102, 4120–4125. [Google Scholar]
- Diamond, D.L.; Jacobs, J.M.; Paeper, B.; Proll, S.C.; Gritsenko, M.A.; Carithers, R.L., Jr.; Larson, A.M.; Yeh, M.M.; Camp, D.G., 2nd; Smith, R.D.; et al. Proteomic profiling of human liver biopsies: Hepatitis C virus-induced fibrosis and mitochondrial dysfunction. Hepatology 2007, 46, 649–657. [Google Scholar] [CrossRef]
- Choi, J.; Lee, K.J.; Zheng, Y.; Yamaga, A.K.; Lai, M.M.; Ou, J.H. Reactive oxygen species suppress hepatitis C virus RNA replication in human hepatoma cells. Hepatology 2004, 39, 81–89. [Google Scholar] [CrossRef]
- Seronello, S.; Montanez, J.; Presleigh, K.; Barlow, M.; Park, S.B.; Choi, J. Ethanol and reactive species increase basal sequence heterogeneity of hepatitis C virus and produce variants with reduced susceptibility to antivirals. PLoS One 2011, 6, e27436. [Google Scholar]
- Forns, X.; Purcell, R.H.; Bukh, J. Quasispecies in viral persistence and pathogenesis of hepatitis C virus. Trends Microbiol. 1999, 7, 402–410. [Google Scholar] [CrossRef]
- Martinez, C.; Garcia-Martin, E.; Ladero, J.M.; Herraez, O.; Ortega, L.; Taxonera, C.; Suarez, A.; Diaz-Rubio, M.; Agundez, J.A. GSTT1 and GSTM1 null genotypes may facilitate hepatitis C virus infection becoming chronic. J. Infect. Dis. 2007, 195, 1320–1323. [Google Scholar] [CrossRef]
- Di Bona, D.; Cippitelli, M.; Fionda, C.; Camma, C.; Licata, A.; Santoni, A.; Craxi, A. Oxidative stress inhibits IFN-alpha-induced antiviral gene expression by blocking the JAK-STAT pathway. J. Hepatol. 2006, 45, 271–279. [Google Scholar] [CrossRef]
- Jack, S.C.; Chan, S.W. The role of PERK and GCN2 in basal and hydrogen peroxide-regulated translation from the hepatitis C virus internal ribosome entry site. Virus Genes 2011, 43, 208–214. [Google Scholar] [CrossRef]
- MacCallum, P.R.; Jack, S.C.; Egan, P.A.; McDermott, B.T.; Elliott, R.M.; Chan, S.W. Cap-dependent and hepatitis C virus internal ribosome entry site-mediated translation are modulated by phosphorylation of eIF2alpha under oxidative stress. J. Gen. Virol. 2006, 87, 3251–3262. [Google Scholar] [CrossRef]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death. Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef]
- Bartosch, B. HCV and Liver Carbohydrate Metabolism. In 7th Annual Meeting the New Visby University Network on Hepatitis C, Tartu, Estonia, 22 February 2010; Isaguliants, M.G., Ed.; 2010. [Google Scholar]
- von Hahn, T.; Lindenbach, B.D.; Boullier, A.; Quehenberger, O.; Paulson, M.; Rice, C.M.; McKeating, J.A. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 2006, 43, 932–942. [Google Scholar] [CrossRef]
- Blasig, I.E.; Bellmann, C.; Cording, J.; Del Vecchio, G.; Zwanziger, D.; Huber, O.; Haseloff, R.F. Occludin protein family: oxidative stress and reducing conditions. Antioxid. Redox Signal. 2011, 15, 1195–1219. [Google Scholar] [CrossRef]
- Overgaard, C.E.; Daugherty, B.L.; Mitchell, L.A.; Koval, M. Claudins: control of barrier function and regulation in response to oxidant stress. Antioxid. Redox Signal. 2011, 15, 1179–1193. [Google Scholar] [CrossRef]
- Stewart, B.J.; Doorn, J.A.; Petersen, D.R. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem. Res. Toxicol. 2007, 20, 1111–1119. [Google Scholar] [CrossRef]
- Roohvand, F.; Maillard, P.; Lavergne, J.P.; Boulant, S.; Walic, M.; Andreo, U.; Goueslain, L.; Helle, F.; Mallet, A.; McLauchlan, J.; et al. Initiation of hepatitis C virus infection requires the dynamic microtubule network: role of the viral nucleocapsid protein. J. Biol. Chem. 2009, 284, 13778–13791. [Google Scholar]
- Counihan, N.A.; Rawlinson, S.M.; Lindenbach, B.D. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 2011, 7, e1002302. [Google Scholar] [CrossRef]
- Souza dos Santos, R.M.; de Bem, A.F.; Colpo, E.; Bertoncello, I.; Nogueira, C.W.; Rocha, J.B. Plasmatic vitamin C in nontreated hepatitis C patients is negatively associated with aspartate aminotransferase. Liver Int. 2008, 28, 54–60. [Google Scholar]
- Dolganiuc, A.; Norkina, O.; Kodys, K.; Catalano, D.; Bakis, G.; Marshall, C.; Mandrekar, P.; Szabo, G. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology 2007, 133, 1627–1636. [Google Scholar] [CrossRef]
- Khansari, N.; Shakiba, Y.; Mahmoudi, M. Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2009, 3, 73–80. [Google Scholar] [CrossRef]
- Szabo, G.; Mandrekar, P.; Dolganiuc, A. Innate immune response and hepatic inflammation. Semin. Liver Dis. 2007, 27, 339–350. [Google Scholar] [CrossRef]
- Heydtmann, M. Macrophages in hepatitis B and hepatitis C virus infections. J. Virol. 2009, 83, 2796–2802. [Google Scholar] [CrossRef]
- Polakos, N.K.; Cornejo, J.C.; Murray, D.A.; Wright, K.O.; Treanor, J.J.; Crispe, I.N.; Topham, D.J.; Pierce, R.H. Kupffer cell-dependent hepatitis occurs during influenza infection. Am. J. Pathol. 2006, 168, 1169–1178. [Google Scholar] [CrossRef]
- Knolle, P.A.; Gerken, G. Local control of the immune response in the liver. Immunol. Rev. 2000, 174, 21–34. [Google Scholar] [CrossRef]
- Tamai, T.; Uto, H.; Takami, Y.; Oda, K.; Saishoji, A.; Hashiguchi, M.; Kumagai, K.; Kure, T.; Mawatari, S.; Moriuchi, A.; et al. Serum manganese superoxide dismutase and thioredoxin are potential prognostic markers for hepatitis C virus-related hepatocellular carcinoma. World J. Gastroenterol 2011, 17, 4890–4898. [Google Scholar] [CrossRef]
- Chuma, M.; Hige, S.; Nakanishi, M.; Ogawa, K.; Natsuizaka, M.; Yamamoto, Y.; Asaka, M. 8-Hydroxy-2'-deoxy-guanosine is a risk factor for development of hepatocellular carcinoma in patients with chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2008, 23, 1431–1436. [Google Scholar] [CrossRef]
- Tanaka, H.; Fujita, N.; Sugimoto, R.; Urawa, N.; Horiike, S.; Kobayashi, Y.; Iwasa, M.; Ma, N.; Kawanishi, S.; Watanabe, S.; et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis C. Br. J. Cancer 2008, 98, 580–586. [Google Scholar] [CrossRef]
- Maki, A.; Kono, H.; Gupta, M.; Asakawa, M.; Suzuki, T.; Matsuda, M.; Fujii, H.; Rusyn, I. Predictive power of biomarkers of oxidative stress and inflammation in patients with hepatitis C virus-associated hepatocellular carcinoma. Ann. Surg. Oncol. 2007, 14, 1182–1190. [Google Scholar]
- Ezzikouri, S.; El Feydi, A.E.; Chafik, A.; Afifi, R.; El Kihal, L.; Benazzouz, M.; Hassar, M.; Pineau, P.; Benjelloun, S. Genetic polymorphism in the manganese superoxide dismutase gene is associated with an increased risk for hepatocellular carcinoma in HCV-infected Moroccan patients. Mutat. Res. 2008, 649, 1–6. [Google Scholar] [CrossRef]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Gonzalez, F.J.; Aoyama, T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Invest. 2008, 118, 683–694. [Google Scholar]
- Moriya, K.; Nakagawa, K.; Santa, T.; Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Miyazawa, T.; Ishibashi, K.; Horie, T.; et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001, 61, 4365–4370. [Google Scholar]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Tachibana, K.; Yamasaki, D.; Ishimoto, K.; Doi, T. The Role of PPARs in Cancer. PPAR Res. 2008, 2008, 102737. [Google Scholar]
- Mankouri, J.; Dallas, M.L.; Hughes, M.E.; Griffin, S.D.; Macdonald, A.; Peers, C.; Harris, M. Suppression of a pro-apoptotic K+ channel as a mechanism for hepatitis C virus persistence. Proc. Natl. Acad. Sci. USA 2009, 106, 15903–15908. [Google Scholar]
- Pal, S.; Hartnett, K.A.; Nerbonne, J.M.; Levitan, E.S.; Aizenman, E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J. Neurosci. 2003, 23, 4798–4802. [Google Scholar]
- Severi, T.; Vander Borght, S.; Libbrecht, L.; VanAelst, L.; Nevens, F.; Roskams, T.; Cassiman, D.; Fevery, J.; Verslype, C.; van Pelt, J.F. HBx or HCV core gene expression in HepG2 human liver cells results in a survival benefit against oxidative stress with possible implications for HCC development. Chem. Biol. Interact. 2007, 168, 128–134. [Google Scholar] [CrossRef]
- Tsukiyama-Kohara, K. Role of oxidative stress in hepatocarcinogenesis induced by hepatitis C virus. Int. J. Mol. Sci. 2012, 13, 15271–15278. [Google Scholar] [CrossRef]
- Suzuki, T.; Harashima, H.; Kamiya, H. Effects of base excision repair proteins on mutagenesis by 8-oxo-7,8-dihydroguanine (8-hydroxyguanine) paired with cytosine and adenine. DNA Repair (Amst) 2010, 9, 542–550. [Google Scholar] [CrossRef]
- Poon, R.T.; Ng, I.O.; Lau, C.; Yu, W.C.; Yang, Z.F.; Fan, S.T.; Wong, J. Tumor microvessel density as a predictor of recurrence after resection of hepatocellular carcinoma: A prospective study. J. Clin. Oncol. 2002, 20, 1775–1785. [Google Scholar] [CrossRef]
- Tanigawa, N.; Lu, C.; Mitsui, T.; Miura, S. Quantitation of sinusoid-like vessels in hepatocellular carcinoma: its clinical and prognostic significance. Hepatology 1997, 26, 1216–1223. [Google Scholar]
- Waris, G.; Siddiqui, A. Hepatitis C virus stimulates the expression of cyclooxygenase-2 via oxidative stress: Role of prostaglandin E2 in RNA replication. J. Virol. 2005, 79, 9725–9734. [Google Scholar] [CrossRef]
- Jahan, S.; Khaliq, S.; Ijaz, B.; Ahmad, W.; Hassan, S. Role of HCV Core gene of genotype 1a and 3a and host gene Cox-2 in HCV-induced pathogenesis. Virol. J. 2011, 8, 155. [Google Scholar] [CrossRef]
- Rahman, M.A.; Kohno, H.; Nagasue, N. COX-2 - a target for preventing hepatic carcinoma? Expert Opin. Ther. Targets 2002, 6, 483–490. [Google Scholar] [CrossRef]
- Chiarugi, V.; Magnelli, L.; Gallo, O. Cox-2, iNOS and p53 as play-makers of tumor angiogenesis (review). Int. J. Mol. Med. 1998, 2, 715–719. [Google Scholar]
- Tang, T.C.; Poon, R.T.; Lau, C.P.; Xie, D.; Fan, S.T. Tumor cyclooxygenase-2 levels correlate with tumor invasiveness in human hepatocellular carcinoma. World J. Gastroenterol. 2005, 11, 1896–1902. [Google Scholar]
- Sumitani, K.; Kamijo, R.; Toyoshima, T.; Nakanishi, Y.; Takizawa, K.; Hatori, M.; Nagumo, M. Specific inhibition of cyclooxygenase-2 results in inhibition of proliferation of oral cancer cell lines via suppression of prostaglandin E2 production. J. Oral Pathol. Med. 2001, 30, 41–47. [Google Scholar]
- Ripoli, M.; D'Aprile, A.; Quarato, G.; Sarasin-Filipowicz, M.; Gouttenoire, J.; Scrima, R.; Cela, O.; Boffoli, D.; Heim, M.H.; Moradpour, D.; et al. Hepatitis C virus-inked mitochondrial dysfunction promotes hypoxia-inducible factor 1 alpha-mediated glycolytic adaptation. J. Virol. 2010, 84, 647–660. [Google Scholar]
- Shlomai, A.; Rechtman, M.M.; Burdelova, E.O.; Zilberberg, A.; Hoffman, S.; Solar, I.; Fishman, S.; Halpern, Z.; Sklan, E.H. The metabolic regulator PGC-1alpha links hepatitis C virus infection to hepatic insulin resistance. J. Hepatol. 2012. [Google Scholar]
- Nagy, M.A. HIF-1 is the Commander of Gateways to Cancer. J. Cancer Sci. Ther. 2011, 3, 35–40. [Google Scholar]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar]
- Mitsuyoshi, H.; Itoh, Y.; Sumida, Y.; Minami, M.; Yasui, K.; Nakashima, T.; Okanoue, T. Evidence of oxidative stress as a cofactor in the development of insulin resistance in patients with chronic hepatitis C. Hepatol. Res. 2008, 38, 348–353. [Google Scholar] [CrossRef]
- Cotler, S.J.; Kallwitz, E.; TenCate, V.; Bhushan, A.; Berkes, J.; Benedetti, E.; Layden-Almer, J.; Layden, T.J.; Valyi-Nagy, T.; Guzman, G. Diabetes and hepatic oxidative damage are associated with hepatitis C progression after liver transplantation. Transplantation 2007, 84, 587–591. [Google Scholar] [CrossRef]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 2010, 138, 2509–2518. [Google Scholar]
- Bataller, R.; Lemon, S.M. Fueling fibrosis in chronic hepatitis C. Proc. Natl. Acad. Sci. USA 2012, 109, 14293–14294. [Google Scholar] [CrossRef]
- Ray, S.; Broor, S.L.; Vaishnav, Y.; Sarkar, C.; Girish, R.; Dar, L.; Seth, P.; Broor, S. Transforming growth factor beta in hepatitis C virus infection: in vivo and in vitro findings. J. Gastroenterol. Hepatol. 2003, 18, 393–403. [Google Scholar] [CrossRef]
- Tsushima, H.; Kawata, S.; Tamura, S.; Ito, N.; Shirai, Y.; Kiso, S.; Doi, Y.; Yamada, A.; Oshikawa, O.; Matsuzawa, Y. Reduced plasma transforming growth factor-beta1 levels in patients with chronic hepatitis C after interferon-alpha therapy: association with regression of hepatic fibrosis. J. Hepatol. 1999, 30, 1–7. [Google Scholar]
- Urtasun, R.; Lopategi, A.; George, J.; Leung, T.M.; Lu, Y.; Wang, X.; Ge, X.; Fiel, M.I.; Nieto, N. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin alpha(V)beta(3) engagement and PI3K/pAkt/NFkappaB signaling. Hepatology 2012, 55, 594–608. [Google Scholar] [CrossRef]
- Gieseler, R.K.; Marquitan, G.; Schlattjan, M.; Sowa, J.P.; Bechmann, L.P.; Timm, J.; Roggendorf, M.; Gerken, G.; Friedman, S.L.; Canbay, A. Hepatocyte apoptotic bodies encasing nonstructural HCV proteins amplify hepatic stellate cell activation: implications for chronic hepatitis C. J. Viral. Hepat. 2011, 18, 760–767. [Google Scholar] [CrossRef]
- Wu, C.F.; Lin, Y.L.; Huang, Y.T. Hepatitis C virus core protein stimulates fibrogenesis in hepatic stellate cells involving the obese receptor. J Cell Biochem 2012. [Google Scholar]
- Mormone, E.; Lu, Y.; Ge, X.; Fiel, M.I.; Nieto, N. Fibromodulin, an oxidative stress-sensitive proteoglycan, regulates the fibrogenic response to liver injury in mice. Gastroenterology 2012, 142, 612–621 e615. [Google Scholar] [CrossRef]
- Novo, E.; Busletta, C.; Bonzo, L.V.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol. 2011, 54, 964–974. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Parise, E.R.; Catarino, R.M.; Lanzoni, V.; Leite-Mor, M.M.; Simon, K.A.; Junqueira, V.B. Insulin resistance and not steatosis is associated with modifications in oxidative stress markers in chronic hepatitis C, non-3 genotype. Free Radic. Res. 2009, 43, 1187–1194. [Google Scholar] [CrossRef]
- Vidali, M.; Tripodi, M.F.; Ivaldi, A.; Zampino, R.; Occhino, G.; Restivo, L.; Sutti, S.; Marrone, A.; Ruggiero, G.; Albano, E.; et al. Interplay between oxidative stress and hepatic steatosis in the progression of chronic hepatitis C. J. Hepatol. 2008, 48, 399–406. [Google Scholar] [CrossRef]
- Tachi, Y.; Katano, Y.; Honda, T.; Hayashi, K.; Ishigami, M.; Itoh, A.; Hirooka, Y.; Nakano, I.; Samejima, Y.; Goto, H. Impact of amino acid substitutions in the hepatitis C virus genotype 1b core region on liver steatosis and hepatic oxidative stress in patients with chronic hepatitis C. Liver Int. 2010, 30, 554–559. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Rhee, J.; Inoue, Y.; Yoon, J.C.; Puigserver, P.; Fan, M.; Gonzalez, F.J.; Spiegelman, B.M. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc. Natl Acad. Sci. USA 2003, 100, 4012–4017. [Google Scholar]
- Kumashiro, N.; Tamura, Y.; Uchida, T.; Ogihara, T.; Fujitani, Y.; Hirose, T.; Mochizuki, H.; Kawamori, R.; Watada, H. Impact of oxidative stress and peroxisome proliferator-activated receptor gamma coactivator-1alpha in hepatic insulin resistance. Diabetes 2008, 57, 2083–2091. [Google Scholar] [CrossRef]
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130. [Google Scholar] [CrossRef]
- Weinreb, O.; Amit, T.; Mandel, S.; Kupershmidt, L.; Youdim, M.B. Neuroprotective multifunctional iron chelators: From redox-sensitive process to novel therapeutic opportunities. Antioxid. Redox Signal. 2010, 13, 919–949. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef]
- Andrews, N.C. Iron homeostasis: insights from genetics and animal models. Nat. Rev. Genet. 2000, 1, 208–217. [Google Scholar]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of Mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef]
- Fiel, M.I.; Schiano, T.D.; Guido, M.; Thung, S.N.; Lindsay, K.L.; Davis, G.L.; Lewis, J.H.; Seeff, L.B.; Bodenheimer, H.C., Jr. Increased hepatic iron deposition resulting from treatment of chronic hepatitis C with ribavirin. Am. J. Clin. Pathol. 2000, 113, 35–39. [Google Scholar] [CrossRef]
- Piperno, A.; Vergani, A.; Malosio, I.; Parma, L.; Fossati, L.; Ricci, A.; Bovo, G.; Boari, G.; Mancia, G. Hepatic iron overload in patients with chronic viral hepatitis: role of HFE gene mutations. Hepatology 1998, 28, 1105–1109. [Google Scholar] [CrossRef]
- Bonkovsky, H.L.; Troy, N.; McNeal, K.; Banner, B.F.; Sharma, A.; Obando, J.; Mehta, S.; Koff, R.S.; Liu, Q.; et al. Iron and HFE or TfR1 mutations as comorbid factors for development and progression of chronic hepatitis C. J. Hepatol. 2002, 37, 848–854. [Google Scholar] [CrossRef]
- Tung, B.Y.; Emond, M.J.; Bronner, M.P.; Raaka, S.D.; Cotler, S.J.; Kowdley, K.V. Hepatitis C, iron status, and disease severity: relationship with HFE mutations. Gastroenterology 2003, 124, 318–326. [Google Scholar]
- Erhardt, A.; Maschner-Olberg, A.; Mellenthin, C.; Kappert, G.; Adams, O.; Donner, A.; Willers, R.; Niederau, C.; Haussinger, D. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J. Hepatol. 2003, 38, 335–342. [Google Scholar]
- Sartori, M.; Andorno, S.; Pagliarulo, M.; Rigamonti, C.; Bozzola, C.; Pergolini, P.; Rolla, R.; Suno, A.; Boldorini, R.; Bellomo, G.; et al. Heterozygous beta-globin gene mutations as a risk factor for iron accumulation and liver fibrosis in chronic hepatitis C. Gut 2007, 56, 693–698. [Google Scholar] [CrossRef]
- Valenti, L.; Pulixi, E.A.; Arosio, P.; Cremonesi, L.; Biasiotto, G.; Dongiovanni, P.; Maggioni, M.; Fargion, S.; Fracanzani, A.L. Relative contribution of iron genes, dysmetabolism and hepatitis C virus (HCV) in the pathogenesis of altered iron regulation in HCV chronic hepatitis. Haematologica 2007, 92, 1037–1042. [Google Scholar] [CrossRef]
- Mifuji, R.; Kobayashi, Y.; Ma, N.; Qiang, Q.L.; Urawa, N.; Horiike, S.; Iwasa, M.; Kaito, M.; Malavasi, F.; Adachi, Y. Role of transferrin receptor 2 in hepatic accumulation of iron in patients with chronic hepatitis C. J. Gastroenterol. Hepatol. 2006, 21, 144–151. [Google Scholar] [CrossRef]
- Saito, H.; Fujimoto, Y.; Ohtake, T.; Suzuki, Y.; Sakurai, S.; Hosoki, Y.; Ikuta, K.; Torimoto, Y.; Kohgo, Y. Up-regulation of transferrin receptor 1 in chronic hepatitis C: Implication in excess hepatic iron accumulation. Hepatol. Res. 2005, 31, 203–210. [Google Scholar] [CrossRef]
- Girelli, D.; Pasino, M.; Goodnough, J.B.; Nemeth, E.; Guido, M.; Castagna, A.; Busti, F.; Campostrini, N.; Martinelli, N.; Vantini, I.; et al. Reduced serum hepcidin levels in patients with chronic hepatitis C. J. Hepatol. 2009, 51, 845–852. [Google Scholar] [CrossRef]
- Fillebeen, C.; Muckenthaler, M.; Andriopoulos, B.; Bisaillon, M.; Mounir, Z.; Hentze, M.W.; Koromilas, A.E.; Pantopoulos, K. Expression of the subgenomic hepatitis C virus replicon alters iron homeostasis in Huh7 cells. J. Hepatol. 2007, 47, 12–22. [Google Scholar] [CrossRef]
- Sebastiani, G.; Vario, A.; Ferrari, A.; Pistis, R.; Noventa, F.; Alberti, A. Hepatic iron, liver steatosis and viral genotypes in patients with chronic hepatitis C. J. Viral. Hepat. 2006, 13, 199–205. [Google Scholar] [CrossRef]
- Aoki, C.A.; Rossaro, L.; Ramsamooj, R.; Brandhagen, D.; Burritt, M.F.; Bowlus, C.L. Liver hepcidin mRNA correlates with iron stores, but not inflammation, in patients with chronic hepatitis C. J. Clin. Gastroenterol. 2005, 39, 71–74. [Google Scholar]
- Fujita, N.; Sugimoto, R.; Takeo, M.; Urawa, N.; Mifuji, R.; Tanaka, H.; Kobayashi, Y.; Iwasa, M.; Watanabe, S.; Adachi, Y.; et al. Hepcidin expression in the liver: relatively low level in patients with chronic hepatitis C. Mol. Med. 2007, 13, 97–104. [Google Scholar]
- Miura, K.; Taura, K.; Kodama, Y.; Schnabl, B.; Brenner, D.A. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 2008, 48, 1420–1429. [Google Scholar] [CrossRef]
- Kakizaki, S.; Takagi, H.; Horiguchi, N.; Toyoda, M.; Takayama, H.; Nagamine, T.; Mori, M.; Kato, N. Iron enhances hepatitis C virus replication in cultured human hepatocytes. Liver 2000, 20, 125–128. [Google Scholar]
- Fillebeen, C.; Rivas-Estilla, A.M.; Bisaillon, M.; Ponka, P.; Muckenthaler, M.; Hentze, M.W.; Koromilas, A.E.; Pantopoulos, K. Iron inactivates the RNA polymerase NS5B and suppresses subgenomic replication of hepatitis C Virus. J. Biol. Chem. 2005, 280, 9049–9057. [Google Scholar]
- Fillebeen, C.; Pantopoulos, K. Iron inhibits replication of infectious hepatitis C virus in permissive Huh7.5.1 cells. J. Hepatol. 2010, 53, 995–999. [Google Scholar] [CrossRef]
- Lehmann, E.; El-Tantawy, W.H.; Ocker, M.; Bartenschlager, R.; Lohmann, V.; Hashemolhosseini, S.; Tiegs, G.; Sass, G. The heme oxygenase 1 product biliverdin interferes with hepatitis C virus replication by increasing antiviral interferon response. Hepatology 2010, 51, 398–404. [Google Scholar] [CrossRef]
- Cho, H.; Lee, H.C.; Jang, S.K.; Kim, Y.K. Iron increases translation initiation directed by internal ribosome entry site of hepatitis C virus. Virus Genes 2008, 37, 154–160. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; An, D.; Diao, H.; Xu, W.; He, X.; Sun, R.; Wei, L.; Li, L. Regulation of hepatitis C virus translation initiation by iron: Role of eIF3 and La protein. Virus Res. 2012, 167, 302–309. [Google Scholar] [CrossRef]
- Zhu, Z.; Wilson, A.T.; Luxon, B.A.; Brown, K.E.; Mathahs, M.M.; Bandyopadhyay, S.; McCaffrey, A.P.; Schmidt, W.N. Biliverdin inhibits hepatitis C virus nonstructural 3/4A protease activity: mechanism for the antiviral effects of heme oxygenase? Hepatology 2010, 52, 1897–1905. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Coelmont, L.; Vliegen, I.; Van hemel, J.; Vandenkerckhove, J.; Peys, E.; Sas, B.; De Clercq, E.; Neyts, J. Hemin potentiates the anti-hepatitis C virus activity of the antimalarial drug artemisinin. Biochem. Biophys. Res. Commun. 2006, 348, 139–144. [Google Scholar] [CrossRef]
- Martinelli, A.L.; Ramalho, L.N.; Zucoloto, S. Hepatic stellate cells in hepatitis C patients: Relationship with liver iron deposits and severity of liver disease. J. Gastroenterol. Hepatol. 2004, 19, 91–98. [Google Scholar] [CrossRef]
- Casaril, M.; Stanzial, A.M.; Tognella, P.; Pantalena, M.; Capra, F.; Colombari, R.; Corrocher, R. Role of iron load on fibrogenesis in chronic hepatitis C. Hepatogastroenterology 2000, 47, 220–225. [Google Scholar]
- Nishina, S.; Korenaga, M.; Hidaka, I.; Shinozaki, A.; Sakai, A.; Gondo, T.; Tabuchi, M.; Kishi, F.; Hino, K. Hepatitis C virus protein and iron overload induce hepatic steatosis through the unfolded protein response in mice. Liver Int. 2010, 30, 683–692. [Google Scholar] [CrossRef]
- Furutani, T.; Hino, K.; Okuda, M.; Gondo, T.; Nishina, S.; Kitase, A.; Korenaga, M.; Xiao, S.Y.; Weinman, S.A.; Lemon, S.M.; et al. Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 2006, 130, 2087–2098. [Google Scholar]
- Sumida, Y.; Kanemasa, K.; Fukumoto, K.; Yoshida, N.; Sakai, K. Hepatic iron accumulation may be associated with insulin resistance in patients with chronic hepatitis C. Hepatol. Res. 2007, 37, 932–940. [Google Scholar]
- D'Souza, R.F.; Feakins, R.; Mears, L.; Sabin, C.A.; Foster, G.R. Relationship between serum ferritin, hepatic iron staining, diabetes mellitus and fibrosis progression in patients with chronic hepatitis C. Aliment. Pharmacol. Ther. 2005, 21, 519–524. [Google Scholar] [CrossRef]
- Darwich, E.; To-Figueras, J.; Molina-Lopez, R.A.; Deulofeu, R.; Olbina, G.; Westerman, M.; Sanchez-Tapias, J.M.; Munoz-Santos, C.; Herrero, C. Increased serum hepcidin levels in patients with porphyria cutanea tarda. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 68–74. [Google Scholar] [CrossRef]
- Hofer, H.; Osterreicher, C.; Jessner, W.; Penz, M.; Steindl-Munda, P.; Wrba, F.; Ferenci, P. Hepatic iron concentration does not predict response to standard and pegylated-IFN/ribavirin therapy in patients with chronic hepatitis C. J. Hepatol. 2004, 40, 1018–1022. [Google Scholar] [CrossRef]
- Pianko, S.; McHutchison, J.G.; Gordon, S.C.; Heaton, S.; Goodman, Z.D.; Patel, K.; Cortese, C.M.; Brunt, E.M.; Bacon, B.R.; Blatt, L.M. Hepatic iron concentration does not influence response to therapy with interferon plus ribavirin in chronic HCV infection. J. Interferon Cytokine Res. 2002, 22, 483–489. [Google Scholar] [CrossRef]
- Carlo, C.; Daniela, P.; Giancarlo, C. Iron depletion and response to interferon in chronic hepatitis C. Hepatogastroenterology 2003, 50, 1467–1471. [Google Scholar]
- Fargion, S.; Fracanzani, A.L.; Sampietro, M.; Molteni, V.; Boldorini, R.; Mattioli, M.; Cesana, B.; Lunghi, G.; Piperno, A.; Valsecchi, C.; et al. Liver iron influences the response to interferon alpha therapy in chronic hepatitis C. Eur. J. Gastroenterol. Hepatol. 1997, 9, 497–503. [Google Scholar] [CrossRef]
- Fujita, N.; Sugimoto, R.; Urawa, N.; Tanaka, H.; Konishi, M.; Kobayashi, Y.; Iwasa, M.; Watanabe, S.; Kaito, M. Influence of phlebotomy on iron-related gene expression levels in the livers of patients with chronic hepatitis C. J. Gastroenterol. 2007, 42, 326–327. [Google Scholar] [CrossRef]
- Yano, M.; Hayashi, H.; Yoshioka, K.; Kohgo, Y.; Saito, H.; Niitsu, Y.; Kato, J.; Iino, S.; Yotsuyanagi, H.; Kobayashi, Y.; et al. A significant reduction in serum alanine aminotransferase levels after 3-month iron reduction therapy for chronic hepatitis C: A multicenter, prospective, randomized, controlled trial in Japan. J. Gastroenterol. 2004, 39, 570–574. [Google Scholar]
- Kaito, M.; Iwasa, M.; Kobayashi, Y.; Fujita, N.; Tanaka, H.; Gabazza, E.C.; Adachi, Y.; Kojima, Y.; Nakagawa, N.; Watanabe, S. Iron reduction therapy by phlebotomy reduces lipid peroxidation and oxidative stress in patients with chronic hepatitis C. J. Gastroenterol. 2006, 41, 921–922. [Google Scholar] [CrossRef]
- Guyader, D.; Boucher, E.; Andre, P.; Even, C.; Cottereau, J.; Bianchi, A.; Gasser, P.; Mendler, M.H.; Deugnier, Y.; Brissot, P. A pilot study of iron depletion as adjuvant therapy in chronic hepatitis C patients not responding to interferon. Am J Gastroenterol 1999, 94, 1696–1698. [Google Scholar]
- Herrera, J.L. Iron depletion is not effective in inducing a virologic response in patients with chronic hepatitis C who failed to respond to interferon therapy. Am. J. Gastroenterol. 1999, 94, 3571–3575. [Google Scholar]
- Di Bisceglie, A.M.; Bonkovsky, H.L.; Chopra, S.; Flamm, S.; Reddy, R.K.; Grace, N.; Killenberg, P.; Hunt, C.; Tamburro, C.; Tavill, A.S.; et al. Iron reduction as an adjuvant to interferon therapy in patients with chronic hepatitis C who have previously not responded to interferon: A multicenter, prospective, randomized, controlled trial. Hepatology 2000, 32, 135–138. [Google Scholar]
- Iwasa, M.; Kaito, M.; Ikoma, J.; Kobayashi, Y.; Tanaka, Y.; Higuchi, K.; Takeuchi, K.; Iwata, K.; Watanabe, S.; Adachi, Y. Dietary iron restriction improves aminotransferase levels in chronic hepatitis C patients. Hepatogastroenterology 2002, 49, 529–531. [Google Scholar]
- Fontana, R.J.; Israel, J.; LeClair, P.; Banner, B.F.; Tortorelli, K.; Grace, N.; Levine, R.A.; Fiarman, G.; Thiim, M.; Tavill, A.S.; et al. Iron reduction before and during interferon therapy of chronic hepatitis C: Results of a multicenter, randomized, controlled trial. Hepatology 2000, 31, 730–736. [Google Scholar]
- Fujita, N.; Horiike, S.; Sugimoto, R.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Hasegawa, K.; Ma, N.; Kawanishi, S.; Adachi, Y.; et al. Hepatic oxidative DNA damage correlates with iron overload in chronic hepatitis C patients. Free Radic. Biol. Med. 2007, 42, 353–362. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and Oxidative Stress in the Liver. Viruses 2013, 5, 439-469. https://doi.org/10.3390/v5020439
Ivanov AV, Bartosch B, Smirnova OA, Isaguliants MG, Kochetkov SN. HCV and Oxidative Stress in the Liver. Viruses. 2013; 5(2):439-469. https://doi.org/10.3390/v5020439
Chicago/Turabian StyleIvanov, Alexander V., Birke Bartosch, Olga A. Smirnova, Maria G. Isaguliants, and Sergey N. Kochetkov. 2013. "HCV and Oxidative Stress in the Liver" Viruses 5, no. 2: 439-469. https://doi.org/10.3390/v5020439